

Abstract
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are part of a disease spectrum associated with TDP-43 pathology. Strong evidence supporting this is the existence of kindreds with family members affected by FTD, ALS or mixed features of FTD and ALS, referred to as FTD-MND. Some of these families have linkage to chromosome 9, with hexanucleotide expansion mutation in a noncoding region of C9ORF72. Discovery of the mutation defines c9FTD/ALS. Prior to discovery of mutations in C9ORF72, it was assumed that TDP-43 pathology in c9FTD/ALS was uniform. In this study, we examined the neuropathology and clinical features of 20 cases of c9FTD/ALS from a brain bank for neurodegenerative disorders. Included are six patients clinically diagnosed with ALS, eight FTD, one FTD-MND and four Alzheimer type dementia. Clinical information was unavailable for one patient. Pathologically, the cases all had TDP-43 pathology, but there were three major pathologic groups: ALS, FTLD-MND and FTLD-TDP. The ALS cases were morphologically similar to typical sporadic ALS with almost no extramotor TDP-43 pathology; all had oligodendroglial cytoplasmic inclusions. The FTLD-MND showed predominantly Mackenzie Type 3 TDP-43 pathology, and all had ALS-like pathology in motor neurons, but more extensive extramotor pathology, with oligodendroglial cytoplasmic inclusions and infrequent hippocampal sclerosis. The FTLD-TDP cases had several features similar to FTLD-TDP due to mutations in the gene for progranulin, including Mackenzie Type 1 TDP-43 pathology with neuronal intranuclear inclusions and hippocampal sclerosis. FTLD-TDP patients were older and some were thought to have Alzheimer type dementia. In addition to the FTD and ALS clinical presentations, the present study shows that c9FTD/ALS can have other presentations, possibly related to age of onset and presence of hippocampal sclerosis. Moreover, there is pathologic heterogeneity not only between ALS and FTLD, but within the FTLD group. Further studies are needed to address the molecular mechanism of clinical and pathological heterogeneity of c9FTD/ALS due to mutations in C9ORF72.
Introduction
Frontotemporal lobar degeneration (FTLD) is a term for a class of neurodegenerative disorders associated with focal cortical degeneration often with a predilection for frontal and temporal multimodal association cortices with variable involvement of parietal lobe, basal ganglia and motor neurons [9, 39]. There are several major clinical presentations of FTLD, including behavioral variant frontotemporal dementia (bvFTD), progressive nonfluent aphasia, semantic dementia and corticobasal syndrome [22], as well as FTD with motor neuron disease (FTD-MND) [40, 51]. Pathologically, FTLD can be classified according to the major protein in neuronal and glial inclusion bodies – tau (FTLD-tau), TDP-43 (FTLD-TDP) and FUS (FTLD-FUS), being the most common subtypes [48].
Attempts to subtype FTLD-TDP have produced several classification schemes [9, 42, 44, 61] based upon variable methods of assessing pathology and variable requirements of anatomical regions than need to be included. Sampathu and colleagues defined FTLD-TDP subtypes using a specific collection of monoclonal antibodies to TDP-43 that had variable reactivity with neuronal cytoplasmic inclusions (NCI) and dystrophic neurites (DN) with respect to cortical laminae [61]; they did not link the classification to clinical phenotype. Mackenzie and colleagues defined subtypes of FTLD-TDP with ubiquitin immunohistochemistry based upon distribution and density of NCI and DN in cerebral cortex and hippocampus, especially the dentate fascia, and linked subtypes to specific clinical syndromes [42]. Recently, these two groups proposed a harmonization between the two classification schemes by introducing yet a third scheme that was different from both previous schemes [47], which was based on assessment of undefined cortical regions, lacked information on operational methodology and did not include TDP-43 associated with motor neuron disease and other disorders, such as Alzheimer's disease and hippocampal sclerosis [1]. Given these deficiencies in the “harmonization,” the present report uses the Mackenzie classification scheme, since we have been able to validate its link to specific clinical syndromes, genetic basis of disease, and since we were able to identify distinct patterns of neuronal vulnerability to TDP-43 pathology not only in cortical regions, but also subcortical regions that differed between the TDP-43 pathologic subtypes [28].
TDP-43 pathology is also characteristic of sporadic motor neuron disease, including disease affecting both upper and lower motor neurons (i.e. amyotrophic lateral sclerosis (ALS) [2, 54]), as well as variants affecting primarily lower motor neurons (i.e. progressive muscular atrophy (PMA) [19]) or primarily upper motor neurons (i.e. primary lateral sclerosis (PLS) [13]). TDP-43 pathology is present in a subset of familial motor neuron diseases due to mutations in TARDBP gene [30, 60, 63], but not in other genetic causes of motor neuron disease [45]. In addition to NCI and DN, TDP-43 inclusions in oligodendroglia are reported in ALS [2, 45, 53, 64, 70]. TDP-43 inclusions have been reported in capillary astrocytic end-feet in FTLD-TDP [38], but not in ALS.
There is overlap in TDP-43 pathology in ALS with that found in FTLD-TDP suggesting that they form a disease spectrum [18, 33]. While most patients with mutations in TARDBP present with ALS, there are some with FTD with or without motor neuron disease [21, 34]. Additional evidence of an FTD-ALS spectrum has come from families with members presenting with ALS, FTD, or FTD-MND. Linkage analysis of these families identified a locus on the short arm of chromosome 9 [3, 4, 20, 36, 41, 50, 65, 66]. The same region was identified in genome-wide association studies in large ALS cohorts [35, 62, 67]. Recently, the genetic basis for the chromosome 9 association of FTD and ALS was discovered to be an expansion of a non-coding GGGGCC hexanucleotide repeat in the gene C9ORF72 [10, 59]. Traditional cloning methods were unable to identify the expansion owing to the size of the expanded allele. Southern blot analysis was needed to identify the expanded alleles, which were not only found in affected individuals in multiplex families, but also identified in some affected individuals with no family history of FTD or ALS [10]. A repeat-primed polymerase chain reaction method has been developed to screen for mutations in C9ORF72, but it cannot accurately quantify the number of repeats. The recommended term to refer to FTD and ALS due to mutations in C9ORF72, c9FTD/ALS, will be used in this report [10]
Information about pathology in c9FTD/ALS is limited, and all available studies are based upon linkage studies with their inherent case selection bias. For example, families with both ALS and FTD would tend to be selected for linkage, while families with other types of neurologic phenotypes, such as Alzheimer type dementia, would not be as likely to be selected for linkage studies. Descriptive data have been reported on severity and distribution of neuronal degeneration, type of TDP-43 pathology and other features, such as involvement of motor neurons and white matter, but results are highly variable between the reports (see Supplementary Table) [4, 20, 36, 41, 49, 50, 56, 68]. All reported cases had TDP-43 pathology and most fit with MacKenzie Type 3 [42], with predominance of NCI over DN and no neuronal intranuclear inclusions (NII). Recently, it has been noted that most cases with chromosome 9-linked FTLD-TDP also have ubiquitin-positive, TDP-43-negative NCI in neurons of the cerebellar internal granular cell layer [4]. This type of cerebellar pathology was first noted in a detailed neuroanatomical investigation of the distribution of ubiquitin and TDP-43 pathology in a series of Finnish cases of FTLD [57]. In that study, ubiquitin-positive, TDP-43-negative inclusions were detected in a subset of FTLD-TDP cases, some of which were later included in the Finnish case series with C9ORF72 mutations [59]. In a subset of cases from Canada, cerebellar granular NCI were also noted in c9FTD/ALS [10]. The present report confirms this finding for the first time in c9FTD/ALS cases from Mayo Clinic. In addition, we present comprehensive pathologic data on a series of 20 cases of c9FTD/ALS ascertained from a single source, the brain bank for neurodegenerative disorders at Mayo Clinic Jacksonville. Neuropathology was assessed in these 20 cases with semi-quantitative methods for a range of parameters, including cortical and subcortical degeneration, motor neuron pathology and distribution and type of TDP-43 pathology. We find that c9FTD/ALS due to C9ORF72 hexanucleotide repeat expansion is clinically and pathologically heterogeneous, and suggest that proposed harmonization of TDP-43 subtypes with respect to genetic associations [47] may have been premature.
Materials and Methods
Case material
All cases from the brain bank for neurodegenerative disorders at Mayo Clinic Jacksonville with frozen tissue for DNA extraction, TDP-43 pathology and a neuropathologic diagnosis of FTLD-TDP or ALS were screened for mutations in C9ORF72 using a repeat-primed polymerase chain reaction method to detect expansions of the GGGGCC hexanucleotide [10]. Cases included in this report are limited to those in which the pathologic evaluation was performed in a standardized manner by a single neuropathologist (DWD) and for which paraffin blocks were available for additional histopathologic studies. Of the 102 cases (74 FTLD-TDP and 28 ALS) screened for mutations in C9ORF72, 20 cases had mutations and fit our inclusion criteria. The cases were from several sources – the State of Florida Alzheimer's Disease Initiative (n=12); ALS Center at Mayo Clinic Florida (n=3); the Memory Disorder Clinic or Movement Disorders Clinic at Mayo Clinic Florida (n=3); and CurePSP l Society of Progressive Supranuclear Palsy brain bank (n=2). A subset of FTLD and ALS cases that were determined to lack c9ORF72 mutations were included for comparison purposes, including 13 cases of FTLD-TDP and 5 cases of ALS. Including these cases, 31 cases (13 FTLD, 18 ALS) were screened for ubiquitin-positive inclusions in the cerebellar granule cell layer.
