

Abstract
Novel monomethylauristatin E (MMAE) prodrug 8 was designed and prepared that bound cell surface glycoprotein integrin αvβ3, and was activated using legumain protease as a catalyst. Upon activation, prodrug 8 strongly induced the death of MDA-MB-435 cells that express integrin αvβ3 on cell surface. Efficacies of prodrug 8 were determined in vivo using animal models of 4T1 murine breast cancer, D121 Lewis lung carcinoma, and MDA-MB-435 human breast cancer. The results demonstrated that prodrug 8 decreased tumor growth and metastasis effectively. In comparison to the parent cytotoxin, MMAE, and prodrug 3, prodrug 8 was less toxic to mould white blood cells. The latter caused no loss in weight gain of mice at a dose 3 mg/kg, which is over 30 times in excess to MMAE (0.1 mg/kg). We hypothesize that overexpression and co-localization of integrin αvβ3 and legumain protease on tumor cells, tumor vasculature, and/or tumor microenvironments can be exploited to enhance the efficacy and selectivity of potent cytotoxins, such as MMAE, which is otherwise too toxic to use for therapy.
Keywords: Monomethyleauristatin E (MMAE), Prodrug, Integrin, Legumain, Prodrug activation
INTRODUCTION
Many potent cytotoxins, including enediynes, epothilones, duocarmycins, dolastatins, tubulysins, etc. possess subnanomolar cytotoxicity in in vitro assays, yet they have only limited therapeutic efficacies in vivo at a dose that is not lethal to animals.1 Fortunately, such potent cytotoxins can be formulated as prodrugs, and delivered to tumor or in tumor microenvironments (TMEs) using various tumor-targeting agents, including monoclonal antibodies (Abs) or a small molecule inhibitors.2 Previously, we have synthesized and examined a series of doxorubicin,3,4 enediyne5 and duocarmycin analog.6 Prodrugs that are activated using the monoclonal aldolase Ab 38C27 or 93F3.8 We have also prepared and evaluated several monomethylauristatin E (MMAE, 1) and didesmethylauristatin E (DDAE, 2) prodrugs, including 3 and 4 (Fig. 1),9 that recruit tumor-associated protease (TAP), legumain, for their activation.10 Based on our in vitro studies with these prodrugs, prodrug 3 was chosen and its efficacy was determined in vivo using animal models of murine breast cancer 4T1; prodrug 3 reduced growth of 4T1 tumor by 57% at 0.5 mg/kg, whereas all animals died when parent compound 1 was used at this dose. We argued that the therapeutic efficacies of prodrug 3 could be further enhanced by directing it, in the form of a small molecule-prodrug or an antibody (Ab)-prodrug conjugate, to tumor cells and in TMEs, where legumain protease will catalyze the release of free drugs.
Fig. 1.

Auristatin E analogs and their prodrugs; MMAE, monomethylauristatin E; DDAE, Dodesmethylauristatin E.
Legumain is an asparaginylendopeptidase with a remarkably restricted specificity for asparagine at P1 site of the substrate sequence.11 It is an evolutionary offshoot of the C13 family of cysteine proteases,12 initially identified in plants as a processing enzyme of storage proteins during seed germination, and later also identified in parasites and in mammals. Legumain is present intracellularly in endosome/lysosome systems (13,14), and associated with intracellular protein degradation, but also overexpressed in a majority of tumors, including carcinomas of the breast, colon, and prostate, in central nervous system neoplasms,10 and secreted to tumor cell surface and in the TMEs. Because legumain is active at low pH, i.e., 4.0-6.5, and gets deactivated at a physiologically neutral pH, it is uniquely set to catalyze prodrug activation in acidic TMEs and in tumor cells. Interestingly, legumain protease co-localize with various integrins, including integrin αvβ3 on cell surface and interact with each other, and both are overexpressed in many tumor cells and/or in tumor vasculature, and are highly implicated in tumor growth and metastases. Integrin αvβ3, a transmembrane glycoprotein composed of α and β subunits, is a major target implicated in tumor angiogenesis.15 In response to a cellular stimulation, the extracellular domain of integrin αvβ3 is activated, thereby changing its conformation from a low-affinity ligand-binding state to a high-affinity state.16,17 Integrin αvβ3 expression level is high and its activation has been detected in different types of tumors, including prostate,18 breast,19 melanomas,20 gliomas,21 and ovary.22 Indeed, αvβ3 integrin has become one of the most valued targets for imaging and drug targeting.23
In this article, we describe in vitro and in vivo evaluation of an integrin inhibitor-MMAE prodrug conjugate 8, which combines properties of prodrug 39 with a previously described low molecular weight inhibitor 6 of integrin αvβ3.24,25 Presumably, prodrug 8 that is prepared using an MMAE derivative 5 and αvβ3 integrin inhibitor 7 (See Supporting Information, SI) can target to integrin αvβ3, and recruit legumain protease to catalyze the prodrug activation selectively in tumor cells or in TMEs. Here, we first examined co-localization of legumain protease with integrins αvβ3 on tumor cell surface, and catalytic activity of the former in various tissues. Inhibitory effects of the inhibitor-prodrug conjugate 8 on cell proliferation, tumor growth and metastasis were determined in vitro and in vivo using 4T1 murine breast cancer and D121 Lewis Lung cancer, and MDA-MB-435 human breast cancer cell lines. Notably, MDA-MB-435 has high expression of integrin αvβ3 on cell surface.26 With these results, we emphasize that targeting cell surface αvβ3 and αvβ5 integrins and recruiting tumor-associated protease for prodrug activation would greatly increase efficacies of a cytotoxin, such as MMAE, which may otherwise be too toxic to have any therapeutic value.
