

Abstract
CNS myelin is strongly inhibitory to growing axons and is thought to be a major contributor to CNS axon regenerative failure. Although a number of proteins present in myelin, including Nogo, MAG, and oligodendrocyte-myelin glycoprotein (OMgp), have been identified as myelin-associated inhibitors, studies of mice lacking these genes suggest that additional inhibitors present in CNS myelin remain to be identified. Here we have investigated the hypothesis that myelin lipids contribute to CNS regenerative failure. We identified sulfatide, a major constituent of CNS myelin, as a novel myelin-associated inhibitor of neurite outgrowth. Sulfatide, but not galactocerebroside or ceramide, strongly inhibited the neurite outgrowth of retinal ganglion cells (RGCs) when used as a purified lipid substrate. The mechanism involved in sulfatide-mediated inhibition may share features with other known inhibitors, because the Rho inhibitor C3 transferase lessened these effects. Myelin in which sulfatide was lacking or blocked using specific antibodies was significantly less inhibitory to RGC neurite outgrowth in vitro than was wild-type myelin, indicating that sulfatide is a major component of the inhibitory activity of CNS myelin. Mice unable to make sulfatide did not regenerate RGC axons more robustly after optic nerve crush than wild-type littermates under normal conditions but did exhibit a small but significant enhancement in the extent of zymosan-induced regeneration. These results demonstrate that specific lipids can powerfully inhibit axon growth, identify sulfatide as a novel myelin-associated axon growth inhibitor, and provide evidence that sulfatide inhibition contributes to axon regenerative failure in vivo.
Introduction
The failure of CNS axons to regenerate after damage has devastating consequences for the sufferers of spinal cord and other CNS injuries. Factors contributing to CNS regenerative failure include the following: the intrinsic state of the injured neuron (Richardson and Issa, 1984; Goldberg et al., 2002; Neumann et al., 2002; Qiu et al., 2002; Park et al., 2008; Moore et al., 2009), the formation of the inhibitory glial scar (McKeon et al., 1991; Davies et al., 1997; Zuo et al., 1998; Fidler et al., 1999; Moon et al., 2001; Bradbury et al., 2002), and the presence of inhibitory myelin debris (McKerracher et al., 1994; Mukhopadhyay et al., 1994; Chen et al., 2000; GrandPré et al., 2000; Prinjha et al., 2000; Wang et al., 2002a; Moreau-Fauvarque et al., 2003; Goldberg et al., 2004; Benson et al., 2005).
Although the inhibitory properties of myelin are well established, removal of individual or combinations of known myelin-associated inhibitors (MAIs) results in inconsistent and often poor regeneration (Cafferty et al., 2010; Lee et al., 2010). Treatments such as intraocular macrophage activation induced by either lens injury or zymosan injection (Leon et al., 2000; Yin et al., 2003), conversely, can stimulate regeneration even in the presence of these MAIs. Likewise, inhibition of RhoA or Rho kinase (ROCK)—downstream components of the axonal response to both the chondroitin sulfate proteoglycans (CSPGs) of the glial scar and many of the MAIs—promotes optic nerve and spinal cord regeneration (Lehmann et al., 1999; Fournier et al., 2003; Monnier et al., 2003). Experiments in which MAIs are blocked or myelin is globally reduced (Savio and Schwab, 1990; Schnell and Schwab, 1990; Keirstead et al., 1992; Huang et al., 1999; Ellezam et al., 2003) have also more consistently elicited long-distance regeneration, suggesting that additional MAIs may remain to be identified. Although the search for such molecules has focused on proteins, lipids constitute 70–75% of CNS myelin by weight (Norton and Poduslo, 1973b). Here, we describe the identification of sulfatide as a novel lipid inhibitor of axon outgrowth.
Sulfatide comprises 4–7% of CNS myelin lipid by weight (Norton and Poduslo, 1973b). It is generated from galactocerebroside by cerebroside sulfotransferase (CST), which catalyzes a sulfate group addition; UDP–galactose:ceramide galactosyltransferase (CGT) transfers galactose to ceramide to generate galactocerebroside (Fig. 1). Mice lacking CST cannot generate sulfatide (Honke et al., 2002); CGT null mice lack sulfatide and galactocerebroside and overproduce glucocerebroside (Coetzee et al., 1996), but glucocerebroside does not compensate for galactolipid function in CGT null myelin (Saadat et al., 2010). Both knock-out mice generate compact myelin that becomes disorganized over time, inducing tremors and progressive ataxia.
Figure 1.

The sulfatide synthetic pathway. Sulfatide is generated from ceramide in a two step process. CGT catalyzes the addition of a galactose group to ceramide to form galactocerebroside (also termed galactosylceramide). CST then catalyzes the addition of a sulfate group to the 3′ carbon of the galactose group to create sulfatide (3-O-sulfogalactosylceramide). Arylsulfatase A (ARSA) catalyzes the removal of the sulfate group from sulfatide to generate galactocerebroside. Ceramide comprises a sphingosine moiety and an additional fatty acid chain. Because this chain can vary in length, the carbon chain has been represented as an R group. The most common species in CNS myelin are C24:0 and C24:1 fatty acids (MacBrinn and O'Brien, 1969), but this balance varies in other tissues such as the pancreas, in which C16:0 and C24:0 are the predominant species (Buschard et al., 2006).
We observed that sulfatide was inhibitory to retinal ganglion cell (RGC) neurite outgrowth in vitro and that the Rho inhibitor C3 transferase lessened these effects. Sulfatide was also required for the inhibitory properties of CNS myelin in vitro. Although sulfatide deficiency did not promote optic nerve regeneration under normal conditions, it did enhance zymosan-induced regeneration. These data suggest that sulfatide is a significant contributing factor to CNS regenerative failure.
Materials and Methods
Retinal ganglion cell culture.
Step-by-step protocols for all methods are available on request. RGCs were harvested from postnatal day 5 (P5) Sprague Dawley rat or C57BL/6 mouse pups and cultured via previously established protocols (Barres et al., 1988; Meyer-Franke et al., 1995). Briefly, retinas were dissociated into a single-cell suspension with papain (LS003126; Worthington Biochemicals), and the resulting cells were immunopanned to purify RGCs. For rat RGC preps, macrophages were removed with negative panning plates using a rabbit anti-rat macrophage antibody (CLAD51240; Cedarlane) and positively selected for with plates coated with supernatant from the T11D7 hybridoma line (TIB-103; American Type Culture Collection), which binds to Thy1.1. For mouse RGC preps, macrophages were removed with Bandeireia lectin (L-1100; Vector Laboratories) and RGCs were selected for with a mouse anti-mouse Thy1.2 IgM antibody (MCA02R; Serotec).
RGCs were cultured in media containing 5 μg/ml insulin (I-6634; Sigma), 10 μg/ml forskolin (F6886; Sigma), 50 ng/ml BDNF (450-02; Peprotech), 100 ng/ml CNTF (450-13; Peprotech), and (for some experiments) 25 ng/ml basic FGF (100-18B; Peprotech) at a density of 500–3000 cells per well (for outgrowth experiments) or 20,000–40,000 cells per well (for survival experiments). Ethanol-washed 12-mm-diameter glass coverslips (633009; Carolina Biological Supply) were coated with poly-d-lysine (P1024 or P6407; Sigma) diluted to 10 μg/ml in water for 30 min, washed, transferred into 24-well tissue culture plates (353047; Falcon), and dried. We generated myelin, lipid, or lipid solvent layers by adding the appropriate quantity of substrate in a small volume to each coverslip and allowing the solvent to completely evaporate. Finally, coverslips were incubated in Neurobasal media (21103; Invitrogen) containing 2 μg/ml mouse laminin (3400-010-01; Cultrex) overnight.
To coat coverslips with purified myelin, myelin aliquots were thawed on ice, mixed, and diluted to the appropriate concentration in sterile water. Each dilution was then sonicated in three 10 s bursts, between which the samples were incubated on ice to prevent overheating. Fifty microliters of each aliquot were then added to each coverslip and allowed to evaporate as described above.