Medical records were available for review in all but one of the cases. The quality of the medical records with respect to completeness and longitudinal information was judged to be good-to-excellent in 12 of the 19 cases, including all patients followed at Mayo Clinic in Jacksonville, FL, and at the Wien Center in Miami Beach, FL. Demographics of the cases are summarized in Table 1. The clinical information abstracted from the medical records is shown for the 19 cases with medical records in Table 2 and include age of onset, age of death, disease duration, and clinical diagnosis. Clinical symptoms described by the subject or caregiver and neurologic signs reported on physical examinations were recorded for each patient. Other pertinent clinical features were recorded, such as presence of motor neuron disease and its type, presence of extrapyramidal signs, psychiatric disturbances (including depression), dementia, memory impairment, focal cortical signs, and frontal-behavioral symptoms. This was not a prospective, longitudinal study with standardized clinical measures from a single center. The clinical data were retrospective and not complete. The diagnosis of dementia was based on the clinical judgment of the physician of record. The diagnosis of motor neuron disease in all ALS cases was confirmed with electrodiagnostic tests, but these tests were not available on patients with FTD. Motor neuron disease was subtyped based upon distribution of clinical signs and symptoms – bulbar if the predominant or only features included dysphagia, dysarthria or hypophonia not related to parkinsonism; presence of facial and tongue muscle atrophy and fasciculations was recorded in some cases. Spinal motor neuron disease cases had muscle weakness with variable atrophy and fasciculations in limb or trunk muscles, but minimal or no bulbar signs. Spinobulbar motor neuron disease indicated involvement of both bulbar muscles and limb/trunk muscles. Long tract signs were recorded and were present in all cases meeting clinical criteria for ALS [8]. Extrapyramidal signs included tremors, bradykinesia, rigidity, gait impairment, and falls. Psychiatric disorders included depression, delusions, hallucinations and paranoia. Dementia diagnoses included Alzheimer's disease, dementia with Lewy bodies, normal pressure hydrocephalus, Pick's disease and FTD. Focal cortical signs included apraxia and significant language impairment or aphasia. Frontal-behavioral signs included apathy, personality change, poor judgment, inappropriate behavior, perseveration, and disinhibition.
Table 1. Summary of Clinical and Pathologic Features.
| ALS N=5 | FTLD-MND N=8 | FTLD-TDP N=7 | ||
|---|---|---|---|---|
| Demographic | ||||
| Sex, count | 1M: 4F | 4M:4F | 6M:1F | p=0.073 |
| Age of onset (years), mean ± SD | 52 ± 6.4 | 57± 6.0 | 71 ± 5.9 * | p<0.001 |
| Disease duration (years) | 2.0 (1.0, 4.5) | 4.5 (3.0, 6.5) | 5.0 (4.0, 7.7) | p=0.201 |
| Age at death (years), mean ± SD | 59 ± 11 | 62± 5.6 | 77 ± 6.1 * | p<0.001 |
| Family history (dementia, parkinsonism or ALS),%positive | 60% | 50% | 71% | p=0.700 |
|
| ||||
| Pathology | ||||
| Macroscopic findings | ||||
| Brain weight (grams), mean ± SD | 1290 ± 206 | 1090 ± 193 | 1052 ± 170 | p=0.106 |
| Frontal cortex atrophy | 0.0 (0.0, 1.0) | 1.5 (0.5, 2.5) | 2.0 (2.0, 2.8) ∫ | p=0.028 |
| Temporal cortex atrophy | 0.0 (0.0, 0.0) | 0.5 (0.0, 1.5) | 1.0 (0.0, 1.9) | p=0.133 |
| Motor cortex atrophy | 1.0 (0.0, 1.0) | 1.0 (0.5, 2.0) | 0.0 (0.0, 1.5) | p=0.300 |
| Parietal cortex atrophy | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.5) | 1.0 (0.0, 1.8) | p=0.116 |
| Ventricular enlargement | 0.0 (0.0, 0.5) | 1.0 (0.2, 2.0) | 2.0 (2.0, 2.8) ∫ | p=0.006 |
| Hippocampal atrophy | 0.0 (0.0, 0.2) | 0.5 (0.0, 1.8) | 2.0 (2.0, 2.5) ∫ | P=0.010 |
| Caudate nucleus atrophy | 0.0 (0.0, 0.0) | 0.0 (0.0, 1.0) | 0.0 (0.0, 0.8) | p=0.311 |
| Alzheimer type pathology: None | SC | PA | AD, count | 3 | 0 | 2 | 0 | 6 | 1 | 0 | 1 | 4 | 1 | 2 | 0 | p=0.513 |
| Braak neurofibrillary tangle stage | II (I, II) | II (I, II) | II (II, III) | p=0.086 |
| Cortical senile plaques | 0.0 (0.0, 24) | 0.0 (0.0, 1.1) | 1.1 (0.0, 20) | p=0.690 |
| Cerebellar granular neuron inclusions | 1.0 (0.8, 2.0) | 2.8 (0.8, 3.5) | 3.0 (1.0, 3.0) | p=0.291 |
| Hippocampal sclerosis score | 0.0 (0.0, 0.0) | 0.8(0.0, 2.0) | 2.0 (1.3, 2.9) ∫ | p=0.009 |
| Substantia nigra degeneration | 0.5 (0.4, 0.6) | 2.0 (1.0, 2.5) | 1.0 (1.0, 3.0) ∫ | p=0.026 |
|
| ||||
| Motor neuron pathology | ||||
| Pyramid myelin pallor (Luxol fast blue) | 2.0 (2.0, 2.0) | 1.2 (0.8, 1.5) | 0.0 (0.0, 0.0) ∫ | p<0.001 |
| Pyramid microgliosis (IBA-1) | 3.0 (2.9, 3.0) | 2.5 (1.8, 2.8) | 0.0 (0.0, 0.5) * | p<0.001 |
| Hypoglossal neuronal loss & gliosis | 2.0 (1.9, 2.1) | 1.8 (0.8, 2.0) | 0.0 (0.0, 0.0) * | p<0.001 |
| Hypoglossal Bunina bodies, % present | 100% | 87% | 29% * | p=0.011 |
|
| ||||
| Focal microscopic atrophy | ||||
| Frontal atrophy | 0.0 (0.0, 0.0) | 1.2 (0.0, 2.2) | 2.0 (1.5, 2.0) ∫ | p=0.014 |
| Temporal atrophy | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.5) | 0.5 (0.0, 1.8) | p=0.143 |
| Parietal atrophy | 0.0 (0.0, 0.0) | 0.5 (0.0, 1.2) | 0.5 (0.0, 1.4) | p=0.141 |
| Motor atrophy | 1.5 (0.9, 2.1) | 0.8 (0.2, 1.5) | 0.5 (0.0, 0.9) | p=0.112 |
| Striatal atrophy | 0.0 (0.0, 0.0) | 1.5 (0.0, 2.0) | 1.0 (0.6, 1.9) ∫ | p=0.022 |
| TDP-43 pathology – general features | ||||
| TDP43 Type: 1 | 2 | 3 | ALS, count | 0 | 0 | 0 | 5 | 1 | 1 | 6 | 0 ∫ | 6 | 1 | 0 | 0 * | p<0.001 |
| Neuronal intranuclear inclusions, % present | 0% † | 38% | 86% | p=0.011 |
| Hippocampal synaptic or fine neurites, % present | 0% † | 63% | 71% | p=0.034 |
p<0.05;
different from ALS;
different from ALS and FTLD-MND;
different from FTLD-MND;
different from FTLD-MND & FTLD-TDP;
Abbreviations: M = male, F = Female, SC = senile change, PA = pathological aging, AD = Alzheimer's disease. Semi-quantitative data from a 4-point scale (none, mild, moderate, severe) is displayed as median (25th, 75th percentile), all other data displayed as indicated. Alzheimer type pathology was classified as none for cases with no cortical senile plaques (SP), SC for cases with sparse cortical SP, PA for cases with enough SP to meet Khachaturian AD criteria, and AD for cases with many cortical SP and Braak NFT stage of IV or greater. Braak stage is less than IV in SC and PA. TDP-43 pathology in Type 1 has widespread NCI and DN involving cortical and subcortical areas; Type 2 has long thick dystrophic neurites and Pick body like NCI with minimal brainstem pathology; Type 3 has NCI and paucity of DN in the cortex, variable NCI in hippocampus and amygdala, and NCI in motor neurons; ALS type has NCI in motor neurons and GCI in the pyramidal tract
Table 2. Demographics and Clinical Features.
| Case # |
Path Type | TDP type |
Sex | FHx | Age at death (years) |
Dur- ation (years) |
Dem- entia |
MND | Terminal MND phenotype |
Extra- pyramidal signs |
Psychiatric (including depression) |
Memory disorder |
Focal cortical signs |
Lang- uage disorder |
Frontal- behavioral disorder |
Initial clinical diagnosis |
Final clinical diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ALS | ALS | F | Yes | 49 | 1 | No | Yes | spinal | ALS | ALS | ||||||
| 2 | ALS | ALS | F | Yes | 50 | 3 | No | Yes | bulbar | + | ALS | ALS | |||||
| 3 | ALS | ALS | M | Yes | 58 | 6 | No | Yes | spinal | + | ALS | ALS | |||||
| 4 | ALS | ALS | F | No | 62 | 1 | No | Yes | bulbar | Dysarthria | Bulbar palsy | ||||||
| 5 | ALS | ALS | F | 76 | “PSP” | ||||||||||||
| 6 | FTLD-MND | 3 | M | No | 58 | 2 | No | Yes | spinobulbar | + | ALS | ALS | |||||
| 7 | FTLD-MND | 3 | M | No | 59 | 3 | Yes | Yes | spinobulbar | + | bvFTD | FTD-MND | |||||
| 8 | FTLD-MND | 3 | M | Yes | 61 | 9 | No | Yes | spinobulbar | ALS | ALS | ||||||
| 9 | FTLD-MND | 3 | F | Yes | 55 | 5 | Yes | No | + | + | + | + | AD v. bvFTD | bvFTD v. Pick's | |||
| 10 | FTLD-MND | 2 | F | No | 61 | 4 | Yes | No | + | + | + | AD | bvFTD v. Pick's | ||||
| 11 | FTLD-MND | 1 | M | Yes | 62 | 5 | Yes | No | + | + | + | + | NPH v. AD | bvFTD | |||
| 12 | FTLD-MND | 3 | F | 62 | 8 | Yes | No | + | + | Depression | bvFTD | ||||||
| 13 | FTLD-TDP | 3 | F | Yes | 74 | 4 | Yes | No | + | + | + | + | AD v. DLB | bvFTD v. DLB | |||
| 14 | FTLD-TDP | 1 | M | No | 70 | 4 | Yes | No | + | + | AD v. FTD | bvFTD | |||||
| 15 | FTLD-TDP | 1 | M | Yes | 72 | 2 | Yes | No | + | AD | AD | ||||||
| 16 | FTLD-TDP | 1 | M | Yes | 74 | 5 | Yes | No | + | + | + | DLB v. bvFTD | bvFTD v. DLB | ||||
| 17 | FTLD-TDP | 2 | M | No | 74 | 11 | Yes | No | + | + | + | AD | AD | ||||
| 18 | FTLD-TDP | 1 | M | Yes | 81 | 7 | Yes | No | + | + | + | + | MCI | bvFTD v. PNFA | |||
| 19 | FTLD-TDP | 1 | F | Yes | 83 | 4 | Yes | No | + | MCI | AD | ||||||
| 20 | FTLD-TDP | 1 | M | Yes | 86 | 8 | Yes | No | + | + | + | + | AD | AD v. DLB |
Cells left blank indicate absence of the clinical feature or lack of information about the particular feature, while “+” indicates positive documentation of the clinical feature. Case 5 had only demographic information and final clinical diagnosis, but was not submitted with any medical records. Abbreviations: M = male, F = Female, MND = motor neuron disease; PSP = progressive supranuclear palsy, v. = versus (two clinical diagnoses in the differential), bvFTD = behavioral variant frontotemporal degeneration, DLB = dementia with Lewy bodies, MCI = mild cognitive impairment, AD = Alzheimer's disease, PNFA = progressive non-fluent aphasia.