MATERIALS AND METHODS
Antibody and Tumor Cell Lines
The mouse 4T1, D121, and RAW264.7, and human MDA-MB-435 cell lines were obtained from ATCC. Human embryonic kidney 293 cells stably expressing human legumain (HEK-293L) were kindly provided by Dr. G. David Roodman (Department of Medicine and Hematology, University of Texas Health Science Center, San Antonio, TX). RAW264.7 cells were maintained in Dulbecco’s modified Eagle’s/Ham’s F12 medium supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotics. 4T1, D121, and MDA-MB-435 cells were grown in DMEM medium with 10% fetal bovine serum. Mouse anti-human legumain (H00005641-MO2) monoclonal Ab was purchased from (Abnova). Rabbit anti-human legumain polyclonal Ab was affinity purified from rabbit serum, which was prepared by immunization with CGMKRASSPVPLPP peptide conjugated to keyhole limpet hemocyanin (KLH).
Integrin αvβ3-targeted legumain activated MMAE prodrug 8
To a mixture of compounds 5 (108 mg, 0.09 mmol) and 7 (50 mg, 0.09 mmol) in DMF (2 ml) were added Cu powder (4 mg, 0.06 mmol) and aqueous CuSO4 (1M, 20 μl, 0.02 mmol), and the mixture was stirred at 40 °C for 24 hrs. After the reaction was complete, as determined using LC-MS, solvents were removed under reduced pressure, and the residue was purified using HPLC affording compound 8 (90 mg, 57%). HRMS (ESI): Calcd. for C94H128N17O9S (M+H)+ 1830.9217; Found, 1830.9117.
Confocal immunoanalysis
Immunofluorescent double staining was performed on hypoxia culture human MDA-MB-435 and HEK-293L cells, which were treated with 10 nM EFG in fibronectin coating flask for 3 days. We dissected MDA-MB-435 tumor to make frozen section (5 μm) for double staining. For staining of integrin, mouse monoclonal Ab to integrin αvβ3 (0.1 μg/ml) were used as first Ab and Texas-red conjugated anti-mouse IgG (Vector Laboratories) was used as the secondary reporting reagent. For legumain identification, Legumain Polyclonal Ab was used at 0.5 μg/ml, followed by FITC-conjugated anti-Rabbit IgG (Vector Laboratories) as the second antibody. Nuclei were stained with DAPI. The slides were analyzed by using Bio-Rad Radiance 2100 Rainbow laser scanning confocal microscope (LSCM). Equal concentrations of mouse IgG, rabbit IgG and secondary antibodies were used as the negative controls.
Proteolytic activities of cells and tissues
To determine the proteolytic activity in the cell culture media, cells were incubated under hypoxic conditions for 3 days, and the cultured media (200μl) were diluted using an activity assay buffer (750 μl) (50 mM citric acid, 121 mM Na2HPO4, pH 6.0, containing 1 mM DTT, 1 mM EDTA, and 0.1% CHAPS). Subsequently, a solution of Z-Ala-Ala-Asn-NHMec (50 μl, final concentration 10 M) was added to the diluted media (950 μl) and incubated at 37 °C for 30mins, and activation was determined by the fluorescence measurement using a Perkin-Elmer LS-50-B spectrofluorometer (excitation: 360 nm; emission: 460 nm). Similarly, to examine the distribution of legumain activity in tumor and various organs, equal weight of 4T1 murine breast cancer tumor and normal tissues were homogenized in OG (octyl glucoside) buffer (50 mM OG, 50 mM Tris, 150 mM NaCl, 1 mM DTT, 1 mM EDTA pH 6.0) to make single cell suspension. After the suspensions were incubated with substrate at room temperature for 1 hour, fluorescence of NH2Mec was determined, and compared to provide the legumain activity in tumor and in organs.
Cytotoxicity assays
Cytotoxicity assays were performed as described previously. Typically, HEK-293L and MDA-MB-435 cells were incubated with serial dilutions of cytotoxic agent and prodrugs (compounds) in a 96-well tissue culture plate for 2 days. Cells were processed using the CellTiter 96TM non-radioactive cell proliferation kit (Promega), as described in the instructions. OD (450 – 630 nM) of the processed cells was measured by using an ELISA micro plate reader (BIO-RAD450). Dose-response curves were plotted as percent of non-exposed control cells.
Maximum tolerated dose
Four six-week-old BALB/c mice were used in each experimental group. The mice were weighed individually, and the average weight of the group was used to define the doses. Mice were given i.p injection of the indicated amounts of MMAE or prodrugs daily for 5 days. The maximum tolerable dose (MTD) was defined as the maximum drug dose administered to non-tumor-bearing mice once daily for 5 consecutive days without mortality.
Blood cell toxicity
250 μL of fresh blood (with EDTA) was collected from each mouse eye. Cells were diluted to 1 to 1000 in PBS. 10 μL cells were applied to a hemocytometer and counted the number of RBC per large square. RBC cell number was: counts/large square × 1,000 dilution × 10 large squares/μL = RBC/μL blood. To perform white blood cell count, 10 μL whole blood was mixed and incubated with 190 μL of RBC lysing reagent for 1 min. Then 10 μL of lysed blood were counted the number of WBC per large square. WBC cell number was: counts/large square × 20 dilution × 10 large squares/μL = WBC / μL blood.