All lipids used were obtained in lyophilized form, dissolved in varying mixtures of chloroform, methanol, and water (C/M/W), and stored in plastic- and rubber-free glass bottles (600470; Avanti Polar Lipids) under a layer of nitrogen gas. Purified sulfatides (131305 from Avanti and 1049 from Matreya) and galactocerebrosides (131303; Avanti Polar Lipids) were dissolved in C/M/W at a ratio of 2:1:0.1. Purified ceramides (860052; Avanti) were dissolved in C/M/W at 98.5:1:0.5. Purified soy-derived glucocerebrosides [131304 from Avanti and 1522 from Matreya; high in hydroxyl fatty acids (HFAs)] and bovine-derived glucocerebrosides (1521 from Matreya; high in non-hydroxyl fatty acids) were dissolved in C/M/W at a ratio of 2:1:0.1. Synthetic sulfatides N-palmitoyl-sulfatide (1875; Matreya) and lyso-sulfatide (1904; Matreya) were dissolved in C/M/W at a ratio of 2:1:0, whereas N-tetracosanoyl-sulfatide (1888; Matreya) was dissolved in C/M/W at 5:1:0, and N-acetyl-sulfatide (2076; Matreya) was dissolved in C/M/W at a ratio of 1:1:0. To coat coverslips with lipids, each lipid was diluted in either the sulfatide solvent or its own solvent (results from both were equivalent for the same lipid), and 50 μl of each dilution was added to each glass coverslip, avoiding contact with the plastic of the plate. Lipid solvents were allowed to evaporate, and then coverslips were coated with laminin overnight as described above. The sulfatide solvent (C/M/W at 2:1:0.1) was used as the solvent control in all lipid-coating experiments.
For human embryonic kidney (HEK) cell experiments, HEK cells were transfected with Lipofectamine 2000 reagent (Invitrogen) according to the specifications of the manufacturer and cultured in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen). Cells were plated at a density of 300,000 HEK293H cells per well of a 24-well plate 1 d before transfection, resulting in ∼90% confluency on the day of transfection. For transfection, 1 μg of CST expression plasmid (mmGAL3ST1–pCMVsport6; Open Biosystems) diluted in 50 μl of OPTIMEM (Invitrogen) and 3 μl of Lipofectamine 2000 diluted in 50 μl of OPTIMEM were incubated at room temperature for 5 min and subsequently combined. After an additional 30 min incubation at room temperature, the Lipofectamine/DNA mixture was added to the cells. After 24 h, medium was refreshed and switched to a 1:1 mixture of DMEM–10% FCS/Neurobasal–B27 medium. Twenty-four hours later, the medium was switched to Neurobasal–B27 medium supplemented with 50 ng/ml BDNF, 100 ng/ml CNTF, and 10 ng/ml forskolin. P5 rat RGCs were then plated on top of the monolayer of mmGAL3ST1–pCMVsport6 transfected or control cells, and outgrowth was assessed after 24 h.
For antibody blocking experiments, coverslips were preincubated with hybridoma supernatants or sera from immunized mice at a dilution of 1:100. For immunodepletion experiments, 6 mg of goat anti-mouse IgG + IgM (Jackson ImmunoResearch) was conjugated to 2 ml of Affi-Gel-10 beads (Bio-Rad) according to the instructions of the manufacturer. IgGs and IgMs from the serum (40 μl diluted 1:25 in PBS) of an immunized mouse were immunodepleted by incubating with 1 ml of the goat anti-mouse antibody-conjugated Affi-Gel-10 beads for 18 h at 4°C. The beads were centrifuged, and the supernatant consisting of the depleted sample was collected. These samples were then used to preincubate myelin-coated substrates used for neurite growth assays. In parallel, RGCs were grown on coverslips coated with myelin that had been preincubated with either nondepleted myelin-immunized serum or nondepleted liver membrane-immunized serum. Liver membrane-coated coverslips preincubated with preimmune serum were used as a control for maximum outgrowth on a permissive substrate. Three thousand RGCs were plated on these substrates, and outgrowth was assessed after 24 h.
Cell-permeable C3 transferase (CT04; Cytoskeleton) was prepared in a 1:1 mixture of 4% tissue-culture-grade BSA (A-4161; Sigma) in Dulbecco's PBS (DPBS) and glycerol and diluted to 50 ng/ml in RGC growth media.
Survival assay and outgrowth assessment.
To assess survival, calcein AM and ethidium homodimer-1 (L-3224; Invitrogen) were added to RGCs at a final concentration of 2 and 1 μm, respectively. Cells were returned to the incubator for 20–30 min, after which the ratio of live (green, calcein AM positive) and dead (red, ethidium homodimer-1 positive) cells was quantified and used to calculate the percentage survival (live/all) in each condition.
For outgrowth assessment, visualization of the neurites of cultured RGCs was performed using either immunofluorescence or calcein AM. For immunofluorescence, cells were fixed for 10–15 min in 4% paraformaldehyde (PFA), washed, blocked for 1 h at room temperature in a 1:1 mixture of antibody buffer/normal goat serum containing 0.4% Triton X-100, washed, and incubated at 4°C overnight with an antibody recognizing β-tubulin III (Tuj1) (MMS-435P; Covance) at 1:500 in 10% goat serum in antibody buffer. The following day, the coverslips were washed, incubated with Alexa Fluor-488 or -594-conjugated goat anti-mouse antibody (A11029 or A11032; Jackson ImmunoResearch) diluted 1:500 in antibody buffer, washed, and mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories). Antibody buffer comprises 150 mm NaCl, 50 mm trizma (tris) base, 100 mm l-lysine monohydride (L5626-100G; Sigma), 1% BSA (A2153; Sigma), and 0.2% sodium azide. For calcein AM visualization, calcein AM (C3100MP or L-3224; Invitrogen) was added to RGCs at a final concentration of 2 μm as described above. Cells were returned to incubator for 20–30 min and imaged immediately afterward. Ethidium homodimer-1 was also occasionally used to confirm that cells without neurites were not dead or dying.
Fifteen to 30 images of random 10× fields of 1–20 RGCs were typically taken per coverslip; two to four coverslips were assessed for each experiment, typically resulting in 100–500 cells per condition. All images within each condition were pooled (although coverslips were also analyzed separately in case of individual variation), and each image was analyzed using the neurite outgrowth module within the MetaMorph software suite (Molecular Devices), which identifies cell bodies and traces all neurites associated with each cell body separately. The average extent of total outgrowth per cell was used as the metric for all outgrowth experiments.
Immunization and serum collection.
For immunization experiments, adult female BALB/c mice were immunized for a total of 5 weeks. One hundred micrograms of liver membranes or CNS myelin was emulsified 1:1 by volume in Freund's incomplete adjuvant (AR002; Millipore Bioscience Research Reagents), and the resulting mixture was subcutaneously injected into each mouse twice weekly. Serum samples were obtained before and after the immunization period by retro-orbital eye bleed using glass capillary tubes. Peripheral blood was chilled at 4°C and spun down, allowing sera to be isolated from the non-coagulated supernatant.
Optic nerve injury and regeneration assessment.
Optic nerve injury was induced by retro-orbital nerve crush. Briefly, CGT null, heterozygous, and wild-type littermate mice were anesthetized using isoflurane, and the nerve was accessed using small scissors (15024-10; Fine Science Tools) and dull, angled forceps (FST 11063-07) to gently push away tissue near the eye, carefully avoiding the orbital venous sinus. Fine angled forceps (FST 11253-25) were used to crush the nerve for 10 s at consistent pressure. Paralube (1621; 1–800-Petmeds) was used to prevent drying of the eye during and after surgery, and carprofen was given as an analgesic.
To induce regeneration via zymosan injection, mice were injected with zymosan and 8-CPT-cAMP 3 d after nerve injury. Zymosan at 12.5 μg/μl (Z4250-250MG; Sigma) was sterilized by boiling for 10 min immediately before injection. After cooling to room temperature, 8-CPT-cAMP (CN130-0020; Enzo Life Sciences) was added to a final concentration of 100 μm. A volume of 0.5 μl of the final mixture was injected into the vitreous of the eye connected to the injured nerve using a 2.5 ml Hamilton syringe (87942; Hamilton) fitted with a custom 33 gauge, ⅜ inch, point style 4 needle (7803-05; Hamilton). Regenerating axons were anterogradely labeled 24 h before harvest with a 0.5 μl injection of 5 μg/μl Alexa Fluor-594-conjugated cholera toxin β (CTβ) (C22842; Invitrogen) into the vitreous of the eye in the same manner as was described for the zymosan injections.