Macroscopic pathology
Data abstracted from neuropathology reports included fixed brain weight (calculated by doubling the weight of the fixed hemibrain), distribution and severity of cortical atrophy, presence and severity of ventricular enlargement, presence and severity of hippocampal atrophy, and presence and severity of caudate nucleus atrophy. Cortical atrophy descriptions, confirmed by review of photographs of the brain, were converted to a 4-point scale: 0 = unremarkable, 1 = mild, 2 = moderate, and 3 = severe. Enlargement of the frontal and temporal horns of the lateral ventricle were converted to a 4-point scale and then averaged. Atrophy of the caudate nucleus was converted to a 4-point scale similar to those used in a previous study [7]: 0 = unremarkable, 1 = mild atrophy, 2 = flattening of the caudate head, and 3 = severe atrophy giving a convex appearance of the caudate head.
Microscopic pathology - methods
Slides of frontal, temporal, parietal and motor cortex, hippocampus, amygdala, basal ganglia, thalamus, midbrain, pons, medulla and cerebellum were reviewed in all cases. Medulla was available for review in all cases and spinal cord was available in 8 cases (ALS: n = 3, FTLD-MND: n = 3, FTLD-TDP: n = 2). In addition to histologic staining methods, including hematoxylin and eosin (H&E), thioflavin-S fluorescent microscopy and Luxol fast blue/periodic acid Schiff of medulla, all cases underwent immunohistochemistry with a monoclonal phospho-TDP antibody (ps409/410, 1:5000, Cosmobio Co., Tokyo). Sections of cerebellum were processed for immunohistochemistry with a monoclonal antibody to ubiquitin (Ubi-1, 1:60,000; Millipore, Billerica, MA.) to detect NCI. Sections of medulla were processed for immunohistochemistry with a monoclonal antibody to ionized binding adaptor molecule-1 (IBA-1) to detect microgliosis (1:3000, Wako Chemicals USA, Richmond, VA). Immunohistochemistry for all cases was performed in batches with positive and negative controls to limit variability between slides using a Universal Autostainer (DAKO, Carpinteria, CA) and DAKO Envision kits with diaminobenzidine as the chromogen.
Microscopic pathology - semiquantitative analyses
Quantitative methods for assessing distribution of neurofibrillary tangles, senile plaques, and amyloid angiopathy using thioflavin S fluorescent microscopy have been previously described [52]. Alzheimer's type pathology was classified based on the density of senile plaques and neurofibrillary tangles: absent, senile changes of Alzheimer type, pathological aging [12], and Alzheimer's disease. Cases with senile changes and pathological aging had cortical senile plaques, but few or no neurofibrillary tangles and a Braak neurofibrillary tangle stage less than IV. Pathological aging cases had enough cortical senile plaques to meet Khachaturian criteria for Alzheimer's disease [32]. Alzheimer's disease cases had many cortical senile plaques and at least some cortical neurofibrillary tangles with a Braak neurofibrillary tangle stage of IV or greater [5]. Presence of Lewy bodies was assessed with α-synuclein immunohistochemistry and classified according to previous methods [16]. Cerebrovascular pathology was assessed on all cases using a simple scoring method [25].
The density of ubiquitin-positive cerebellar inclusions in the internal granular cell layer was graded on a semi-quantitative 4-point scale: 0 = none, 1 = mild, 2 = moderate, and 3 = severe. The presence and severity of neuronal loss and gliosis in the hippocampus was used to score severity of hippocampal sclerosis. Hippocampal pathology was assessed on H&E stained sections of hippocampus at the level of the lateral geniculate and scored on a semi-quantitative 4-point scale: 0 = absence of neuronal loss in the subiculum or CA1, 1 = neuronal loss and gliosis in the subiculum, 2 = mild-to-moderate neuronal loss and gliosis in both the subiculum and CA1, and 3 = severe neuronal loss and gliosis in both the subiculum and CA1 [27]. The presence and severity of neuronal loss and gliosis in the substantia nigra was assessed on H&E stained sections with a semi-quantitative 3-point scale: 0 = preservation of neuromelanin-containing neurons throughout the substantia nigra, 1 = mild neuronal loss with some extraneuronal pigment, 2 = moderate neuronal loss with gliosis and extraneuronal pigment, 3 = severe neuronal loss and gliosis with extensive extraneuronal pigment.
Severity of motor neuron pathology was evaluated in the medullary pyramid and hypoglossal nucleus in the medulla, as well as the corticospinal tract and anterior horn cells in spinal cord, if available. Degeneration of the corticospinal tract was evaluated for myelin pallor and microgliosis on LFB and IBA-1 stained sections, respectively. Severity was measured on a 4-point grading scale: 0 = none, 1 = mild, 2 = moderate, and 3 = severe. Hypoglossal nucleus and/or anterior horn cells in the spinal cord were assessed for neuronal loss, gliosis, and the presence or absence of Bunina bodies on H&E. If the hypoglossal nucleus was not available due to an off center sagittal cut of the hemi-brainstem, the nucleus ambiguus was evaluated for signs of motor neuron degeneration (Case 5, 8, 13, 14, 15). Cases with no Bunina bodies on original medulla or spinal cord sections were evaluated by taking additional non-overlapping sections, with the number of additional sections ranging from 5 to 20 depending upon if spinal cord or more than one level of medulla was included in the paraffin blocks. H&E stains from frontal, temporal, motor, and parietal cortices were used to evaluate the severity of neuronal loss, spongiosis, and gliosis associated with cortical degeneration. Semi-quantitative grading methods for cortical degeneration, which assessed neuronal loss, gliosis and spongiosis, and of striatal atrophy were similar to methods previously described [26].
Methods for assigning TDP-43 pathology type was according to our validation of the Mackenzie subtyping scheme [28]. Briefly, Type 1 cases have at least some lentiform NII as well as granular or crescent shaped NCI and short, curvilinear DN most dense in layer II of the cortex. They also have granular, small dense or crescent shaped NCI in the hippocampal dentate fascia and frequently fine thin neurites in CA1, usually associated with hippocampal sclerosis. The TDP-43 pathology in Type 1 is widespread, and involves amygdala, basal ganglia, midbrain and medulla (e.g., inferior olivary nucleus). Microvascular astrocytic inclusions [37] are detected in the cortex, amygdala and basal forebrain. Type 2 cases lack NII and have sparse cortical NCI, but demonstrate sometimes numerous Pick-body like NCI in the dentate fascia, amygdala and basal ganglia. Cortical DN are thick and long and can be detected in all cortical layers. Type 2 cases have minimal TDP-43 pathology (e.g., sparse DN) in brainstem. Type 3 cases have NCI (granular or “pre-inclusion” type [6]) with a paucity of DN in the cortex. They have variable NCI in hippocampal dentate fascia, and often many NCI in the amygdala. NCI are sparse in midbrain and medulla, but significantly more NCI are detected in hypoglossal nucleus than Types 1 and 2 [28]. An ALS type was also defined in this study. It required skein-like, Lewy body-like or pre-inclusion type NCI in motor neurons, more often in hypoglossal nucleus, nucleus ambiguus and anterior horn cells than in the Betz cells of the motor cortex [11], as well as oligodendroglial cytoplasmic inclusions (GCI) in the pyramidal tract [11, 53].
The presence and density of NCI, GCI, DN and perivascular (PV) astrocytic inclusions were assessed semiquantitatively in 25 brain regions with TDP-43 immunohistochemistry, including middle frontal cortex, motor cortex, dentate fascia of the hippocampus, CA1-subiculum of the hippocampus, entorhinal cortex, inferior temporal gyrus, putamen, globus pallidus, basal nucleus of Meynert, hypothalamus, amygdala, midbrain tectum, periaqueductal gray matter, oculomotor nucleus, red nucleus, substantia nigra, hypoglossal nucleus, medullary tegmentum (including nucleus ambiguus), inferior olivary nucleus, anterior horn of the spinal cord; as well as five areas of white matter – paracentral cerebral white matter, anterior limb of internal capsule, cerebral peduncle, medullary pyramid, and corticospinal tract of the spinal cord. NCI, DN, GCI and PV were each scored independently on a 4-point scale (0 = none, 1 = mild, 2 = moderate, 3 = severe) in each region.