Mouse tumor models
The 4T1 murine breast carcinoma model, D121 Lewis Lung cancer and MDA-MB-435 human breast cancer models were generated and maintained at The Scripps Research Institute. 4T1, D121, and MDA-MB-435 cells (1 × 106) were injected separately s.c. on the back of 4 to 8 weeks old BALB/c, C57BL/6J, and Hsd:Athymic nude mice, repectively. Starting on day 5, when the tumors were approximately 100 mm3 (for 4T1 and D121 model), the tumor bearing mice were i.p. injected with saline alone (control group) and prodrug 8 with the indicated dosage and treatment schedule (n = 6 per group). Tumor growth and signs of physical discomfort were monitored daily including for any gross evidence of tumor necrosis, local tumor ulceration, as well as evidence of toxicity including the mobility of animals, response to stimulus, piloerection, eating, and weight. Tumor volumes of treated animals were measured every three days in 4T1 and D121 models or five days in MDA-MB-435 model by microcaliper (volume = length × width × width/2). As soon as the tumor volume reached 1500 mm3 in the control groups (500 mm3 in the control groups for MDA-MB-435 model), the tumor were removed. All mice were euthanized and removed lungs to the Bouin’s solution after two weeks of removing tumor. Spontaneous lung metastases were counted under anatomy microscope. All experiments were conducted at The Scripps Research Institute facilities using the protocols reviewed and approved by the Institutional Animal Care and Use Committee.
Statistical analysis
Results are expressed as means±s.e.m. Student paired t-test was used to analyze the difference between two groups. Values were regarded significant at P < 0.05.
Results
Integrin αvβ3 and legumain protease are Co-localized on MDA-MB-435 Human Breast Cancer Cells
To confirm that integrin αvβ3 and the cysteine protease legumain are indeed co-localized on tumor cells, we used MDA-MB-435 human breast cancer cells, and determined the legumain and integrin expressions using the confocal immunoanalysis after the cells were cultured under the hypoxia condition. Integrin expression was determined using the anti-αvβ3 integrin mouse monoclonal antibody and the anti-legumain polyclonal rabbit antibody together with the appropriate secondary antibodies. As shown in Fig. 2A, legumain protease was most abundantly visualized in lysosome and endosome. This is in consistent with the fact that legumain is involved in the processing of various intracellular proteins and proteases27 and the delivery of membranous vesicles containing proteases, actin-binding proteins and adhesion molecules toward the leading edge of migratory cells has been implicated in cell locomotion.28 However, legumain is also present on cell surface of MDA-MB-435 in lamellipodia where it associates and co-localizes with the adhesion protein integrin αvβ3 (Fig. 2A). Presumably, association of the legumain with integrin αvβ3 takes place through the RGD domain present in former to the RGD-dependent integrin.13 On the other hand, studies with HEK 293L cells show that they do not express integrinαvβ3, nor there are any colocalization of integrin αvβ3 with legumain, as expected (Fig. 2B). Co-localization of legumain with integrin αvβ3 is also accompanied by the appearance of legumain activity in MDA-MB-435 culture medium (Fig. 2C), but not in HEK 293L culture medium.
Fig. 2. Legumain is exclusively colocalized with integrin αvβ3 on the MDA-MB-435 cancer cell surface.

(A) Legumain (green) is detected in intracellular vesicles and prominently co-localized with integrin αvβ3 (red) under hypoxia. Cell nuclei are stained with DAPI (blue). Double staining is performed with anti-integrin antibodies and anti-legumain antibody. Stained cells are imaged by confocal microscopy, and the slice closest to the coverslip is presented for each cell. Note that legumain is localized on MDA-MB-435 cell surface (green, white arrow). (B) High expression legumain is only detected in intracellular vesicles of human HEK-293L cells. HEK-293 cells do not express legumain and integrin αvβ3 on cell surface. (C) MDA-MB-435 cells secret more active legumain into the culture medium than human HEK-293L does. The experiments were repeated three times (p<0.001).
Distribution of Active Legumain Protease is high in Tumor cells and tissues
Legumain protease is an essential enzyme found in all cells, however the catalytic activity of tumor tissues is invariably high in most tumors and TMEs than it is in normal tissues. This is primarily because legumain protease is overexpressed in/on tumor cells and in TMEs, and that the intracellular compartments of the tumor cells and the TMEs are more acidic than the normal cells. It should be noted that the legumain protease is active at low pH, and virtually inactive at the physiologic pH.29 To determine and compare the active legumain protease distribution, tumor tissues and tissues of various organs from a tumor-bearing mice were used and the protease activity was determined using Z-Ala-Ala-Asn-NHMec as a substrate. The latter undergoes legumain-catalyzed hydrolysis affording the fluorescent 7-aminomethylcoumarin (Mec-NH2). As shown later in Fig. 4D, the legumain activity was indeed high in tumor tissues as compared to that in normal tissues of various organs indicating that the legumain-catalyzed activation of the MMAE prodrugs would take place primarily in the tumor tissue.
Fig. 4.