Mice were perfused with PBS and then 4% PFA in PBS before nerve harvest. Nerves were postfixed for 2 h in 4% PFA, washed in PBS, and transferred into 30% sucrose in PBS for at least 24 h. Before sectioning, tissue surrounding the nerve was removed, and nerves were transferred into a 2:1 mixture of 30% sucrose in PBS and Optimal Cutting Temperature liquid (OCT). To cut longitudinal sections through the nerve on the cryostat, a frozen block of OCT was shaven flat, an individual nerve was laid flat across the freshly cut plane, and the nerve was covered with 2:1 30% sucrose/OCT. Nerves were cut into 10-μm-thick sections, and the nerves chosen to be analyzed were mounted with Vectashield containing DAPI (H-1200; Vector Laboratories).
In each longitudinal section, the crush site was identified by both the end of the wave front of CTβ tracing and the accompanying changes in nerve morphology that can be observed in DAPI-stained nerves. Three representative sections were identified in which both the crush site and the most regenerating axons could be visualized. These nerves were mounted and imaged at 10× or 20×. A line perpendicular to the nerve was drawn at the crush site, and additional lines were added every 100 μm from the crush site along the nerve within the first millimeter from the site of injury. The number of regenerating axons crossing each line was quantified for each image. As the width of each nerve varied between sections, the width of the nerve at each distance was also quantified. The number of regenerating axons per millimeter of section was calculated by dividing the number of axons at a distance by the width of the nerve in millimeters at that distance. The number of regenerating axons at each distance was averaged for each nerve, and averages for individual nerves (rather than values per section) were used as data points for statistical analysis. Because the CGT null phenotype is visible by eye, none of the surgeries could be performed blind. Samples were blinded after harvest, however, and all analysis was performed blind to the genotypes of the animals.
Myelin isolation.
Myelin was harvested from adult Sprague Dawley wild-type rats or CGT wild-type, heterozygous, and homozygous null mice following the methods described by Norton and Podulso (1973a). Rat brains and spinal cords were harvested from PBS-perfused rats and used for myelin isolation immediately after harvest. Brain and spinal cord tissue was isolated from nonperfused CGT littermates between P28 and P35, snap frozen in liquid nitrogen immediately after tissue harvest, and stored at −80°C until enough samples had been collected to perform the myelin preparation. Brain and spinal cord samples from at least four animals per genotype were pooled for each myelin preparation. For the second preparation, samples from four mice were used in each pool, and only one matching littermate of each genotype was used.
All steps detailed below, including spins, were performed at 4°C in solutions containing one Complete, EDTA-free protease inhibitor cocktail tablet (11 873 580; Roche) dissolved per 25 ml of solution. Samples were thawed on ice in prechilled 0.32 m sucrose, homogenized in a 30 ml, motor-driven Wheaton Potter-Elvehjem Tissue Grinder (62400-788; VWR), and then brought to a final volume of 20 ml with 0.32 m sucrose. Homogenate was layered over 19 ml of 0.85 m sucrose, and the discontinuous gradient was spun for 30 min at 75,000 × g at 4°C in an SW28Ti rotor (342217; Beckman). The interface between the sucrose layers containing the membrane fraction was collected, washed with sterile water, and spun for 20 min at 100,000 × g in an SW28 rotor to remove excess sucrose. The pellet was collected, resuspended again in water, and subjected to two osmotic shock spins, each for 10 min at 10,000 × g in an SW40Ti rotor (331302; Beckman). The pellet was then resuspended in 0.32 m sucrose, again layered in a discontinuous gradient over 0.85 m sucrose as described above, and spun for 1 h at 75,000 × g in an SW28 rotor. The sucrose washout spin and two osmotic shock spins were repeated as described above. The final pellets were resuspended in 500 μl of sterile water containing protease inhibitors, aliquoted, snap frozen in liquid nitrogen, and stored at −80°C.
Liver membrane isolation.
Liver samples were harvested from PBS-perfused adult Sprague Dawley rats. All steps were performed at 4°C with solutions containing protease inhibitors as described above. The tissue was washed and collected in 0.32 m sucrose on ice, then cut into pieces and homogenized in a manner similar to the brain and spinal cord samples. Homogenate was spun down for 30 min at 500 × g at 4°C. The pellet was discarded, and the supernatant was then spun at for 1 h at 100,000 × g at 4°C in an SW40Ti rotor (331302; Beckman). The pellet was resuspended in 1 ml of sterile water containing protease inhibitors, aliquoted, snap frozen in liquid nitrogen, and stored at −80°C.
Sphingostrips.
Sphingostrips (S23753; Invitrogen) were incubated with sera from CNS myelin-immunized mice and liver membrane-immunized mice, as well as preimmune sera from those mice by diluting the sera 1:100 into TBS containing Tween 20 and 3% fatty acid free BSA (Sigma). Antibody binding was detected with a mouse anti-mouse IgG–HRP secondary antibody (AP124P; Millipore) and standard chemiluminescence (RPN2106V1/2; GE Healthcare).
RhoA activation assay.
One million to 1.5 million P5 Sprague Dawley rat RGCs were plated per condition in 10-cm-diameter tissue culture plates and cultured for 14–24 h. Media included 0.1 U/ml calpeptin (CN01; Cytoskeleton) or DMSO vehicle control, 50 ng/ml C3 transferase (CT04; Cytoskeleton) or 1:1 4% BSA/glycerol vehicle control (described above), and 10 μg/ml sulfatide (131305; Avanti) or vehicle control. Sulfatide was solubilized by evaporating the chloroform, methanol, and water solvent (described above) under nitrogen gas from a small sample of sulfatide and resuspending to a concentration of 10 mg/ml in a 95:5 (v/v) mixture of DMSO/1N HCl. Sample was immediately diluted with 4% endotoxin-free, tissue-culture-grade BSA (A-4161; Sigma) in DPBS to a concentration of 500 μg/ml and subsequently added to RGC growth media at a concentration of 10 μg/ml. To generate vehicle control sample, we performed the same steps with chloroform/methanol/water solvent alone rather than sulfatide. The three vehicle controls were tested both alone and in combination for effects on RGC neurite outgrowth, and no inhibition was observed (data not shown). For harvest, cells were washed once in ice-cold PBS, lysed in 150–250 μl of ice-cold lysis buffer (provided in kit), scraped off plate, cleared by centrifugation for 2 min at 10,000 rpm at 4°C, aliquoted, and immediately snap frozen for storage at −80°C. RhoA activation assay (“G-LISA”; BK124; Cytoskeleton) was subsequently performed according to the instructions of the manufacturer. RGCs from each harvest were also plated in 24-well plates containing the same media combinations, and outgrowth was assessed to confirm that the treatments had been effective in each experiment.
Results
Immunization with CNS myelin generated antibodies recognizing the myelin lipid sulfatide
Huang et al. (1999) demonstrated that, when mice were immunized with CNS myelin, they generated antibodies that promoted axon regeneration after injury in vivo and lessened the inhibitory effect of myelin on neurite outgrowth in vitro. Although these antibodies presumably acted by binding to and blocking inhibitory epitopes present in myelin, they did not recognize MAG, Nogo, or CSPGs (Ellezam et al., 2003), implying that they might bind to previously unidentified myelin-associated inhibitors. Thus, to determine whether any myelin lipids contribute to the inhibitory activity of myelin, we first asked whether immunization with myelin generated antibodies recognizing any myelin lipids. We repeated the aforementioned experiments by immunizing mice with either CNS myelin or liver membranes for a total of 5 weeks and similarly observed that the sera of CNS myelin-immunized mice was sufficient to reduce the outgrowth inhibition of RGCs grown on CNS myelin, whereas the sera of liver membrane-immunized mice had no effect (Fig. 2A–C). Immunodepletion of myelin-immunized sera abrogated its ability to promote outgrowth on CNS myelin, implying that the effect was indeed a result of the antibodies present in the sera. Sphingostrips—nitrocellulose membranes containing spots of major myelin lipids—were then probed with preimmune sera (data not shown), sera of mice immunized with liver membranes, and sera of mice immunized with CNS myelin (Fig. 2D–F). We consistently observed a strong response to sulfatide, as well as an occasional weak response to disialoganglioside GD3, in strips probed with myelin-immunized sera but saw no reactivity with liver-immunized sera. Thus, immunization with myelin, but not immunization with liver membranes, induced the generation of antibodies recognizing sulfatide and, to a lesser extent, disialoganglioside GD3.