Genetic analyses
Genomic DNA was extracted from samples using standard procedures. For each case, DNA was screened for the presence of the expanded hexanucleotide repeat in C9ORF72 using the repeat primed polymerase chain reaction method as previously described [10]. In short, 100 ng/μl of DNA was amplified by polymerase chain reaction in a 20 μl reaction containing 1M betaine (Sigma), 5% dimethyl sulfoxide (Sigma), 5 mM of dCTP, dATP, dTTP (Promega) and 7-deaza-2-deoxy GTP (Roche) in complete substitution for dGTP. The cycling program included an initial denaturation at 98°C for 10 minutes followed by 10 cycles of denaturation at 97°C for 35 seconds, annealing at 64°C for 2 minutes and extension at 68°C for 8 minutes, followed by 25 cycles of denaturation at 97°C for 35 seconds, annealing at 64°C for 2 minutes and extension at 68°C for 8 minutes plus 20 additional seconds each cycle.
Statistical analyses
SigmaPlot (version 11) was used for all statistical analyses. The difference in mean values of normally distributed data (i.e. age onset, age at death, and brain weight) was calculated with Analysis of Variance (ANOVA) tests, and post-hoc pairwise comparisons were performed with a t-test. A nonparametric Kruskal-Wallis one-way ANOVA on Ranks was used on disease duration and all semi-quantitative pathologic data. Post-hoc pairwise comparisons of nonparametric data were performed using the Holm-Sidak method. For categorical data (i.e. family history and presence/absence of pathology), a χ2 test was used to establish whether the proportions of observations differed between groups. Correlative analyses were performed using a Spearman rank order correlation. Significance was considered to be p<0.05.
Results
Demographics and clinical presentation
c9FTD/ALS cases fell into three groups based on their pathologic characteristics: ALS (n=5), FTLD-MND (n=8), and FTLD-TDP (n=7). ALS cases had motor neuron degeneration, but minimal extramotor TDP-43 pathology. FTLD-TDP had no motor neuron degeneration and widespread TDP-43 pathology. FTLD-MND had both motor neuron degeneration and widespread TDP-43 pathology. The source of patients in this pathologic series were Mayo Clinic Florida (ALS n=2, FTLD-MND n=3, FTLD-TDP n=1), State of Florida Alzheimer Disease Initiative (ALS n=1, FTLD-MND n=5, FTLD-TDP n=6) and the CurePSPlSociety of Progressive Supranuclear Palsy brain bank (ALS n=2, FTLD-MND n=0, FTLD-TDP n=0). Demographics and clinical features are summarized in Table 1, with further detailed information on individual cases provided in Table 2 and Table 3. Patients thought to have Alzheimer type dementia, dementia with Lewy bodies or FTD were mostly from Florida memory disorder clinics, while most of the ALS cases came from clinics specializing in ALS. Two of the ALS cases had progressive supranuclear palsy in the differential diagnosis due to severe dysarthria and dysphagia. On closer inspection of the clinical information, they lacked other diagnostic clinical characteristics of progressive supranuclear palsy. There were fewer men in the ALS (1 of 5) group than either FTLD-MND (4 of 8) or FTLD-TDP (6 of 7) groups, although this only approached statistical significance. Cases in the ALS and FTLD-MND groups were younger than in the FTLD-TDP group at symptom onset and death. There were no differences in frequency of family history of neurodegenerative disorder by referral source or by c9FTD/ALS pathologic group. For cases in which family history was clearly documented, 33% (6/18) did not have a first-degree relative with ALS, parkinsonism or dementia.
Table 3. Pathologic Features.
| Case # | Path Type | Spinal cord | Bunina bodies | Lewy bodies | Braak NFT Stage | SN score | HpScl score |
|---|---|---|---|---|---|---|---|
| 1 | ALS | 0 | 1 | 0 | II | 0-1 | 0 |
| 2 | ALS | 1 | 1 | 0 | 0-I | 0-1 | 0 |
| 3 | ALS | 1 | 1 | 0 | I | 0 | 0 |
| 4 | ALS | 0 | 1 | 0 | II | 1 | 0 |
| 5 | ALS | 1 | 1† | 0 | III | 0-1 | 0 |
| 6 | FTLD-MND | 0 | 1 | 0 | I-II | 1 | 0 |
| 7 | FTLD-MND | 1 | 1 | 0 | II | 1 | 0 |
| 8 | FTLD-MND | 1 | 0 | 0 | II | 0-1 | 0-1 |
| 9 | FTLD-MND | 0 | 1 | 0 | II | 3 | 1 |
| 10 | FTLD-MND | 1 | 1 | 0 | V-VI | 2 | 3 |
| 11 | FTLD-MND | 0 | 1 | 0 | II | 2 | 3 |
| 12 | FTLD-MND | 0 | 1 | 0 | 0 | 2 | 1 |
| 13 | FTLD-TDP | 0 | 1† | 0 | 0-I | 3 | 0 |
| 14 | FTLD-TDP | 0 | 1 | TLBD | IV | 3 | 3 |
| 15 | FTLD-TDP | 1 | 0† | 0 | II | 1 | 0-1 |
| 16 | FTLD-TDP | 0 | 0 | 0 | II-III | 1 | 1 |
| 17 | FTLD-TDP | 0 | 1 | 0 | II-III | 0-1 | 2-3 |
| 18 | FTLD-TDP | 0 | 0 | 0 | II | 3 | 2 |
| 19 | FTLD-TDP | 1 | 0 | BLBD | III | 1 | 2 |
| 20 | FTLD-TDP | 1 | 0 | 0 | II-III | 3 | 3 |
The presence or absence of Bunina bodies in motor nuclei in the medulla (hypoglossal nucleus and/or †nucleus ambiguous if section was cut off midline precluding evaluation of hypoglossal nucleus) and/or spinal cord. Presence and type of Lewy bodies detected with α-synuclein immunohistochemistry. Semi-quantitative data are displayed from a 4-point scale (none, mild, moderate, severe). Abbreviations: NFT = neurofibrillary tangle, SN = substantia nigra, HpScl = hippocampal sclerosis, TLBD = transitional Lewy body disease, BLBD = brainstem Lewy body disease.
Initial symptoms and clinical diagnoses were consistent with ALS in the ALS pathologic group; except for Case 4 who was initially diagnosed with dysarthria thought to be due to multiple sclerosis because of white matter lesions on neuroimaging. A diagnosis of bulbar palsy consistent with ALS was made on the last available clinical evaluation before death. The patients in the ALS pathologic group had no cognitive or behavioral disturbances, but psychiatric symptoms, most often depression, were not uncommon in ALS (2 of 4) and FTLD-MND (4 of 8) groups. Patients in the FTLD-MND pathologic group were heterogeneous in initial clinical presentations, including initial diagnoses of ALS (n=2), Alzheimer's disease (versus bvFTD, normal pressure hydrocephalus, or dementia with Lewy bodies) (n=4), depression (n=1), and bvFTD (n=1). The frequency of frontal-behavioral features was similar in FTLD-MND (6 of 8) and FTLD-TDP (3 of 7) pathologic groups (p=0.047). Patients in the FTLD-TDP group were more often given an initial diagnosis of Alzheimer's disease, mild cognitive impairment or dementia with Lewy bodies than those in the FTLD-MND group, but they were also older and mostly evaluated in memory disorder clinics. Extrapyramidal signs and memory impairment were relatively common in c9FTD/ALS, but they did not differ significantly between FTLD-MND and FTLD-TDP pathologic groups. Although not significant, focal cortical signs were more frequent in FTLD-TDP (5 of 7) compared with FTLD-MND (3 of 8), including one patient whose final clinical diagnosis included the differential between progressive nonfluent aphasia and bvFTD due to presence of mixed language and frontal-behavioral features. This was the only case (Case 18) with clinically significant nonfluent aphasia and no c9FTD/ALS patient was considered to have semantic dementia or corticobasal syndrome.
Pathologic findings
The brain weight did not differ amongst the three pathological groups. Macroscopic atrophy of the frontal cortex, however, tended to be more severe on average in FTLD-TDP, while atrophy was less in most cases of FTLD-MND (Figure 1). Frontal or temporal atrophy was not present in ALS, but mild atrophy of the motor cortex was sometimes noted in ALS and more often in FTLD-MND. Parietal cortices were minimally affected, while occipital cortices were spared in all three groups. Hippocampal atrophy was prominent in FTLD-TDP and to a lesser extent FTLD-MND, compared to ALS. Atrophy of the caudate did not differ across groups; however, mild-to-moderate atrophy was occasionally observed in FTLD-MND (Cases 9, 10, &13) and FTLD-TDP (Cases 16 & 17).
Figure 1.
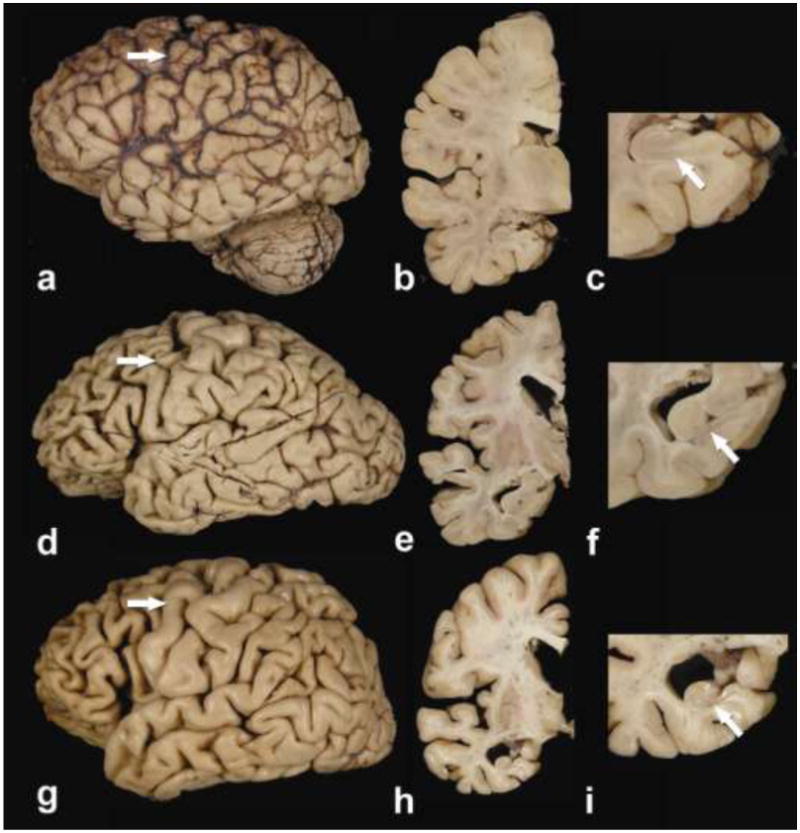
Macroscopic findings in c9FTD/ALS (Case 1: a, b & c; Case 9: d, e & f; Case 14: g, h & i)). ALS case with mild peri-Rolandic cortical atrophy (a, arrow), but no ventricular enlargement on a coronal section at level of the subthalamic nucleus (b) and no gross hippocampal atrophy (c). FTLD-MND case (Mackenzie Type 3) with moderate peri-Rolandic cortical atrophy (arrow) and moderate frontal-predominant atrophy (d). Enlargement of the frontal, but not temporal horn of the lateral ventricle can be seen on a coronal section at level of the subthalamic nucleus (e) and mild hippocampal atrophy of the subiculum (f). FTLD-TDP case (Mackenzie Type 1) with no peri-Rolandic atrophy (arrow), but moderate frontal and temporal atrophy (g), as well as enlargement of both frontal and temporal horns of the lateral ventricle on a coronal section at level of the subthalamic nucleus (h). Hippocampal sclerosis was severe and correlates with atrophy in CA1 and the subiculum (arrow) (i).