Prodrug 8 suppresses 4T1 tumor growth and lung metastasis. (A) In vivo effect of prodrug 8 on 4T1 mammary carcinoma. Starting on day 5, when the tumors averaged ≈ 4 mm in diameter, the 4T1 tumor mice were treated with 0.5, 1.5 and 3 mg/kg of the prodrug 8 separately on day 5, 8, 11, 14 and 17. without any toxicity, but 3 mg/kg dosage of prodrug 3 caused mouse death. 6 mice were treated in each group; (p<0.001). (B) Prodrug 8 treatment induces cell death in TUNEL staining of 4T1 tumor sections. (C) Prodrug 8 suppresses spontaneous lung metastasis in 4T1 mouse breast cancer models (p<0.001). (D) Legumain activity of tumor tissue is significantly higher than that of normal tissues. The experiments were repeated in three mice (p<0.01).
Targeting integrin αvβ3 enhances tumor-selectivity of the MMAE prodrug 8
We have earlier shown that both cytotoxin 1 and prodrug 3 possess subnanomolar to low nanomolar activities to MDA-MB-435 cells (using 72 hrs incubation period).9 We anticipated that the integrin-targeted MMAE prodrug 8 should mediate higher and selective antitumor effects than both the parent cytotoxin 1 and prodrug 3 to MDA-MB-435 cells, which overexpress both legumain protease and αvβ3 integrin, through localizing in tumor cells and TMEs, in vivo. In addition, there shouldn’t be a minimum difference in the activity of prodrugs 3 and 8 to HEK-293L cells that donot express cell surface αvβ3 integrin, but have high level of legumain protease stably expressed, and the activity of the prodrugs should approach to the cytotoxicity of the parent cytotoxin 1. To assess our hypothesis, first we determined the in vitro cytotoxicity of 1, 3 and 8 to both MDA-MB-435 and HEK-293L cells (using 48 hrs incubation period). The results are shown in Figs. 3A and 3C. Evidently, cytotoxin 1 was more potent than the prodrugs 3 and 8 to both cell lines, and prodrug 8 was also less cytotoxic than 3 to MDA-MB-435 cells, whereas they were equipotent to HEK-293L cells. Next, to mimic the in vivo condition and determine the integrin-targeting effects of the prodrug 8, we examined its activity to MDA-MB-435 cells under a modified cytotoxicity assay condition. Here, cells were incubated with 1, 3 and 8 at different concentrations at 37 °C for 1 hour, and media were replaced with the fresh one with no additional cytotoxin or prodrug and incubation was continued for an additional 47 hrs. The results are shown in Fig. 3B. Under this condition, prodrug 8 was found more effective and superior than both the parent cytotoxin 1 as well as its prodrug 3.
Fig. 3.

Prodrug 8 enhances enhances cytotoxicity to MDA-MB-435 cancer cell (A) HEK 293L cell cytotoxicity assay of MMAE, prodrug 3 and prodrug 8 incubation for 48 h. (B) MDA-MB-435 cell cytotoxicity assay of MMAE, prodrug 3 and prodrug 8 incubation for 1 h and then removing the unbound compound by change medium.
Prodrug 8 has low toxicity and is stable in serum
Before carrying out the in vitro and in vivo assays, we assessed the stability of prodrug 8 by mixing the compound with mouse blood, and analyzing the reaction mixture after it was incubated at 37 °C for 16 h using LC-MS. There were no degradation products observed showing that the prodrug was stable in serum. Prodrug 8 is significantly less toxic than prodrug 3 when evaluated in vivo. Prodrug 8 had a much higher cumulative MTD and reduced LD50 compared with prodrug 3 (Table 1A), when given by five repeat i.p. administrations. For a comparison, the previously-described prodrug 3 and cytotoxin 1 were administered at 0.1, 0.5 and 1.5 mg/kg. Buffer alone was used as a control. All animals were weighed periodically starting on day 1 for 3 weeks. The results shown in (Table 1B) confirmed that the prodrug 8 (3 mg/kg) had no toxicity, whereas the previously-described legumain activated prodrug 3 possessed some toxicity (1.5 mg/kg). Again cytotoxin 1 was extremely toxic at (0.1 mg/kg), and comparable to the previously reported results. We further evaluated the toxicity of compound 8 and compared to 3 by examining their effects on white blood cells and red blood cells (Table 1C). Whereas both compounds had no appreciable effect on red blood cells when animals were treated with a single injection of the compounds at 9 mg/kg, WBC counts were significantly reduced for the prodrug 3, but not for compound 8, which showed no appreciable reduction in the WBC counts at the same dose.
Table 1.
In vitro and in vivo toxicity of Prodrug 8 compared with MMAE and Prodrug 3a
| A. Estimated MTD and LD50 of prodrugs 3 and 8 (mg/kg) in BALB/c mice | ||||
| 3 | 8 | |||
| MTD | LD50 | MTD | LD50 | |
| I.p. comm. | 15 | 12 | >60 | >60 |
| I.p. 5x | 3 | 1.5 | >6 | >6 |
| B. Comparison of gross toxicity of mice; i.p. 5 x | |||||
| I.p. 5x Dose (mg/kg) | 0.1 | 0.5 | 1.5 | 3 | |
| MMAE, 1 | Wt. loss (%) | 33.4 | |||
| Death (%) | 20 | 100 | 100 | ||
| Prodrug 3 | Wt. loss (%) | 10.8 | 12.7 | 38.7 | |
| Death (%) | 0 | 0 | 40 | 100 | |
| Prodrug 8 | Wt. loss (%) | 0 | 3.4 | 8.2 | 14.9 |
| Death (%) | 0 | 0 | 0 | 10 | |
| C. Comparison of blood cell density of mice; i.p. 1 x Dose, 9 mg/kg | |||
| day 0 | day 4 | ||
| PBS | WBC (109/L) RBC (1012/L) |
8.1±1.7 9.1±1.5 |
7.8±1.5 9.3±1.4 |
| Prodrug 3 | WBC (109/L) RBC (1012/L) |
7.8±1.7 9.0±1.4 |
4.4±2.3 8.1±1.9 |
| Prodrug 8 | WBC (109/L) RBC (1012/L) |
8.2±1.8 9.4±1.4 |
7.6±1.1 9.2±2.0 |
Key: See Materials and methods for the experimental details.