Figure 2.

Immunization with myelin generated sulfatide-specific antibodies. Eight-week-old female BALB/c mice were immunized with either liver membranes or CNS myelin emulsified in Freund's incomplete adjuvant twice weekly for a total of 5 weeks (n = 4 mice per condition). Serum samples were harvested from peripheral blood samples both before and after immunization. A–C, P5 rat RGCs were cultured on a myelin (M) or liver membrane (L) substrate in the presence of a variety of serum samples: preimmune sera (Pre), sera harvested from mice immunized with either liver membranes (L) or CNS myelin (M), or immunodepleted CNS myelin-immunized sera (M–D). Representative cells cultured on a CNS myelin substrate in the presence of preimmune serum (A) or CNS myelin-immunized serum (B) were incubated with calcein AM (green) to visualize RGC neurites and ethidium homodimer-1 (red) to exclude dead or dying cells. The extent of outgrowth in each condition was quantified in C. Sera from myelin-immunized mice was sufficient to lessen the inhibitory effect of CNS myelin in an antibody-dependent manner. Scale bar, 50 μm. Similar results were observed in at least three independent experiments; quantification is of one representative experiment. Error bars are ± SEM. *p < 0.05 by one-way ANOVA with a Bonferroni's post hoc test compared with the preimmune sera condition. D–F, Sphingostrip membranes were incubated with either liver-immunized (D) or CNS myelin-immunized (E) sera. The extent of binding was visualized using a goat anti-mouse IgG–HRP secondary antibody and chemiluminescence. Strong and consistent binding to spot 11 (red arrows), which contained sulfatide, was observed in multiple experiments. Reactivity to spot 10, which contained disialoganglioside GD3, was also observed but was less consistent. No reactivity was observed after incubation with sera of liver-immunized mice. The layout of the entire strip (F) was as follows: 1, sphingosine; 2, sphingosine 1-phosphate; 3, phytosphingosine; 4, ceramide; 5, sphingomyelin; 6, sphingosylphosphocholine; 7, lysophosphatidic acid; 8, myriocin; 9, monoganglioside GM1; 10, disialoganglioside GD3; 11, sulfatide; 12, sphingosylgalactoside (psychosine); 13, cholesterol; 14, lysophosphatidylcholine; 15, phosphatidylcholine; 16, blank. One hundred picomoles of lipid per spot.
Sulfatide was sufficient to inhibit RGC neurite outgrowth in vitro
To determine whether sulfatide was sufficient to inhibit RGC neurite outgrowth, we began by overexpressing the enzyme CST, which catalyzes the addition of the sulfate group to galactocerebroside (Honke et al., 2002), in HEK cells. Transfection of HEK cells with CST induced the generation and surface localization of high levels of sulfatide (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). RGCs cultured on a layer of CST-expressing HEK cells exhibited significantly inhibited neurite outgrowth compared with RGCs grown on control HEK cells (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material).
To confirm that the sulfatide molecule itself, rather than a sulfatide-binding partner or an unknown byproduct of CST expression, was inhibitory, we obtained purified brain sulfatide, as well as galactocerebroside and ceramide, the two immediate precursors of sulfatide. When adsorbed onto glass coverslips together with our standard substrate of poly-d-lysine and mouse laminin, 1 μg of sulfatide was sufficient to strongly and significantly inhibit rat RGC neurite outgrowth, whereas even higher doses of galactocerebroside and ceramide were not inhibitory (Fig. 3A–E). This effect was also observed when mouse RGCs were cultured under the same conditions (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), as well as with rat RGCs obtained at embryonic day 20 (data not shown).
Figure 3.
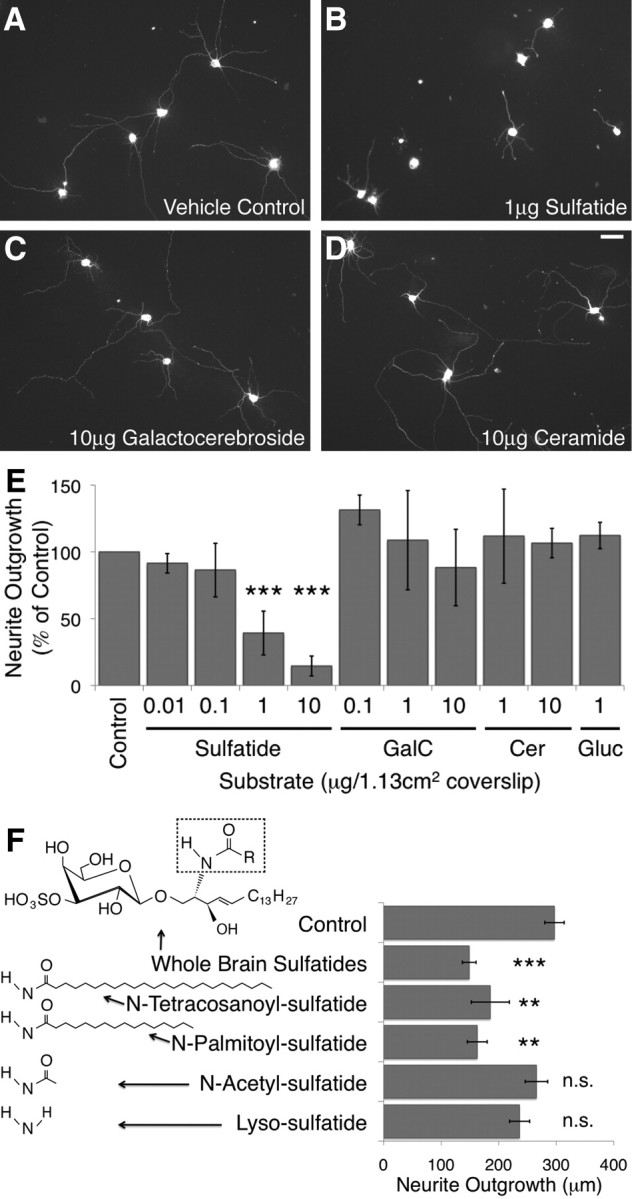
Sulfatide was sufficient to inhibit neurite outgrowth in vitro. Postnatal day 5 rat retinal ganglion cells were cultured for 18–24 h at low density on glass coverslips coated with poly-d-lysine and mouse laminin, as well as purified brain lipids, synthetic sulfatide isoforms, or lipid solvent. Neuronal processes were imaged with either calcein AM or immunofluorescent staining for β-tubulin III, and outgrowth of all processes was quantified using the MetaMorph neurite outgrowth application. A–E, Sulfatide significantly inhibited RGC neurite outgrowth at densities of 1 μg/coverslip and above. Galactocerebroside (GalC) and ceramide (Cer), the direct precursors of sulfatide, did not significantly affect the extent of RGC neurite outgrowth at any concentration tested. HFA–glucocerebroside (Gluc), which is generated at abnormally high concentrations in the absence of the CGT enzyme, also had no effect on RGC neurite outgrowth. Representative fields of RGCs grown on coverslips coated with sulfatide solvent (A), 1 μg of sulfatide (B), 10 μg of galactocerebroside (C), and 10 μg of ceramide (D) are shown. Scale bar, 50 μm. The average extent of outgrowth per cell, normalized to the control in each experiment, averaged across all experiments, is shown in E. All bars represent an average of at least three independent experiments. All quantities listed were plated on 12-mm-diameter glass coverslips with an area of 1.13 cm2. Results are graphed ± SEM. ***p < 0.001 by one-way ANOVA with a Dunnett's post hoc test compared with lipid solvent control levels. F, Synthetic sulfatide isoforms lyso-sulfatide (1904; Matreya) and N-acetyl-sulfatide (2076; Matreya), which lack a fatty acid chain, as well as N-palmitoyl-sulfatide (1875; Matreya), which contains a C16:0 side chain, and N-tetracosanoyl-sulfatide (1888; Matreya), which contains a C24:0 side chain, were used as substrates in our neurite outgrowth assay. The chemical structure for the portion of the sulfatide molecule that differs between these isoforms is indicated to the left of the name of each isoform. The generic structure for sulfatide is illustrated at the top (arrow from whole-brain sulfatides) and contains a dashed box indicating the portion of sulfatide that is pictured for each isoform below. Synthetic sulfatides containing 24 (N-tetracosanoyl-sulfatide) and 16 (N-palmitoyl-sulfatide) carbon chain fatty acids inhibited RGC neurite outgrowth, whereas those without a second carbon chain (lyso-sulfatide and N-acetyl-sulfatide) did not. Results are averages of five independent experiments and graphed ± SEM. **p < 0.01, ***p < 0.001 compared with solvent control by a repeated measures one-way ANOVA with a Dunnett's post hoc test compared with lipid solvent controls.