Hippocampal sclerosis (Table 3) was not detected in any ALS case, but was present in FTLD-MND (5 of 8) and FTLD-TDP (7 of 7). Hippocampal sclerosis was moderate-to-severe in five of the FTLD-TDP, but only two of the FTLD-MND cases. All of the cases with moderate-to-severe hippocampal sclerosis (n=7) presented with memory impairment and had a clinical diagnosis of Alzheimer's disease either initially or in their final clinical diagnosis. Mild hippocampal sclerosis had a variable impact on memory dysfunction and clinical diagnosis of Alzheimer's disease (2 of 5). Macroscopic hippocampal atrophy and microscopic assessment of hippocampal sclerosis were highly correlated (r=0.88, p<0.0001).
Alzheimer type pathology was minimal to absent in most cases, and sufficient to diagnose Alzheimer's disease in one FTLD-MND case (Case 10), 61-year old woman with many cortical senile plaques and a Braak neurofibrillary tangles stage of V-VI. Lewy bodies were detected in only two cases, both FTLD-TDP. Case 19, an 83-year-old man with no extrapyramidal signs, had Lewy bodies in the brainstem and basal forebrain not associated with neuronal loss and gliosis, consistent with an incidental finding. Case 14, a 73-year-old man with no extrapyramidal signs, had Lewy bodies in brainstem, basal forebrain, hippocampus and limbic cortices, but minimal neuronal loss in the vulnerable brainstem nuclei, except the substantia nigra. Substantia nigra degeneration was variable and was moderate-to-severe in three of the FTLD-TDP, four of the FTLD-MND, but in none of the ALS group. Of the five cases with the most severe degeneration of the substantia nigra, four had extrapyramidal signs and two of these had dementia with Lewy bodies as an antemortem clinical diagnostic consideration, including an 86-year-old man (Case 20) enrolled in a prospective, longitudinal study of dementia with Lewy bodies. None of the patients with parkinsonism and severe substantia nigra degeneration had Lewy bodies.
All cases had ubiquitin-positive, TDP-43-negative NCI in cerebellar internal granular cell layer (Figure 2). There was no difference in the density of cerebellar inclusions across the three groups, although they tended to be less numerous in ALS. Of the 31 additional FTLD and ALS cases that were negative for C9ORF72 mutations, no ubiquitin-positive NCI were detected in the cerebellar granule cells, although there were often age-related ubiquitin-positive structures in white matter and the Purkinje cell layer [14].
Figure 2.

Ubiquitin immunohistochemistry of cerebellum of c9FTD/ALS shows many NCI in internal granular cell layer in FTLD-TDP (a) and fewer in ALS (b). Insets show higher magnification of NCI. (bar = 30 μm for a & b and 90 μm for insets)
Motor neuron degeneration
Severity of pyramidal myelin pallor and microgliosis was greater in ALS and FTLD-MND than in FTLD-TDP, which had no myelin pallor in any of the cases and showed only mild microgliosis in three of seven cases. Moderate-to-severe hypoglossal neuronal loss and gliosis was detected in all ALS and in four of the FTLD-MND cases, but was absent in FTLD-TDP. The remaining four FTLD-MND cases showed mild neuronal loss and gliosis. Bunina bodies were found in the hypoglossal nucleus and/or anterior horn cells in all ALS; however, Bunina bodies in Cases 1 and 2 were found only after evaluation of additional levels. Seven of eight FTLD-MND cases had Bunina bodies, including a case (Case 13) in which they were detected only after additional sections were examined. Despite additional sampling, there were few neurons left to detect Bunina bodies in Case 8. Bunina bodies were originally not detected in any of the FTLD-TDP cases, but after evaluation of additional non-overlapping sections of medulla and/or spinal cord if available, isolated Bunina bodies were sometimes detected – one Bunina body was detected in Case 14 and two Bunina bodies in Case 17. In neither case were they associated with neuronal loss or TDP-43 positive NCI in motor neurons.
Cortical neuronal loss and gliosis
Mild-to-severe frontal cortical neuronal loss and gliosis was present in five cases of FTLD-MND and all seven cases of FTLD-TDP, compared to ALS where no significant cortical neuronal loss or gliosis was found. Temporal cortical neuronal loss and gliosis was absent in ALS, present in only two cases of FTLD-MND and to some degree in four cases of FTLD-TDP. Motor cortex neuronal loss and gliosis was found to some degree in all ALS and almost all FTLD-MND (6 of 8), but in only four FTLD-TDP (4 of 7) cases. Mild-to-moderate striatal atrophy was present in five cases of FTLD-MND and all seven cases of FTLD-TDP, but was not detected in any case of ALS. In no case was striatal atrophy severe.
TDP-43-pathology
Table 1 summarizes general features of TDP-43 pathology; including the TDP-43 type, the presence of NII, and the presence of hippocampal synaptic-like immunoreactivity or fine thin neurites in CA1. TDP-43 types were classified based on our previous study [28], which validated the scheme originally proposed by Mackenzie and colleagues [42] (Figure 3). In the present study, a TDP-43 type for ALS was also denoted, since it had distinct features from the other types. The ALS type was associated with NCI (e.g., skein-like, Lewy body like or pre-inclusion type) in motor neurons (i.e. Betz cells in motor cortex, neurons of hypoglossal nucleus and/or anterior horn cells of the spinal cord), GCI in motor cortex and the corticospinal tract, but minimal extramotor TDP-43 pathology.
Figure 3.

Phospho-TDP-43 immunohistochemistry of c9FTD/ALS cases with range of TDP-43 pathology. (a) ALS cortex has many GCI in lower cortical layers. (b) Type 1 cases have many NCI and DN in the superficial cortical layers, with lentiform NII (upper left inset) and dense, globular PV astrocytic inclusions (lower right inset). (c) Type 2 cases have large thick dystrophic neurites in lower (in this figure) and upper cortical layers, with dense round Pick body like NCI in the hippocampal dentate fascia (upper left inset) (d) Type 3 cases have many granular or pre-inclusion type NCI with sparse or absent DN in cortex and hippocampal dentate fascia (upper left inset). (bar = 30 μm for a, b, c & d and 90 μm for insets)
Type 1 cases had the oldest age of onset and death (age onset=70 years, age death=75 years) compared to Type 2 (58, 68 years), Type 3 (57, 62 years), and ALS type (52, 59 years) (respectively: p=0.006, p=0.020). Disease duration, however, was longest in Type 2 (10 years) compared to Type 1 (4.8 years), Type 3 (4.3 years), and ALS type (3.3 years) (p=0.025). FTLD cases with NII (n=9) were more likely to have a positive family history as those without NII (n=6) (89% vs. 17%, p=0.011).
Table 2 shows the TDP-43 type for each case and Table 4 summarizes the range of TDP-43 pathology found in the 25 brain regions reviewed for GCI, NCI, DN, and PV astrocytic inclusions. Table 4 shows only the regions with statistically significant findings (except for motor neurons, which were included for purpose of comparison) and is organized to illustrate pathologic patterns of overlap with regard to GCI (ALS overlaps with FTLD-MND), NCI (FTLD-MND, FTLD-TDP), and DN (FTLD-MND, FTLD-TDP). PV astrocytic inclusions were relatively limited to FTLD-TDP. The majority of FTLD-MND cases (6 of 8) were Type 3, with one Type 1 and one Type 2 detected (Table 2). Conversely, most of the FTLD-TDP cases (6 of 7) were Type 1, with one Type 2 detected (Table 2).