The integrin inhibitor-targeted prodrug 8 suppresses tumor growth and metastasis effectively
Next, we examined the antitumor effects of the integrin-targeted prodrug 8 using the primary tumor models of murine 4T1 mammary carcinoma and D121 Levis lung carcinoma cell lines. Both 4T1 and D121 cells have high legumain expression.30,31 4T1 tumor models were generated by s.c. injection of 1 × 106 cells in the right flank of six-week-old BALB/c mice. A dose-response assessment shows that administration of compound 8 (0.5, 1.5 or 3.0 mg/kg) on day 5, 8, 11, 14 and 17 suppresses tumor growth (Fig. 4A). Prodrug 8 has a better effer in suppression tumor growth than prodrug 3 in dosage of 0.5mg/kg. High dosage of prodrug 8 (3mg/kg) can be applied without any toxicity, but 1.5 mg/kg dosage of prodrug 3 caused mouse death. TUNEL assay, which is based on in situ labeling of DNA fragmentation sites in nuclei of intact fixed cells on tumor sections, shows that the treatment with prodrug 8 inducd tumor cell death (Fig. 4B). Significantly, the treatment with prodrug 8 also blocked spontaneous metastasis of 4T1 tumor to lungs (Fig. 4C). Similarly, to determine the effect of compound 8 on growth of the D121 Levis lung tumor model, tumors were generated by s.c. injection of 1 × 106 D121 cells in the right flank of six-week-old C57BL/6J mice, and compound 8 (3.0 mg/kg) was i.p. administered on day 5, 8, 11, 14 and 17. Evidently, compound 8 suppressed tumor growth significantly and blocked the spontaneous metastasis to lungs (Figs. 5A and 5B).
Fig. 5. Prodrug 8 suppresses D121 tumor growth and lung metastasis.
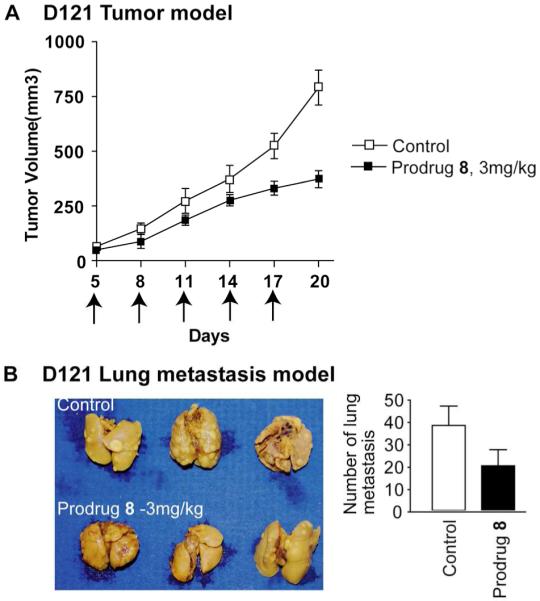
(A) In vivo effect of Prodrug 8 on D121 Lewis lung carcinoma. Starting on day 5, when the tumors averaged ≈4 mm in diameter, the D121 tumor mice were treated with 3 mg/kg separately on day 5, 8, 11, 14 and 17. 6 mice were used in each group; (p<0.001). (B) Prodrug 8 suppresses spontaneous lung metastasis in D121 mouse models; (p<0.001).
Prodrug 8 eradicates tumors of human breast carcinoma
The expression and presence of co-localization of legumain and integrin αvβ3 is supported by cofocal analysis of MDA-MB435 tumor double staining sections (Fig. 6A). We further examined the efficacy of prodrug 8 using human MDA-MB-435 tumor model in Hsd:Athymic nude mice, which showed a significant growth inhibition in animals treated with 3 mg/kg with the prodrug (Fig. 6B, Left). Animals showed no weight loss or any apparent signs of toxicity, and were live during the whole period of the in vivo study. In contrast, animals treated with only vehicle had large tumors, and died within 65 days (Fig. 6B, Right).
Fig. 6. Tumoricidal effect of Prodrug 8 in MDA-MB-435 carcinoma in vivo.

(A) Confocal analysis of legumain (green) and integrin αvβ3 (red) in MDA-MB-435 tumor tissue. (B) In vivo effect of Prodrug 8 on MDA-MB435 carcinoma. Starting on day 15, when the tumors averaged ≈5 mm in diameter, the MDA-MB435 tumor mice were treated with Saline alone, and Prodrug 8 (3mg/kg) on day 15, 20, 25, 30, 35 and 40; The experiment were repeated twice and 6 mice were used in each treated group; (p<0.01). (b) Kaplan-Meier survival curves of mice bearing MDA-MB435 tumors in Control and Prodrug 8 groups. The survival was based on the primary tumor diameter (>1.5 cm) and natural death.