We also tested the effects of glucocerebroside, the molecule that is generated at abnormally high levels in CGT null mice, on RGC neurite outgrowth (Coetzee et al., 1996; Dupree et al., 1998b). The glucocerebrosides upregulated in mice lacking galactosphingolipids contain predominantly HFA chains and are thus a form of glucocerebroside not normally expressed at high levels in the brain (Coetzee et al., 1996). Thus, we tested the effects of both glucocerebrosides containing predominantly HFAs (from two sources) and those containing predominantly non-HFAs. None of the three glucocerebroside preparations tested inhibited RGC neurite outgrowth after 1 d in culture; the average outgrowth of RGC neurites on a substrate containing HFA–glucocerebrosides from one representative source is depicted (Fig. 3E).
We observed that RGCs cultured on coverslips coated with high doses of sulfatide appeared unhealthy, suggesting that some changes in RGC outgrowth in the presence of sulfatide might be attributable to toxicity. Thus, we performed survival assays both after 1 d in vitro, our normal time point for measuring outgrowth, and after 2 d. RGC survival was significantly decreased in the presence of 10 μg of sulfatide per coverslip but unaffected by the presence of 1 μg of sulfatide after either 1 d (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) or 2 d (data not shown) in vitro. These results suggested that the striking effects on RGC neurite outgrowth observed at a dose of 1 μg/coverslip were attributable to specific cellular responses rather than simply a matter of poor cell health and toxicity. Thus, this dose was chosen for all future experiments.
Sulfatide is known to bind strongly to laminin, a component of our standard substrate, during basement membrane assembly (Li et al., 2005). Thus, it was possible that the effect of sulfatide on neurite outgrowth was attributable to its ability to bind laminin rather than its direct interaction with RGC neurites. To test this possibility, we repeated our dose–response curve with RGCs cultured on collagen IV rather than on mouse laminin. RGCs grown on collagen IV were inhibited by sulfatide at a similar concentration to those on laminin (data not shown), suggesting that the ability of sulfatide to inhibit RGCs is not laminin specific.
The lack of inhibition observed in the presence of galactocerebroside, which differs from sulfatide only by its lack of a sulfate group, suggested that the sulfate group was required for the inhibitory properties of sulfatide. The sulfatides that we tested in these experiments were a mixed population purified from neural tissue with multiple isoforms that generally differed in the length of the fatty acid chain within the ceramide backbone of the molecule (Eckhardt, 2008). Given that specific isoforms of sulfatide have been shown to have unique biological functions and the distribution of isoforms is known to vary across tissues (Vos et al., 1994; Fredman et al., 2000; Buschard et al., 2006; Isaac et al., 2006), we wanted to know whether the same specificity might be observed in the ability of sulfatide to inhibit RGC neurite outgrowth. Also, the fact that the addition of a sulfate group to galactocerebroside dramatically alters the charge and polarity of the molecule suggested the possibility that the sulfate group was the most important moiety and that any sulfated lipid might be sufficient to inhibit outgrowth. Thus, we were also curious to learn whether removing any piece of the sulfatide molecule other than the sulfate group would be sufficient to reduce the inhibitory effects of sulfatide on neurite outgrowth.
We obtained multiple synthetically generated isoforms of sulfatide: one containing a long fatty acid chain (C24:0), one containing a shorter fatty acid chain (C18:0), and two that lacked a fatty acid chain entirely. We tested the effect of these isoforms as substrates for RGC outgrowth as described above at 1 μg/coverslip, the concentration that was consistently effective for the mixed, purified sulfatides discussed previously. Interestingly, isoforms containing either long or short fatty acid chains were both sufficient to significantly inhibit RGC outgrowth in vitro to the same degree as purified sulfatides (Fig. 3F). Neither of the two isoforms lacking the fatty acid chain, however, had a significant effect on the extent of RGC neurite outgrowth in culture. These results suggested that a fatty acid chain was required for the inhibitory properties of sulfatide but that the effect was not isoform specific. The ability of both fatty acid lengths to inhibit outgrowth is interesting given that long chains of 24 carbons constitute the vast majority of sulfatide isoforms within myelin, whereas shorter chains predominate in other tissues of the body (Fredman et al., 2000); however, given that the end of the fatty acid tail is normally buried in the outer leaflet of the plasma membrane (Eckhardt, 2008), it is perhaps not surprising. More generally, these results also suggest that multiple moieties of the sulfatide molecule, rather than just the sulfate group, are required for its inhibitory properties.
Together, these experiments demonstrated that sulfatide was strongly inhibitory to neurite outgrowth in vitro, either in the context of cell membranes or as a purified substrate. The effect was not attributable to toxicity or substrate interaction and was observed in postnatal rat and mouse RGCs, as well as embryonic rat RGCs. The inhibitory signal presented by sulfatide demonstrated a strong degree of specificity, because both the sulfate group and a fatty acid moiety were required for its effects.
Sulfatide was necessary for myelin inhibition in vitro
Sulfatide constitutes 4–7% of myelin lipid, or 3–5% of the entire weight of CNS myelin (Norton and Poduslo, 1973b). Because lipids on a molar basis constitute >99% of molecules in myelin (Vance and Vance, 2002), the molar amount of sulfatide is far greater than the previously identified protein MAIs. This, combined with the ability of sulfatide to inhibit RGC neurite outgrowth, suggested that it might be a necessary inhibitory component of CNS myelin. To test this possibility, we asked whether myelin in which sulfatide was lacking or blocked using specific monoclonal antibodies was less inhibitory than wild-type myelin. To prevent sulfatide within wild-type myelin from interacting with RGC neurites, we preincubated myelin substrates with the O4 antibody, which specifically recognizes sulfatide (Sommer and Schachner, 1981, 1982). When RGCs were cultured on a substrate of CNS myelin in the presence of the O4 antibody, the inhibitory effect of myelin on RGC neurite outgrowth was significantly lessened (Fig. 4A). The presence of O1, an antibody recognizing galactocerebroside (Sommer and Schachner, 1981, 1982), had no effect on the extent of inhibition. These results suggest that sulfatide, but not galactocerebroside, is a major component of the inhibitory material of myelin.
Figure 4.