Table 4. Semiquantitative TDP-43 pathology.
| ALS | FTLD-MND | FTLD-TDP | ||
|---|---|---|---|---|
| Oligodendroglial cytoplasmic inclusions | ||||
| Motor cortex | 3.0 (2.5, 4.0) | 2.0 (1.5, 2.5) | 0.0 (0.0, 0.0) * | P<0.001 |
| Central white matter | 2.0 (1.8, 2.2) | 1.0 (1.0, 2.0) | 0.0 (0.0, 0.0) * | p=0.001 |
| Cerebral peduncle | 2.0 (1.8, 2.1) | 1.8 (1.0, 2.0) | 0.0 (0.0, 0.8) ∫ | p=0.004 |
| Hypoglossal nucleus | 2.0 (1.5, 2.5) | 1.0 (0.5, 1.0) | 0.0 (0.0, 0.0) ∫ | p=0.003 |
| Medullary tegmentum | 2.0 (1.8, 2.1) | 1.5 (1.0, 2.0) | 0.0 (0.0, 0.0) * | p=0.003 |
| Medullary pyramid | 2.0 (1.0, 2.1) | 1.0 (0.5, 2.0) | 0.0 (0.0, 0.0) ∫ | p=0.003 |
| Spinal cord anterior horn | 2.0 (2.0, 2.0) | 2.0 (1.2, 2.0) | 0.0 (0.0, 0.0) | p=0.121 |
| Spinal cord white matter | 1.0 (1.0, 1.8) | 1.0 (1.0, 1.0) | 0.0 (0.0, 0.0) | p=0.121 |
| Average (25 brain regions) | 0.62 (0.54, 1.0) | 0.71 (0.53, 0.89) | 0.0 (0.0, 0.10) | p= 0.002 |
|
| ||||
| Neuronal cytoplasmic inclusions | ||||
| Middle frontal gyrus | 0.0 (0.0, 0.1) † | 3.0 (2.5, 3.0) | 2.0 (2.0, 3.0) | p=0.005 |
| Dentate fascia | 0.0 (0.0, 0.2) † | 3.3 (2.8, 3.8) | 2.0 (2.0, 3.0) | p=0.002 |
| Hippocampus (CA1-subiculum) | 0.0 (0.0, 0.2) | 2.0 (1.8, 2.3) ∫ | 1.0 (1.0, 2.0) | p=0.003 |
| Entorhinal cortex | 0.0 (0.0, 1.0) | 3.0 (2.3, 3.0) ∫ | 1.0 (1.0, 2.0) | p=0.003 |
| Inferior temporal | 0.0 (0.0, 0.2) † | 3.0 (1.5, 3.0) | 2.0 (1.0, 3.0) | p=0.005 |
| Hypothalamus | 0.0 (0.0, 0.2) † | 2.0 (1.5, 2.5) | 1.0 (1.0, 2.5) | p=0.005 |
| Amygdala | 1.0 (0.0, 1.0) † | 3.3 (3.0, 3.8) | 2.50 (2.0, 3.0) | p=0.002 |
| Substantia nigra | 0.0 (0.0, 1.0) † | 2.0 (1.5, 2.2) ∫ | 1.0 (1.0, 1.8) | p=0.018 |
| Average (20 brain regions) | 0.58 (0.40, 0.66) | 1.0 (1.6, 2.0) | 1.0 (1.0, 1.2) ∫ | p<0.001 |
|
| ||||
| Dystrophic neurites | ||||
| Middle frontal gyrus | 0.0 (0.0, 0.0) | 1.5 (0.5, 2.0) | 2.0 (1.2, 2.0) ∫ | p=0.008 |
| Entorhinal cortex | 0.0 (0.0, 0.2) | 1.0 (1.0, 2.0) | 2.0 (2.0, 2.8) ∫ | p=0.003 |
| Inferior temporal cortex | 0.0 (0.0, 0.2) | 1.0 (0.0, 2.0) | 2.0 (2.0, 2.8) ∫ | p=0.006 |
| Amygdala | 0.0 (0.0, 0.2) | 1.0 (0.0, 1.5) | 1.0 (1.0, 2.0) ∫ | p=0.029 |
| Substantia nigra | 0.0 (0.0, 0.2) | 1.0 (0.5, 1.0) | 1.0 (1.0, 1.0) | p=0.024 |
| Spinal cord anterior horn | 1.0 (0.2, 1.0) | 1.0 (0.2, 1.0) | 0.0 (0.0, 0.0) | p=0.511 |
| Average (25 brain regions) | 0.17 (0.03, 0.23) | 0.67 (0.17, 0.85) | 1.1 (0.89, 1.2) ∫ | p=0.006 |
|
| ||||
| Perivascular astrocytic inclusions | ||||
| Hippocampus | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.8) | 2.0 (1.3, 2.0) *∫ | p=0.002 |
| Entorhinal cortex | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.8) | 1.0 (1.0, 2.0) ∫ | p=0.006 |
| Hypothalamus | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 2.0 (1.0, 2.0) * | p<0.001 |
| Amygdala | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 2.0 (1.0, 2.0) * | p=0.002 |
| Midbrain tectum | 0.0 (0.0, 0.0) | 0.0 (0.0, 1.0) | 2.0 (1.0, 2.0) ∫ | p=0.006 |
| Substantia nigra | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.5) | 1.0 (1.0, 2.0) * | p<0.001 |
| Average (25 brain regions) | 0.0 (0.0, 0.0) | 0.04 (0.0, 0.22) | 0.87 (0.55, 1.2) * | p=0.002 |
p<0.05;
different from ALS;
different from ALS and FTLD-MND;
different from FTLD-MND;
different from FTLD-MND & FTLD-TDP;
Semi-quantitative data from a 4-point scale (none, mild, moderate, severe) are displayed as median (25th, 75th percentile).
All cases had TDP-43 pathology, but there was heterogeneity in the mix of neuronal and glial lesions and in the distribution of TDP-43 pathology (Figure 3). The heterogeneity was not random, but showed clear patterns into three pathology groups – ALS, FTLD-MND and FTLD-TDP (Table 4). GCI were more common in ALS and FTLD-MND than in FTLD-TDP. The highest density of GCI was observed in the motor cortex, which correlated with severe neuronal loss and spongiosis (p=0.020). The density of GCI tended to be greater in ALS than FTLD-MND within the hypoglossal nucleus, medullary tegmentum, and medullary pyramid. Of the cases where spinal cord was available, the anterior horn was moderately affected in ALS and FTLD-MND. GCI were found in the paracentral white matter of ALS and FTLD-MND, but in only one FTLD-TDP case.
Cortical NCI were common in FTLD-TDP and FTLD-MND, but nearly absent in ALS outside of the motor cortex. In addition to characteristic skein-like and Lewy body-like inclusions in motor neurons in ALS, isolated NCI were inconsistently detected in amygdala, red nucleus and medullary tegmentum. Moderate-to-severe NCI were observed in the cortical and limbic structures of FTLD-MND and FTLD-TDP. FTLD-MND showed more brainstem NCI in the midbrain tectum and periaqueductal gray matter compared to both ALS and FTLD-TDP.
DN were infrequent and sparse in ALS. In contrast, motor cortex and hypoglossal nucleus had more numerous DN in FTLD-MND compared with ALS. Moderate-to-severe DN were observed in corticolimbic regions in FTLD-TDP and to a lesser extent in FTLD-MND, the latter driven almost exclusively by the one Mackenzie Type 2 case in the FTLD-MND group. FTLD-TDP tended to have more severe pathology in both cortical and limbic regions than FTLD-MND. PV astrocytic inclusions were frequent in FTLD-TDP, but sparse in FTLD-MND and ALS. This observation correlated with increased Type 1 pathology in FTLD-TDP and the association of PV inclusions with Type 1 pathology [37]. The single FTLD-MND with Type 1 TDP-43 pathology (Case 11) had mild PV astrocytic inclusions.
TDP-43 pathology in Type 1 cases was predominant in superficial cortical layers and characterized by NCI and short and thin DN, while Type 2 cases had long and thick DN that showed no predilection for any cortical layer. Morphologic, as well as regional differences of NCI could be observed across types. Type 1 NCI were granular and dense in appearance, and more often found in cortex, basal ganglia, hypothalamus and hippocampus. The two Type 2 cases had a Pick body like appearance and were most numerous in dentate fascia of the hippocampus, amygdala, and basal ganglia. Skein-like NCI were observed in both Type 1 and Type 2 in isolated neurons of the substantia nigra and red nucleus. Type 3 NCI often had the appearance of diffuse cytoplasmic immunoreactivity consistent with pre-inclusions and they also had more synaptic-like TDP-43 immunostaining in the cortex, hippocampus, amygdala and globus pallidus.
Evaluation of the hippocampus demonstrated unusual TDP-43-positive, synaptic-like pathology that was most abundant in CA4 to CA2 sectors, which has not been emphasized in previous studies of FTLD-TDP (Figure 4). In some cases, this synaptic pattern of TDP-43 pathology was also noted in the amygdala, cortex and the wooly fibers of the globus pallidus. Synaptic TDP-43 pathology was noted in five of eight FTLD-MND and one of seven FTLD-TDP, almost all of which (4 of 6) were also consistent with TDP-43 Type 3. Many small, thin TDP-43 positive DN similar to those originally reported by Hatanpaa, et al. [23] were detected in CA1-subiculum of the hippocampus of nine cases, all seven FTLD-TDP and two FTLD-MND. All but two cases were TDP-43 Type 1 (one Type 3 and one Type 2). All but one of the cases with fine DN in the hippocampal pyramidal layer had hippocampal sclerosis. None of the ALS cases had abnormal TDP-43 synaptic pathology or fine DN in the hippocampus.
Figure 4.

Phospho-TDP-43 immunohistochemistry of c9FTD/ALS shows unusual neuritic and synaptic pathology. Fine neurites in CA1 of hippocampus (a); Synaptic-like TDP-43 grains in CA3 of hippocampus (b), superficial cortical layer (c) and globus pallidus (d). (bar = 30 μm for a, b, c & d)
TDP-43-positive NII were infrequent in FTLD-MND, absent in ALS, but significantly more frequent in FTLD-TDP. Only three FTLD-MND had NII. In FTLD-TDP, NII were most often lentiform shaped, but the few detected in FTLD-MND were more often round.
Comparison of c9FTD/ALS to FTLD and ALS cases without c9ORF72 mutations
A series of 18 cases of FTLD-TDP (TDP-43 Type 1 (n=3), Type 2 (n=4) Type 3 (n=6) and ALS (n=5) cases that were negative for c9ORF72 mutations were processed for TDP-43 immunohistochemistry , as well as Luxol fast blue and IBA-1 immunohistochemistry identical to that used for c9FTD/ALS cases. There were no distinguishing features that differentiated c9FTD/ALS cases. For example, synaptic-like TDP-43 immunoreactivity was detected in some of the Type 3 cases in the hippocampus, globus pallidus and substantia nigra. This pathology was minimal with commercial TDP-43 antibodies and clearly greater with the phospho-TDP (409/410) antibody used in present study. GCI, which were prominent in ALS and FTLD-MND cases in c9FTD/ALS, were also present in sporadic ALS cases. Incidental Bunina bodies were also detected in hypoglossal nucleus of a case of FTLD-TDP in which there was no significant motor neuron loss.