Discussion
Cell-targeted prodrug therapy approach, involving cell surface adhesion molecules to direct a prodrug to tumor cells and tumor-associated proteases for the prodrug activation, is likely to enhance the therapeutic indexes of many anticancer agents that are otherwise toxic and not suitable for therapy. Prior studies with MMAE and analogous cytotoxin prodrugs that are activated by cathepsin B protease and conjugated to monoclonal antibodies or the RGD peptides revealed that tumor-targeting is essential to achieve high efficacies with these prodrugs.32-34 The concept of such dual targeted prodrugs an further benefit by the readily available technologies, including the positional gene expression profiling of tumor tissues and imaging studies, which can identify suitable tumor-associated cell surface receptors and proteases for the prodrug targeting and activation.35 Indeed, the MMAE prodrug 8 was designed after we established that integrin αvβ3 and the legumain protease are not only overexpressed, but they also co-localized on certain cancer cells, including MDA-MB-435 cells (Fig. 2). As expected, prodrug 8 possessed the tripeptide linker (P1-Asn, P2-Ala, P3-Ala) identical to that in prodrug 3 (Fig. 1), which is in keeping with other legumain-activatable prodrugs, and both prodrugs 8 and 3 were fully activated upon overnight incubation with a catalytic amount of legumain.9
The cytotoxicity studies using MMAE and its prodrugs, 3 and 8, revealed that the dual targeted prodrug 8 was least toxic and more effective than both MMAE and prodrug 3. It should be noted that prodrug 8 was indeed more potent than MMAE and prodrug 3 when the cell culture media were exchanged after a brief incubation (Fig. 3B), which supports the hypothesis that targeting integrin αvβ3 enhances the accumulation of prodrug 8 in tumor, tumor vasculature and/or TMEs in high concentration, where legumain converts the prodrug to the active drug MMAE, 1. In turn, the latter induces tumor cell death and prevents cancer cell invasion and distant metastasis. The fact that prodrug 3 showed higher cytotoxicity than prodrug 8 under the normal cytotoxicity evaluation conditions to MDA-MB-435 cells is however interesting. This could be explained, if the prodrugs 8 is weakly activated than 3, which appears less likely as the both prodrugs showed comparable cytotoxicity to HEK-293L cells that expresses neither integrin αvβ3 nor the legumain protease on cell surface. We argue that prodrug 8 functions both as an integrin inhibitor and as a prodrug, and the selective toxicity due to prodrug activation is realized only after the prodrug gets internalized. In contrast, prodrug 3 is also activated extracellularly. It should be noted that the legumain protease is secreted to cell surface in MDA-MB-435 cells, but not in HEK-293L cells.
Integrin αvβ3, which is overexpressed and activated on invasive tumor cells and on angiogenic blood vessels in tumor tissues, fulfills growth promoting and invasive enhancement of the sprouting endothelial cells.36 As expected, an inhibition of αvβ3 integrin using low molecular weight inhibitors and Abs, including cyclic RGD peptide inhibitors and a humanized anti-αvβ3 Ab MEDI-522, have shown positive effects in patients. Thus, in addition to selective targeting and prodrug activation, prodrug 8 could also have inhibited tumor growth and metastases through inhibiting integrin αvβ3 owing to the inhibitor component of the molecule. Importantly, the integrin-targeting MMAE prodrug 8 is also likely to mediate highly selective anti-tumor effect through localizing in tumor cells and TMEs, but not in normal tissues, especially from kidney, liver, and lung, which also possess considerable protease activity.30
Previous examples of the prodrugs that are activated using legumain protease include those derived from doxorubicin30 and camptothecin.37 For cardiac tissue, previous legumain activated doxorubicin prodrugs has shown reduced accumulation >15-fold and has a notable advantage of less cardiotoxic. In this study, prodrug 8 is also a tumor microenvironment activated prodrug that is catalytically converted to end product MMAE in the tumor microenvironment. The activation of prodrug 8 is not found in any significant amounts in normal tissues presumably as a result of no active legumain location on healthy native cell surface. Based on LD50, the toxicity of prodrug 8 in mouse was reduced >30-fold compared with MMAE. Prodrug 8 is stable in plasma, and has little effect on cells of myeloid lineage, as mice showed negligible reduction in peripheral blood or marrow myeloid cells at elevated therapeutic doses (Table 1).
Based on the reduced toxicity, larger cumulative dosage of prodrug 8 can be administered more rapidly, and significant tumor growth and metastasis inhibition were observed, as shown in Fig. 4. In contrast, both MMAE and prodrug 3 were found considerably toxic beyond 0.1 mg/kg and 0.5 mg/kg to mice. Consequently, in the 4T1 mouse breast carcinoma model, a significant reduced tumor growth was obtained after 4T1 tumor cell inoculation into mice, followed by treatment with the prodrug 8. Fewer lung metastases were found in treatment group as compared to the control group in spontaneous lung metastases experiments. Similar results were obtained with other two tumor models, D121 non-small cell lung carcinoma and human MDA-MB-435 carcinoma. There were greater effects on tumor of MDA-MB-435 carcinoma, which completely stopped and started to shrink upon continued treatment. All animals were alive after 70 days in the treatment group. These data strengthen our contention that integrin αvβ3 targeted and legumain activated prodrug approach could be a useful antitumor strategy for suppressing tumor cell invasion and metastases of many cancers.