Sulfatide was necessary for complete inhibition of RGC neurite outgrowth by CNS myelin in vitro. Postnatal day 5 rat retinal ganglion cells were cultured for 18–24 h at low density on glass coverslips coated with poly-d-lysine and mouse laminin, as well as various CNS myelin or liver membrane samples. Neuronal processes were imaged with either calcein AM or immunofluorescent staining for β-tubulin III, and outgrowth of all processes was quantified using the MetaMorph neurite outgrowth application. A, P5 rat RGCs were cultured on a substrate of rat liver membranes or rat CNS myelin at a density of 2 μg of protein per 1.13 cm2 coverslip. Each coverslip was preincubated with hybridoma media, supernatant from the sulfatide-specific O4 hybridoma, or supernatant from the galactocerebroside-specific O1 hybridoma. Liver membranes did not affect the extent of RGC neurite outgrowth, whereas CNS myelin strongly inhibited neurite outgrowth. Preincubation with the O4 antibody lessened the inhibitory effect of CNS myelin on RGC neurite outgrowth, whereas preincubation with the O1 antibody had no effect. Similar results were observed in at least three independent experiments; results shown are the average extent of outgrowth ± SEM in one representative experiment. **p < 0.01 compared with CNS myelin/no antibody condition by one-way ANOVA with a Bonferroni's post hoc test. B, P5 rat RGCs were cultured on 1 μg/1.13 cm2 coverslip of myelin substrates isolated from brain and spinal cord samples collected from CGT+/+, CGT+/−, and CGT−/− mice. When normalized to protein levels, which should not change substantially between the samples, myelin isolated from CGT null (KO; CGT−/−) mice was significantly less inhibitory than myelin isolated from wild-type (WT; CGT+/+) or heterozygote (Het; CGT+/−) littermates. Myelin was isolated from two distinct biological replicate samples of each genotype on two different days. Both sets of samples gave similar results in at least four experiments, and the results of one representative experiment are graphed ± SEM. ***p < 0.001 compared with CGT null (CGT−/−) condition by one-way ANOVA with a Bonferroni's post hoc test.
To confirm that this effect was not attributable to off-target binding of the O4 antibody or steric interference with sulfatide-binding partners, we next tested whether myelin that lacked sulfatide was less inhibitory to neurite outgrowth than wild-type myelin. CGT null mice cannot make either galactocerebroside or sulfatide (Coetzee et al., 1996). We isolated myelin from CGT null homozygote, heterozygote, and wild-type littermates and tested the ability of equivalent amounts of each myelin sample to inhibit RGC neurite outgrowth. Myelin lacking sulfatide and galactocerebroside was consistently and significantly less inhibitory than myelin isolated from wild-type and heterozygous littermates (Fig. 4B). Sulfatide and galactocerebroside together constitute 27% of the lipid fraction of myelin by weight (Norton and Poduslo, 1973b), and their loss should dramatically alter myelin lipid levels; thus, we used myelin protein concentration as a means of normalizing loading between samples. Equal amounts of protein were added in each condition, and the composition of CGT null myelin has been shown to be similar to that of wild-type myelin (Dupree et al., 1998a). In addition, Western blots probing for MBP and myelin oligodendrocyte glycoprotein confirmed that the levels of myelin protein were equivalent between all samples (data not shown).
The inability of the O1 antibody to block the inhibition induced by wild-type myelin, as well as the lack of inhibition observed in the presence of purified galactocerebroside, suggested that the results observed with CGT null myelin were attributable to the lack of sulfatide rather than the loss of galactocerebroside. Additionally, the lack of inhibition observed in the presence of HFA–glucocerebroside suggests that, although this species is overproduced in animals lacking the CGT enzyme, its overabundance likely does not affect the inhibitory properties of myelin derived from CGT null mice. Given the strong phenotype of the CGT null mice, it was possible that other unidentified changes in the composition of myelin in these animals might account for the observed decrease in inhibitory activity. The ability of the O4 antibody to lessen the extent of myelin inhibition when added acutely to wild-type myelin, however, decreases the likelihood of this possibility. Conversely, the CGT null myelin results suggest that the effects observed in the antibody blocking experiments are not attributable to either off-target antigens or to steric interference with inhibitory sulfatide-binding partners and are instead attributable to the blocking of sulfatide specifically. Together, these results demonstrate that sulfatide contributes to the inhibitory properties of CNS myelin.
Sulfatide contributed to CNS regenerative failure in vivo
Our in vitro experiments suggested that sulfatide was an important inhibitory component of CNS myelin. To test whether mice lacking sulfatide were capable of enhanced regeneration after CNS injury, we performed optic nerve crush injuries on mice lacking the CGT enzyme. We saw no regenerating axons in CGT null homozygotes, heterozygotes, or wild-type littermate controls (Fig. 5A,B). Given the continued presence of the multitude of other inhibitory factors discussed previously, however, it was not surprising that the absence of sulfatide alone did not result in robust regeneration.
Figure 5.

Sulfatide contributed to CNS regenerative failure in vivo. CGT null (CGT−/−), heterozygous (CGT+/−), and wild-type (CGT+/+) littermates were injured by retro-orbital optic nerve crush at ages P30–P43 either without (A, B) or with (C–E) intraocular injection of zymosan and 8-CPT-cAMP 3 d after injury to promote regeneration. Regenerating axons were traced using an intraocular injection of Alexa Fluor-594-conjugated cholera toxin β 24 h before harvest. The extent of regeneration was assessed 16 d after injury. A, B, Limited or no regeneration past the crush site (arrows) was observed in mice lacking CGT (B), heterozygote, or wild-type littermate controls (A). C–E, Substantial regeneration was observed in optic nerves of CGT null (CGT−/−, purple line, n = 5), heterozygous (CGT+/−, orange line, n = 15), and wild-type (CGT+/+, blue line, n = 5) littermates after zymosan and CPT-cAMP injection. Although all optic nerves exhibited axon regeneration, RGC axons of CGT null mice regenerated more extensively on average than those of heterozygous and wild-type mice. In particular, two of five CGT null animals (D) exhibited much more extensive regeneration than any wild-type or heterozygous (C) animals. The number of axons per millimeter of nerve width was quantified by counting the number of axons crossing lines at intervals of 100 μm between the crush site and 1 mm distal to site of injury and dividing by the width of the nerve at each distance. The average extent of outgrowth at each distance for each genotype is graphed ± SEM (E). Statistical significance was performed by two-way ANOVA in two different ways (see Results), both of which indicated that genotype significantly affected the extent of regeneration (p < 0.01 and p < 0.0001). Significant differences observed between CGT null and wild-type mice are indicated at distances of 100, 200, and 300 μm from the site of injury. WT, Wild type; Het, heterozygous; KO, knock-out. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar, 100 μm.
It remained possible, however, that a subtle in vivo role for sulfatide in CNS regenerative failure could be observed by testing its requirement in a more sensitized system. Thus, we used the ability of the yeast cell wall extract zymosan to promote optic nerve regeneration by inducing intraocular inflammation (Yin et al., 2003) to ask whether RGCs that had been induced to regenerate would do so more robustly in the absence of sulfatide. Fortunately, the CGT null mutation was generated in the 129 strain background—one of the strains that has proven the most responsive to zymosan. We performed optic nerve crush experiments similar to those described above, with the addition of an intraocular injection of zymosan and 8-CPT-cAMP 3 d after crush to promote optic nerve regeneration. Injection of zymosan promoted RGC axon regeneration in wild-type, heterozygous, and null homozygous animals (Fig. 5C–E). We observed a small but statistically significant increase in the extent of regeneration of CGT null mice in response to zymosan and 8-CPT-cAMP treatment when compared with wild-type and heterozygous animals. This effect was particularly striking in 40% of the CGT null mice analyzed, which exhibited extremely robust regeneration—more so than that observed for any heterozygous or wild-type mice. We quantified the extent of regeneration by counting the number of axon segments crossing lines drawn every 100 μm between the site of injury and 1 mm distal to the crush site. To account for section-to-section differences in nerve width, these fiber counts were then divided by the width of the nerve at each point measured (in millimeters) to obtain the number of axons per millimeter.