Discussion
This is a detailed pathologic description of c9FTLD/ALS due to mutations in C9ORF72. The cases were both clinically and pathologically heterogeneous and fell into three pathologic categories: ALS, FTLD-MND, and FTLD-TDP. ALS cases had motor neuron degeneration, but minimal extramotor TDP-43 pathology. They all had clinical presentation consistent with ALS. FTLD-TDP had no motor neuron degeneration and widespread TDP-43 pathology. They were clinically heterogeneous. FTLD-MND had both motor neuron degeneration and widespread TDP-43 pathology. Only some of the FTLD-MND had clinically recognized evidence of motor neuron disease. There were age of onset and age at death differences between the groups. ALS and FTLD-MND were younger on average than FTLD-TDP. There was no significant difference in disease duration amongst the three pathological groups.
Two caveats should be emphasized with respect to the clinical heterogeneity observed in this series of cases. First, the clinical information in this report is not from a prospective, longitudinal study. Second, the cases were obtained from diverse clinical sources, including tertiary academic medical centers as well as non-specialty community hospitals. As previously reported, clinical presentations of c9FTD/ALS include ALS and FTD, as well as patients with mixed clinical features of FTD-MND [4]. All of these clinical syndromes tend to be symmetrical and associated with predominance of frontal lobe involvement [41]. Asymmetrical or focal cortical syndromes were uncommon in the present autopsy series; only one patient had prominent aphasia. A previously unrecognized clinical presentation of c9FTD/ALS in this autopsy series is amnestic dementia resembling Alzheimer's disease, sometimes clearly preceded by amnestic mild cognitive impairment. Amnestic dementia cases were relatively frequent in our cohort. They were often evaluated in Florida memory disorder clinics, and they tended to be older at death than those presenting with FTD or ALS. Almost all cases with amnestic dementia suggestive of Alzheimer's disease had hippocampal sclerosis, which supports previous findings in the elderly with hippocampal sclerosis who may clinically mimic Alzheimer's disease rather than FTD [55]. FTLD cases with final clinical diagnoses of bvFTD tended to be younger. There was a strong correlation between microscopic evidence of hippocampal sclerosis and macroscopic evidence of hippocampal atrophy, which would make antemortem differential diagnosis difficult in these elderly patients in the absence of other more specific biomarkers. Cases with the most severe macroscopic and microscopic hippocampal involvement had FTLD-TDP compared to mild pathology in FTLD-MND. This fits with the lower frequency of hippocampal sclerosis noted in a previous series of FTLD-MND compared with FTLD-TDP [27].
Although all ALS cases had clinical features compatible with this diagnosis, there was variability in the distribution of motor neuron degeneration, with half showing predominantly terminal bulbar signs and half with predominantly spinal signs. There was a greater range of clinical presentations amongst the FTLD subgroups. As noted in much larger clinical and pathological series [10], not all c9FTD/ALS in this pathology series had a family history of neurological disease. Whether this represents true sporadic disease or incomplete ascertainment remains to be determined. Interestingly, NII were more common in cases with a positive family history, as previously reported [28, 46]. On the other hand, most cases of c9FTD/ALS with NII had Mackenzie Type 1 pathology, not Type 3 as originally suggested in the recent “harmonization” of the two major FTLD-TDP classification schemes [47].
Macroscopic examination of the brain revealed frontal cortical atrophy in both FTLD-TDP and FTLD-MND that was greater than in ALS; atrophy of motor cortex was more frequent in ALS and FTLD-MND than in FTLD-TDP. Ventricular enlargement was greater in FTLD-TDP than both ALS and FTLD-MND. Other pathologies (i.e. Alzheimer type pathology, Lewy bodies, and vascular pathology) were infrequent and did not differ between the groups.
All cases had ubiquitin-positive, TDP-43-negative NCI in cerebellar granular layer. Cerebellar NCI were not detected in 13 FTLD-TDP and 18 ALS cases that were negative for C9ORF72 mutations, although age-related ubiquitin pathology could be observed in the white matter in all cases [14]. Although cerebellar NCI were not detected in cases negative for C9ORF72, caution is advised in over-interpretation of age-related ubiquitin changes that have been previously reported in the granule cell layer of middle-age and older adults [14].
FTLD cases in the present series had heterogeneity regarding the degree and pattern of cortical atrophy, hippocampal sclerosis, striatal atrophy, SN degeneration, motor neuron pathology, and TDP-43 type. Previous studies of chromosome 9p linked FTLD had suggested that TDP-43 pathology in c9FTD/ALS was most often consistent with Mackenzie Type 3 [4, 20, 36, 41, 56]. Our study demonstrates that this is not the case, and that even amongst cases with FTLD-MND, not all had Type 3. Given the larger number of FTLD-TDP cases in this series, the most frequent type was actually Mackenzie Type 1. Some of the Type 1 cases were atypical in the frequency of pre-inclusion type neuronal lesions, a common feature of Type 3. The ALS cases had very little extramotor TDP-43 pathology, and even then, GCI were largely confined to the corticospinal tract. These observations are somewhat contrary to the observations of Geser et al. [17], who implied that extramotor pathology was almost inevitable in ALS.
Most FTLD cases fell into two TDP-43 types: either Mackenzie Type 1 (half with FTD and Alzheimer type dementia) or Mackenzie type 3 (half with FTD and half with clinical evidence of motor neuron disease). There were only two Mackenzie Type 2 cases (one with bvFTD and the other with Alzheimer type dementia). One of the Type 2 cases had evidence of motor neuron degeneration predominantly affecting upper motor neurons and the corticospinal tract. Neither had semantic dementia, which is a common clinical correlate of non-c9FTD/ALS Type 2 cases [28, 42].
FTLD cases differed in degree and pattern of cortical atrophy, with FTLD-TDP having more of a frontotemporal pattern of atrophy compared to FTLD-MND with mild frontal and motor involvement. Frontal cortical atrophy was often consistent with frontal-behavioral clinical presentations; whereas there was a general lack of peri-Sylvian and superior parasagittal atrophy supporting similar lack of focal presentations, such as nonfluent aphasia or corticobasal syndrome. Symmetry could not be evaluated since only one hemibrain was available for histologic studies; the other hemibrain having been frozen for biochemical and genetic studies. On the other hand, there was almost no clinical evidence of asymmetry, with only one patient having a prominent aphasia. This contrasts with asymmetrical clinical presentations (e.g., progressive nonfluent aphasia) that are not uncommon in FTLD-TDP do to GRN mutations [26, 43, 58].
Striatal degeneration was present in some FTLD cases, but it was relatively mild compared to the severe striatal atrophy seen in FTLD-FUS [29]. Severe striatal atrophy may be used to distinguish c9FTD/ALS from FTLD-FUS. Striatal degeneration is variable in FTLD-TDP due to GRN mutations, but can be a predominant feature in some cases [69].
Substantia nigra degeneration was relatively common in c9FTD/ALS and sometimes associated with parkinsonian clinical features. Of the five cases with severe nigral degeneration, four had extrapyramidal signs, including two patients thought to have dementia with Lewy bodies. One of these patients (Case 20) was enrolled in a prospective longitudinal study of dementia with Lewy bodies. Substantia nigra neuronal loss was detected in FTLD-MND and FTLD-TDP, but not ALS. Substantia nigra degeneration is also frequent in FTLD-TDP due to GRN mutations, detected in about 80% of cases [26], but it does not always correlate with overt clinical parkinsonism. Parkinsonism is relatively common in FTLD [15], and is present in as many as 67% in FTLD-TDP due to GRN mutations [31]. Parkinsonism in c9FTD/ALS was less frequent (30% for all cases and 40% for only cases with FTLD). A direct comparison of c9FTD/ALS and GRN-related FTLD-TDP is an objective of future studies. Available evidence, however, would suggest that FTLD-TDP due to GRN mutations is less heterogeneous that c9FTD/ALS, since virtually all reported cases have had Mackenzie Type 1 pathology [26, 44]
Spinobulbar signs were present in three of eight FTLD-MND cases, and of the five patients without clinical evidence of motor neuron degeneration (cases 9-13), two had a family history of ALS. Pyramidal tract degeneration and motor neuron neuronal loss and gliosis was variable in these five cases and severe in only one (case 11), a 62-year-old woman with mild dysphagia and a sister with ALS. Subclinical motor neuron degeneration is increasingly recognized in FTD [40], and electrodiagnostic tests may be needed to reveal subtle evidence of motor neuron degeneration. In this retrospective series, only one of the FTLD cases (case 7, a 59-year-old man with a clinical diagnosis of FTLD-MND) had electromyography and nerve conduction velocity studies. Despite lack of antemortem evidence of motor neuron disease in some cases, the present study shows significant pathologic overlap between FTLD-MND (n = 8) and ALS (n = 5) cases. The major areas of neuropathologic overlap between FTLD-MND and ALS are presence of NCI in motor neurons, prominent GCI in motor cortex and corticospinal tract, and presence of Bunina bodies in lower motor neurons. Bunina bodies were detected in all cases of ALS (5 of 5) and all but one case of FTLD-MND (7 of 8). The case without Bunina bodies had severe motor neuron loss precluding their detection, which is similar to paucity of Lewy bodies in the substantia nigra in end stage Lewy body disease. Two FTLD-TDP cases with very sparse Bunina bodies were likely an incidental finding since they were not associated with motor neuron loss or NCI with TDP-43 immunohistochemistry. The presence of GCI was a characteristic feature of TDP-43 pathology in FTLD-MND and ALS, which was much less common in FTLD-TDP. Oligodendroglial inclusions have been noted previously in ALS and FTLD-TDP [2, 45, 53, 70], and the present study suggest that this histologic feature marks overlap between a subset of c9FTD/ALS with motor neuron degeneration, but that it is not specific to c9FTD/ALS. In fact, other than cerebellar inclusions, there was no specific pathologic feature that distinguishes c9FTD/ALS.