In summary, novel dual-targeted MMAE prodrug 8 designed to bind cell surface glycoprotein integrin αvβ3 and activate using legumain protease as the catalyst, was prepared and evaluated both in vitro and in vivo. The antitumor efficacy of prodrug 8 was critically evaluated by using mouse models of three different cancers, which demonstrated that prodrug activation took place in tumor tissues that effectively decreased tumor growth and metastasis. This strategy greatly reduces toxicity to healthy body cells by synergism of integrin binding αvβ3 and legumain activation function, indicating that this anti-tumor strategy could be widely applicable and relevant for possible cancer therapy. We are also using integrin αvβ3 and the legumain protease here as the target and catalyst, respectively, of our first small molecule-prodrug conjugate, because their combination has never been utilized to realize the efficacy of a cytotoxin. In addition, the fact that a non-peptidic small molecule integrin inhibitor, such as 7, is used to direct the MMAE prodrug to tumor cells will broaden the pool of procytoxins, which can be delivered to tumor cells and tumor vasculature through the integrin αvβ3-targeting.
Supplementary Material
ACKNOWLEDGMENT
Authors thank to the US National Cancer Institute (CA120289 to SCS, and CA127535 to CL) and the US Department of Defense (W81XWH-09-1-0690 to SCS, and W81XWH-07-1-0389 to CL) for the funding support.
Footnotes
SUPPORTING INFORMATION. Synthesis and physical data of prodrug 8. This information is available free of charge via the Internet at http://pubs.acs.org/
Conflict of Interests.
Yuan Liu, None.
Krishna Mohan Bajjuri, None.
Cheng Liu, None at the time of the Research Performed.
Subhash C. Sinha, None.
REFERENCES
- (1).Druker BJ. Perspectives on the development of a molecularly targeted agent. Cancer Cell. 2002;1:31–6. doi: 10.1016/s1535-6108(02)00025-9. [DOI] [PubMed] [Google Scholar]
- (2).Singh Y, Palombo M, Sinko PJ. Recent trends in targeted anticancer prodrug and conjugate design. Curr. Med. Chem. 2008;15:1802–26. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sinha SC, Li LS, Watanabe S, Kaltgrad E, Tanaka F, Rader C, Lerner RA, Barbas CF., III Aldolase antibody activation of prodrugs of potent aldehyde-containing cytotoxics for selective chemotherapy. Chemistry. 2004;10:5467–72. doi: 10.1002/chem.200400419. [DOI] [PubMed] [Google Scholar]
- (4).Abraham S, Guo F, Li L-S, Rader C, Liu C, Barbas CF, III, Lerner RA, Sinha SC. Synthesis of the next-generation therapeutic antibodies that combine cell targeting and antibody-catalyzed prodrug activation. Proc. Natl. Acad. Sci. USA. 2007;104:5584–9. doi: 10.1073/pnas.0700223104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sinha SC, Li LS, Miller GP, Dutta S, Rader C, Lerner RA. Prodrugs of dynemicin analogs for selective chemotherapy mediated by an aldolase catalytic Ab. Proc. Natl. Acad. Sci. USA. 2004;101:3095–9. doi: 10.1073/pnas.0307319101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Li LS, Sinha SC. Studies toward the duocarmycin prodrugs for the antibody prodrug therapy approach. Tetrahedron Lett. 2009;50:2933–5. doi: 10.1016/j.tetlet.2009.03.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wagner J, Lerner RA, Barbas CF., III Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science. 1995;270:1797–800. doi: 10.1126/science.270.5243.1797. [DOI] [PubMed] [Google Scholar]
- (8).Zhong G, Lerner RA, Barbas CF., III Broadening the aldolase catalytic antibody repertoire by combining reactive immunization and transition state theory: new enantio- and diastereoselectivities. Angew. Chem. Int. Ed. 1999;38:3738. doi: 10.1002/(sici)1521-3773(19991216)38:24<3738::aid-anie3738>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- (9).Bajjuri KM, Liu Y, Liu C, Sinha SC. The legumain protease-activated auristatin prodrugs suppress tumor growth and metastasis without toxicity. ChemMedChem. 2011;6:54–9. doi: 10.1002/cmdc.201000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liu C, Sun C, Huang H, Janda K, Edgington T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res. 2003;63:2957–64. [PubMed] [Google Scholar]
- (11).Chen JM, Dando PM, Rawlings ND, Brown MA, Young NE, Stevens RA, Hewitt E, Watts C, Barrett AJ. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J. Biol. Chem. 1997;272:8090–8. doi: 10.1074/jbc.272.12.8090. [DOI] [PubMed] [Google Scholar]
- (12).Ishii S. Legumain: asparaginyl endopeptidase. Meth. Enzymol. 1994;244:604–15. doi: 10.1016/0076-6879(94)44044-1. [DOI] [PubMed] [Google Scholar]
- (13).Chen JM, Dando PM, Stevens RAE, Fortunato M, Barrett AJ. Cloning and expression of mouse legumain, a lysosomal endopeptidase. Biochem. J. 1998;335:111–7. doi: 10.1042/bj3350111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shirahama-Noda K, Yamamoto A, Sugihara K, Hashimoto N, Asano M, Nishimura M, Hara-Nishimura I. Biosynthetic processing of cathepsins and lysosomal degradation are abolished in asparaginyl endopeptidase-deficient Mice. J. Biol. Chem. 2003;278:33194–9. doi: 10.1074/jbc.M302742200. [DOI] [PubMed] [Google Scholar]