We performed the statistical analysis two different ways, both of which demonstrated that the genotype of the animal significantly affected the extent of outgrowth. When the average, SD, and sample size for each genotype was calculated and the effect of genotype was compared across all the distances by two-way ANOVA using these values, genotype contributed significantly to the variation (p < 0.0001). Bonferroni's post hoc tests indicated that wild-type and CGT null optic nerves differed significantly at distances of 100 μm (p < 0.001), 200 μm (p < 0.01), and 300 μm (p < 0.05) from the crush site. CGT heterozygous and null optic nerves differed significantly at distances of 100 μm (p < 0.05) and 200 μm (p < 0.01) from the crush site, whereas wild-type and heterozygous animals showed no significant differences at any distance. The second method of performing statistical analysis involved pooling all animals within the same litter into two groups: CGT null and both wild-type and heterozygous individuals. When the resulting averages for each litter (null vs non-null) were analyzed by repeated measures two-way ANOVA, the analysis again demonstrated that genotype contributed significantly to the variation (p = 0.0034), although significant differences at individual distances were not observed. This method of analysis resulted in the lowering of our effective sample size as well as the artificial lowering of variation within the non-null group due to the averaging of more individuals than the CGT null group but allowed the significance of matching between individuals of the same litter to be calculated. The matching of subjects contributed significantly to the variation (p = 0.0104), indicating that, when significance is quantified by pooling all individuals without matching between litters, important information is lost. Because of differences in sample sizes between the groups, we cannot perform a two-way ANOVA that allows for matching within litters but does not artificially lower our sample size by preaveraging; thus, it is likely that both measures slightly underestimate the significance of the effect of genotype on the extent of optic nerve regeneration.
These experiments demonstrated that, although loss of the sulfatide-synthetic enzyme CGT did not allow for robust regeneration under normal conditions, it was able to enhance regeneration that had been induced by other means. Although galactocerebroside was also absent in these experiments, our in vitro experiments indicated that any regenerative phenotype observed in CGT null animals is likely to be attributable to the loss of sulfatide rather than that of galactocerebroside. Similarly, the lack of an effect of HFA–glucocerebroside on RGC neurite outgrowth in culture suggests that the compensatory overproduction of this molecule in CGT null animals was unlikely to affect their regenerative phenotypes. Thus, the ability of RGCs undergoing zymosan and cAMP-induced regeneration to grow more extensively in the absence of CGT suggests that sulfatide contributes at least to some extent to CNS regenerative failure.
C3 transferase alleviated sulfatide-mediated inhibition
Stabilization of the GTP-bound form of RhoA has been shown to be a necessary component of the downstream signaling pathway activated by many known myelin inhibitors (MAG, Nogo, and OMgp), as well as by the CSPGs present in the glial scar (Lehmann et al., 1999; Bito et al., 2000; Dergham et al., 2002; Wang et al., 2002b; Winton et al., 2002; Dubreuil et al., 2003; Fournier et al., 2003; Monnier et al., 2003; Yamashita and Tohyama, 2003; Fischer et al., 2004b; Jain et al., 2004; Bertrand et al., 2005; Park et al., 2005; Shao et al., 2005). Thus, to examine the mechanisms involved in sulfatide-mediated inhibition, we began by asking whether Rho stabilization was required for the inhibitory effects of sulfatide. C3 transferase is an exoenzyme that binds to and ADP-ribosylates RhoA, RhoB, and RhoC, and has been used previously to promote neurite outgrowth on inhibitory substrates in vitro and in vivo (Lehmann et al., 1999). We tested the ability of a membrane-permeable version of C3 transferase to lessen the response of RGCs to sulfatide. When applied at a dose of 50 ng/ml, C3 transferase lessened the inhibition of outgrowth of RGCs grown on both CNS myelin and sulfatide to similar degrees (Fig. 6A). Although the actions of RhoA, RhoB, and RhoC, as well as those of other unknown C3 transferase targets could not be distinguished by these experiments, they did suggest that the activity of GTP-bound Rho may be a downstream component required for sulfatide-mediated inhibition.
Figure 6.

C3 transferase alleviated sulfatide-mediated inhibition of RGC neurite outgrowth in vitro. A, P5 rat RGCs were cultured for 18–24 h on coverslips coated with poly-d-lysine, mouse laminin, and either mouse CNS myelin (3 μg of protein) or 1 μg of sulfatide per 1.13 cm2 coverslip. Cells were imaged using calcein AM, and total outgrowth was assessed using the MetaMorph neurite outgrowth application. Cell-permeable C3 transferase added to the media at a concentration of 50 ng/ml significantly lessened the inhibitory effect of both myelin and sulfatide on RGC neurite outgrowth. Results shown are the average of five independent experiments and graphed ± SEM. *p < 0.05, **p < 0.01 by repeated measures one-way ANOVA with a Bonferroni's post hoc test comparing each C3 transferase condition with the vehicle-treated condition on the same substrate. B, P5 rat RGCs were cultured for 14–24 h on tissue culture plates coated with poly-d-lysine and mouse laminin. The Rho activator calpeptin, the Rho inhibitor C3 transferase, and sulfatide were added to the media either alone or in combination. Cell lysates were harvested, and the relative level of RhoA–GTP was measured using an ELISA-based RhoA activation assay. Neither calpeptin nor sulfatide promoted the stabilization of GTP-bound RhoA above control levels. Treatment with C3 transferase, however, significantly reduced the level of RhoA–GTP present in cells treated with either sulfatide or calpeptin. Results shown are the average of four independent experiments and graphed ± SEM. *p < 0.05, by one-way ANOVA with a Bonferroni's post hoc test comparing each condition with the C3 transferase-treated condition, as well as calpeptin and sulfatide alone with the vehicle control condition.
To determine whether sulfatide required RhoA specifically for its ability to inhibit outgrowth, we performed RhoA activation assays using the Rhotekin Rho binding domain, which binds specifically to GTP-bound Rho (Aspenström, 1999; Ren et al., 1999), in an ELISA-based format. We used this assay to test two questions: first, whether sulfatide induced elevated levels of RhoA–GTP in cultured RGCs; second, whether C3 transferase lowered levels of RhoA–GTP in the presence of sulfatide, a result that would indicate effective reduction of RhoA signaling in our experiments. To answer these questions, we tested the effect of sulfatide, the Rho activator calpeptin, and the Rho inhibitor C3 transferase on the levels of RhoA-GTP in retinal ganglion cells cultured for 14–24 h, both alone and in combination. We did not observe an increase in RhoA–GTP levels in RGCs treated with either calpeptin or sulfatide (Fig. 6B), despite the fact that both of these treatments strongly inhibited RGC neurite outgrowth in each experiment (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). We did, however, observe a consistent decrease in the levels of RhoA–GTP in conditions treated with C3 transferase (Fig. 6B). This effect was significant when comparing both calpeptin and sulfatide with and without C3 transferase. Thus, although neither sulfatide nor the Rho activator calpeptin was sufficient to increase RhoA–GTP levels above control, the Rho inhibitor C3 transferase did significantly decrease RhoA–GTP levels. The fact that no increase was seen in the calpeptin positive control despite its effects on neurite outgrowth suggests that we would be unlikely to observe any increase in the presence of sulfatide under these conditions, and thus we cannot determine the effect of sulfatide on RhoA–GTP from these experiments. They do, however, indicate that addition of C3 transferase, the treatment that abrogated sulfatide-mediated inhibition, is sufficient to decrease RhoA–GTP levels under the same conditions in which inhibition is alleviated. This result is consistent with a role for RhoA in mediating the inhibitory effects of sulfatide but does not definitively answer the question of whether sulfatide is sufficient to activate RhoA or whether the effects of C3 transferase on sulfatide-mediated inhibition can be attributed to its effects on RhoA.
Discussion
The lipid sulfatide is a myelin-associated axon growth inhibitor
We have demonstrated that the myelin lipid sulfatide was both sufficient to inhibit RGC neurite outgrowth and necessary for inhibition by CNS myelin in vitro. The Rho inhibitor C3 transferase lessened sulfatide-mediated inhibition. Optic nerve regeneration in animals lacking sulfatide and galactocerebroside was not enhanced under normal conditions. RGCs induced to regenerate by zymosan injection, however, did so more extensively than did their wild-type and heterozygous littermates, indicating that sulfatide does play a role in CNS regenerative failure in vivo.
Together, these results demonstrate that sulfatide is a novel myelin-associated inhibitor of CNS axon regeneration. This is the first demonstration of a lipid contributing to the inhibitory properties of myelin. Although abundant myelin lipids such as galactocerebroside and ceramide were not inhibitory, it remains possible that other lipids also contribute to the inhibitory properties of myelin. Given that these lipids constitute 70–75% of myelin by weight (Norton and Poduslo, 1973b) and are even more abundant on a molar basis (Vance and Vance, 2002), they could significantly contribute to the inhibitory properties of myelin; thus, they represent an important direction for additional research.