There are strengths and weaknesses in this study. A strength of the study is that all cases were referred to a single brain bank where neuropathologic procedures are standardized. All cases underwent an in-depth neuropathologic examination by one neuropathologist, including semiquantitative assessment of a range measures using immunohistochemistry with a sensitive and specific phospho-TDP-43 monoclonal antibody [24], processed in batch with an automated tissue stainer to assure uniformity. All cases were also processed with sensitive methods for detecting motor neuron degeneration, and all cases had assessment of characteristic ubiquitin-immunoreactive cerebellar NCI. The major weakness is that cases were obtained from various sources, and there was a wide range in the quality of the medical documentation. Most cases had far from complete family histories. Only the Mayo Clinic patients were enrolled in prospective longitudinal studies, the others were a retrospective cohort.
Summary
This pathologic study of c9FTD/ALS demonstrates that although all cases had TDP-43 pathology, there is greater clinical and pathological heterogeneity than previously thought, including amnestic type dementia in an elderly cohort with hippocampal sclerosis and bvFTD in a younger cohort without motor neuron degeneration. The length of the hexanucleotide expansion in C9ORF72 is unknown in our cases. It remains to be determined if the expansion length can influence the clinical presentation or the nature of neuropathologic findings. This is an objective for future studies, as well as comparison of c9FTD/ALS with sporadic FTLD cases matched for TDP-43 Types and with the other relatively common genetically-determined FTLD-TDP, that due to mutations in GRN [26, 44]. This study further suggests that the harmonization of TDP-43 types proposed for FTLD-TDP [47] may have been premature, at least with respect to genetic associations, since not all c9FTD/ALS are Mackenzie Type 3 (harmonization Type B), and in the present series, Mackenzie Type 1 (harmonization Type A) was actually more common, especially in those patients who presented with dementia (amnestic or frontal-behavioral variants) and no evidence of motor neuron degeneration.
Supplementary Material
Acknowledgments
We are grateful to all patients, family members, and caregivers who agreed to brain donation, without which these studies would have been impossible. We also acknowledge expert technical assistance of Monica Casey-Castanedes for immunohistochemistry, Linda Rousseau and Virginia Phillips for histology and John Gonzalez, Beth Marten, Pamela Desaro and Amelia Johnston for brain banking. This research was funded by Mayo Foundation (Jacoby Professorship of Alzheimer Research, Research Committee CR Program; ALS Center donor funds); National Institutes of Health (P50-AG16574, P50-NS72187, P01-AG03949, P01-AG17216, R01-AG37491, R01-NS65782 and R01-AG26251), the ALS Association; CurePSP∣The Society for Progressive Supranuclear Palsy; and the State of Florida Alzheimer Disease Initiative.
References
- 1.Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 3.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 4.Boxer AL, Mackenzie IR, Boeve BF, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011;82:196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 6.Brandmeir NJ, Geser F, Kwong LK, et al. Severe subcortical TDP-43 pathology in sporadic frontotemporal degeneration with motor neuron disease. Acta Neuropathol. 2008;115:123–131. doi: 10.1007/s00401-007-0315-5. [DOI] [PubMed] [Google Scholar]
- 7.Broe M, Hodges JR, Schofield E, et al. Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology. 2003;60:1005–1011. doi: 10.1212/01.wnl.0000052685.09194.39. [DOI] [PubMed] [Google Scholar]
- 8.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 9.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dickson D, Josephs K, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 2007;114:71–79. doi: 10.1007/s00401-007-0234-5. [DOI] [PubMed] [Google Scholar]
- 12.Dickson DW, Crystal HA, Mattiace LA, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 13.Dickson DW, Josephs KA, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol. 2007;114:71–79. doi: 10.1007/s00401-007-0234-5. [DOI] [PubMed] [Google Scholar]
- 14.Dickson DW, Wertkin A, Kress Y, Ksiezak-Reding H, Yen SH. Ubiquitin immunoreactive structures in normal human brains. Distribution and developmental aspects. Lab Invest. 1990;63:87–99. [PubMed] [Google Scholar]
- 15.Fujioka S, Wszolek ZK. Clinical Aspects of Familial Forms of Frontotemporal Dementia Associated with Parkinsonis. J Mol Neurosci. 2011 doi: 10.1007/s12031-011-9568-5. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujishiro H, Ferman TJ, Boeve BF, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67:649–656. doi: 10.1097/NEN.0b013e31817d7a1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geser F, Brandmeir NJ, Kwong LK, et al. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65:636–641. doi: 10.1001/archneur.65.5.636. [DOI] [PubMed] [Google Scholar]
- 18.Geser F, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. 2010;30:103–112. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geser F, Stein B, Partain M, et al. Motor neuron disease clinically limited to the lower motor neuron is a diffuse TDP-43 proteinopathy. Acta Neuropathol. 2011;121:509–517. doi: 10.1007/s00401-011-0797-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gijselinck I, Engelborghs S, Maes G, et al. Identification of 2 Loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–616. doi: 10.1001/archneurol.2010.82. [DOI] [PubMed] [Google Scholar]
- 21.Gitcho MA, Bigio EH, Mishra M, et al. TARDBP 3′-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 2009;118:633–645. doi: 10.1007/s00401-009-0571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27:48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- 23.Hatanpaa KJ, Bigio EH, Cairns NJ, et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67:271–279. doi: 10.1097/NEN.0b013e31816a12a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inukai Y, Nonaka T, Arai T, et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Letters. 2008;582:2899–2904. doi: 10.1016/j.febslet.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 25.Jellinger KA, Attems J. Incidence of cerebrovascular lesions in Alzheimer's disease: a postmortem study. Acta Neuropathol. 2003;105:14–17. doi: 10.1007/s00401-002-0634-5. [DOI] [PubMed] [Google Scholar]
- 26.Josephs KA, Ahmed Z, Katsuse O, et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007;66:142–151. doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- 27.Josephs KA, Dickson DW. Hippocampal sclerosis in tau-negative frontotemporal lobar degeneration. Neurobiol Aging. 2007;28:1718–1722. doi: 10.1016/j.neurobiolaging.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118:349–358. doi: 10.1007/s00401-009-0547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Josephs KA, Whitwell JL, Parisi JE, et al. Caudate atrophy on MRI is a characteristic feature of FTLD-FUS. Eur J Neurol. 2010;17:969–975. doi: 10.1111/j.1468-1331.2010.02975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 31.Kelley BJ, Haidar W, Boeve BF, et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol Aging. 2009;30:739–751. doi: 10.1016/j.neurobiolaging.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi Z, Tsuchiya K, Arai T, et al. Clinicopathological characteristics of FTLD-TDP showing corticospinal tract degeneration but lacking lower motor neuron loss. J Neurol Sci. 2010;298:70–77. doi: 10.1016/j.jns.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 34.Kovacs GG, Murrell JR, Horvath S, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–1847. doi: 10.1002/mds.22697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laaksovirta H, Peuralinna T, Schymick JC, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Ber I, Camuzat A, Berger E, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- 37.Lin WL, Castanedes-Casey M, Dickson DW. Transactivation response DNA-binding protein 43 microvasculopathy in frontotemporal degeneration and familial Lewy body disease. J Neuropathol Exp Neurol. 2009;68:1167–1176. doi: 10.1097/NEN.0b013e3181baacec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am J Pathol. 2003;162:213–218. doi: 10.1016/S0002-9440(10)63812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipton AM, White CL, Bigio EH., 3rd Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 40.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 41.Luty AA, Kwok JB, Thompson EM, et al. Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackenzie I, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathologica. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackenzie IR. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114:49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- 44.Mackenzie IR, Baker M, Pickering-Brown S, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 45.Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 46.Mackenzie IR, Feldman H. Neuronal intranuclear inclusions distinguish familial FTD-MND type from sporadic cases. Dement Geriatr Cogn Disord. 2004;17:333–336. doi: 10.1159/000077166. [DOI] [PubMed] [Google Scholar]
- 47.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Momeni P, Schymick J, Jain S, et al. Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol. 2006;6:44. doi: 10.1186/1471-2377-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morita M, Al-Chalabi A, Andersen PM, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 51.Murphy JM, Henry RG, Langmore S, et al. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530–534. doi: 10.1001/archneur.64.4.530. [DOI] [PubMed] [Google Scholar]
- 52.Murray ME, Graff-Radford NR, Ross OA, et al. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neumann M, Kwong LK, Truax AC, et al. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol. 2007;66:177–183. doi: 10.1097/01.jnen.0000248554.45456.58. [DOI] [PubMed] [Google Scholar]
- 54.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 55.Pao WC, Dickson DW, Crook JE, et al. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011 doi: 10.1097/WAD.0b013e31820f8f50. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearson JP, Williams NM, Majounie E, et al. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011;258:647–655. doi: 10.1007/s00415-010-5815-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pikkarainen M, Hartikainen P, Alafuzoff I. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions visualized with ubiquitin-binding protein p62 immunohistochemistry. J Neuropathol Exp Neurol. 2008;67:280–298. doi: 10.1097/NEN.0b013e31816a1da2. [DOI] [PubMed] [Google Scholar]
- 58.Rademakers R, Baker M, Gass J, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C-->T (Arg493X) mutation: an international initiative. Lancet Neurol. 2007;6:857–868. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 59.Renton AE, Majounie E, Waite A, et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rutherford NJ, Zhang YJ, Baker M, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sampathu DM, Neumann M, Kwong LK, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shatunov A, Mok K, Newhouse S, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan CF, Eguchi H, Tagawa A, et al. TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 2007;113:535–542. doi: 10.1007/s00401-007-0206-9. [DOI] [PubMed] [Google Scholar]
- 65.Valdmanis PN, Dupre N, Bouchard JP, et al. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64:240–245. doi: 10.1001/archneur.64.2.240. [DOI] [PubMed] [Google Scholar]
- 66.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Es MA, Veldink JH, Saris CG, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- 68.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 69.Wider C, Uitti RJ, Wszolek ZK, et al. Progranulin gene mutation with an unusual clinical and neuropathologic presentation. Mov Disord. 2008;23:1168–1173. doi: 10.1002/mds.22065. [DOI] [PubMed] [Google Scholar]
- 70.Zhang H, Tan CF, Mori F, et al. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;115:115–122. doi: 10.1007/s00401-007-0285-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.