- (15).Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature Rev. Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alphaVbeta3. Science. 2001;294:339–45. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Guo W, Giancotti FG. Integrin signalling during tumour progression. Nature Rev. Mol. Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- (18).Cooper CR, Chay CH, Pienta KJ. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia. 2002;4:191–4. doi: 10.1038/sj.neo.7900224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liapis H, Flath A, Kitazawa S. Integrin alpha V beta 3 expression by bone-residing breast cancer metastases. Diag. Mol. Pathol. 1996;5:127–35. doi: 10.1097/00019606-199606000-00008. [DOI] [PubMed] [Google Scholar]
- (20).Albelda SM, Mette SA, Elder DE, Stewart R, Damjanovich L, Herlyn M, Buck CA. Integrin distribution in malignant melanoma: association of the beta 3 subunit with tumor progression. Cancer Res. 1990;50:6757–64. [PubMed] [Google Scholar]
- (21).Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. J Neuroimmunol. 1995;57:143–53. doi: 10.1016/0165-5728(94)00178-q. [DOI] [PubMed] [Google Scholar]
- (22).Carreiras F, Denoux Y, Staedel C, Lehmann M, Sichel F, Gauduchon P. Expression and localization of alpha v integrins and their ligand vitronectin in normal ovarian epithelium and in ovarian carcinoma. Gynecol. Oncol. 1996;62:260–7. doi: 10.1006/gyno.1996.0225. [DOI] [PubMed] [Google Scholar]
- (23).Chen K, Chen X. Integrin targeted delivery of chemotherapeutics. Theranostics. 2011;1:189–200. doi: 10.7150/thno/v01p0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Duggan ME, Duong LT, Fisher JE, Hamill TG, Hoffman WF, Huff JR, Ihle NC, Leu CT, Nagy RM, Perkins JJ, Rodan SB, Wesolowski G, Whitman DB, Zartman AE, Rodan GA, Hartman GD. Nonpeptide αvβ3 Antagonists 1. Transformation of a Potent, Integrin-Selective αIIbα3 Antagonist into a Potent αvβ3 Antagonist. J. Med. Chem. 2000;43:3736–45. doi: 10.1021/jm000133v. [DOI] [PubMed] [Google Scholar]
- (25).Li LS, Rader C, Matsushita M, Das S, Barbas CF, III, Lerner RA, Sinha SC. Chemical-adaptor immunotherapy: design, synthesis and evaluation of novel integrin-targeting devices. J. Med. Chem. 2004;47:5630–40. doi: 10.1021/jm049666k. [DOI] [PubMed] [Google Scholar]
- (26).Furger KA, Allan AL, Wilson SM, Hota C, Vantyghem SA, Postenka CO, Al-Katib W, Chambers AF, Tuck AB. β3 Integrin Expression Increases Breast Carcinoma Cell Responsiveness to the Malignancy-Enhancing Effects of Osteopontin. Mol. Cancer Res. 2003;1:810–9. [PubMed] [Google Scholar]
- (27).Chen JM, Fortunato M, Stevens RA, Barrett AJ. Activation of progelatinase A by mammalian legumain, a recently discovered cysteine proteinase. Biol. Chem. 2001;382:777–83. doi: 10.1515/BC.2001.093. [DOI] [PubMed] [Google Scholar]
- (28).Wolk K, Grutz G, Witte K, Volk HD, Sabat R. The expression of legumain, an asparaginyl endopeptidase that controls antigen processing, is reduced in endotoxin-tolerant monocytes. Genes Immun. 2005;6:452–6. doi: 10.1038/sj.gene.6364224. [DOI] [PubMed] [Google Scholar]
- (29).Li DN, Matthews SP, Antoniou AN, Mazzeo D, Watts C. Multistep autoactivation of asparaginyl endopeptidase in vitro and in vivo. J. Biol. Chem. 2003;278:38980–90. doi: 10.1074/jbc.M305930200. [DOI] [PubMed] [Google Scholar]
- (30).Wu W, Luo Y, Sun C, Liu Y, Kuo P, Varga J, Xiang R, Reisfeld R, Janda KD, Edgington TS, Liu C. Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res. 2006;66:970–80. doi: 10.1158/0008-5472.CAN-05-2591. [DOI] [PubMed] [Google Scholar]
- (31).Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA, Xiang R. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J. Clin. Invest. 2006;116:2132–41. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Temming K, Meyer DL, Zabinski R, Dijkers EC, Poelstra K, Molema G, Kok RJ. Evaluation of RGD-targeted albumin carriers for specific delivery of auristatin E to tumor blood vessels. Bioconjug. Chem. 2006;17:1385–94. doi: 10.1021/bc060087z. [DOI] [PubMed] [Google Scholar]
- (33).Temming K, Meyer DL, Zabinski R, Senter PD, Poelstra K, Molema G, Kok RJ. Improved efficacy of alphavbeta3-targeted albumin conjugates by conjugation of a novel auristatin derivative. Mol. Pharm. 2007;4:686–94. doi: 10.1021/mp0700312. [DOI] [PubMed] [Google Scholar]
- (34).Alley SC, Okeley NM, Senter PD. Antibody-drug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010;14:529–37. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- (35).Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: design and clinical applications. Nat. Rev. Drug. Discov. 2008;7:255–70. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- (36).Byzova TV, Rabbani R, D’Souza SE, Plow EF. Role of integrin alpha(v)beta3 in vascular biology. Thromb. Haemost. 1998;80:726–34. [PubMed] [Google Scholar]
- (37).Stern L, Perry R, Ofek P, Many A, Shabat D, Satchi-Fainaro R. A novel antitumor prodrug platform designed to be cleaved by the endoprotease legumain. Bioconjugate Chem. 2009;20:500–10. doi: 10.1021/bc800448u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.