We identified sulfatide as an antigen of the antibodies generated by immunizing mice with CNS myelin. Evidence in the literature suggests that the immune response to sulfatide may be attributable to more than just chance, because the manipulations that promote experimental autoimmune encephalomyelitis can generate antibodies recognizing sulfatide despite the absence of the molecule in the immunizing material (Kanter et al., 2006). Indeed, when sulfatide, but not galactocerebroside, sphingomyelin, or GM1, is presented by CD1d, it is capable of inducing specific populations of NK cells to proliferate and generate cytokines (Halder et al., 2007; Podbielska and Hogan, 2009). Our experiments do not address whether sulfatide-specific antibodies contribute to the ability of CNS myelin-immunized sera to neutralize myelin inhibition. The strong immunogenic properties of sulfatide, however, suggest that immunization with CNS myelin may frequently induce sulfatide-specific antibodies, thus allowing it to be blocked recurrently in these immunization experiments.
Given that the effective inhibitory dose of sulfatide in our experiments is 1 μg/coverslip whereas myelin is inhibitory at 1–3 μg/coverslip, it might seem at first glance that sulfatide is only two to three times as potent a neurite outgrowth inhibitor as CNS myelin, which contains many non-inhibitory molecules in addition to the known inhibitors. It should be noted, however, that all myelin concentrations were quantified in micrograms of protein rather than total weight. Given that myelin is 70–75% lipid (Norton and Poduslo, 1973b), a sample containing 2 μg of protein would also contain 5–6 μg of lipid, ∼4–7% of which is sulfatide (Norton and Poduslo, 1973b). Thus, our myelin-coated coverslips contain ∼0.2–0.4 μg of sulfatide per coverslip, a concentration only slightly lower than the effective dose of sulfatide used in all experiments.
How does sulfatide inhibit axon growth?
The inability of either the direct precursors of sulfatide or sulfatide variants lacking a fatty acid chain to inhibit outgrowth suggests that the axonal response to sulfatide is quite specific and not attributable to a preponderance of either negative charge or non-polar fatty acids. The specificity of this response is not unprecedented, because sulfatide is known to be required for specific extracellular signaling processes such as the initiation of basement membrane assembly by binding to laminin (Li et al., 2005), the inhibition of platelet aggregation by binding to von Willebrand factor (Borthakur et al., 2003), and the promotion of metastasis by binding to P-selectin (Garcia et al., 2007). The specificity of these interactions has been studied extensively (Roberts et al., 1986; Vos et al., 1994), and the sulfatide-binding domain of von Willebrand factor has been mapped in detail (Christophe et al., 1991).
The ability of C3 transferase to lessen the inhibitory effects of sulfatide also suggests that sulfatide inhibits outgrowth by activating a specific signaling cascade rather than inducing poor cell health. We would not expect that C3 transferase, which typically affects Rho and consequently the actin cytoskeleton, would promote outgrowth by rescuing a general unhealthiness phenotype.
Given our RhoA activation data, we cannot conclude that sulfatide stabilizes RhoA. The inability of known Rho activator calpeptin to do so despite its effects on RGC neurite outgrowth, however, suggests that observing stabilization of RhoA–GTP under these conditions is extremely difficult. It will be informative to repeat these experiments with higher doses of sulfatide over shorter time periods to determine whether RhoA–GTP stabilization can be observed. Regardless, the fact that C3 transferase so consistently lowers RhoA–GTP levels in the presence of sulfatide indicates that the Rho inhibitor is capable of blocking RhoA–GTP in RGCs at the dose effective in promoting outgrowth, consistent with a potential role for the molecule in mediating sulfatide-induced inhibition of neurite outgrowth.
It will be interesting to determine whether other common components of the axonal response to inhibitory signaling, such as the Nogo receptor complex and PirB (Fournier et al., 2001; Wang et al., 2002b; Mi et al., 2004; Park et al., 2005; Shao et al., 2005; Atwal et al., 2008), are also required for sulfatide-mediated inhibition. Sulfatide, but not galactocerebroside, has been shown to activate protein kinase C (PKC) (Farooqui et al., 1988). Because PKC mediates inhibitory effects of both myelin and CSPGs on axonal regeneration (Sivasankaran et al., 2004), this is another possible point of convergence between sulfatide and other known inhibitors. In addition, it is also possible that known sulfatide-binding partners identified outside of the nervous system such as P-selectin or von Willebrand factor (Borthakur et al., 2003; Li et al., 2005), or proteins with domains similar to the sulfatide-binding domain of von Willebrand factor (Christophe et al., 1991), may play a role in the axonal response to sulfatide. If so, they might receive signals from sulfatide exclusively, or they, like NgR and PirB, might also respond to a wide variety of inhibitory molecules.
What is the role of sulfatide in myelin in vivo?
Like MAG, sulfatide is present at high levels in both PNS and CNS myelin, both of which are inhibitory to axon regeneration (Bedi et al., 1992; Vargas et al., 2010). Interestingly however, evidence suggests that sulfatide may be downregulated after PNS nerve injury even more quickly than the normal course of myelin clearance during Wallerian degeneration. Fourteen days after sciatic nerve injury, phospholipid and galactocerebroside levels decrease twofold, whereas sulfatide levels decrease fivefold (Alberghina et al., 1983). Additionally, Schwann cells downregulate sulfatide within 2 d when placed in culture, whereas oligodendrocytes maintain expression indefinitely (Mirsky et al., 1980). Galactocerebroside is also downregulated but can still be detected after 4 d in culture. It is possible that this transition from myelinating to cultured Schwann cell recapitulates aspects of Schwann cell behavior after injury. If so, this could account for the greater decrease in sulfatide levels observed at day 14 after injury. Together, these findings suggest that, although sulfatide is present in PNS myelin, it is removed quickly after injury and is thus unable to inhibit peripheral nerve regeneration.
Although sulfatide is a proportionally large component of CNS myelin, it is not surprising that its genetic removal enhances RGC axon regeneration after injury in vivo only when combined with intraocular inflammation. Genetic deletion of Nogo, MAG, and OMgp has been shown to have extremely variable effects on axon regeneration in spinal cord injury paradigms, even in animals in which all three genes have been genetically deleted (Cafferty et al., 2010; Lee et al., 2010). In the optic nerve, fewer reports of axon regeneration as a result of targeting an individual MAI have been published (Weibel et al., 1994). More often, robust optic nerve regeneration has been observed only when MAIs are targeted in combination with other growth-promoting conditions, such as lens injury, zymosan injection, or growth factor treatment (Cui et al., 2004; Fischer et al., 2004a; Chen et al., 2009; Kurimoto et al., 2010), or when common downstream signaling components such as Rho and ROCK are targeted (Lehmann et al., 1999; Fischer et al., 2004b; Bertrand et al., 2005). Why significantly enhanced optic nerve regeneration is observed in CGT null mice only in combination with intraocular inflammation is not addressed in our study. One could speculate, however, that although inflammation promoted axonal outgrowth, the axons remained responsive enough to their environment to sense the removal of one of its many inhibitory cues. Unstimulated RGCs, conversely, might be either so responsive to inhibitors or in such a non-regenerative state that the removal of one of many cues would not be sufficient to promote regeneration.
Deletion of many of these MAIs or targeting of common components of the downstream signaling cascade, as well as simultaneous targeting of the glial scar and promotion of intrinsic growth programs will likely be required to effectively promote robust regeneration (Kadoya et al., 2009; Kurimoto et al., 2010). In any case, our findings add to the growing evidence that myelin contains a large number of powerful neurite outgrowth inhibitors and provide additional support for the proposal that a normal function of myelin is to inhibit axonal plasticity (Colello and Schwab, 1994; Lee et al., 2004).
Footnotes
This work was supported by National Institutes of Health (NIH) Grant EY11310 and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. A.M.W. was supported by NIH Training Grant 5T32GM07790 and a Donald E. and Delia B. Baxter Foundation Graduate Fellowship. W.J.M. was supported by National Multiple Sclerosis Society Postdoctoral Fellowship FG 1434-A-1 and Netherlands Organization for Scientific Research TALENT Stipend S 93-380. M.Z.S. was supported by a Stanford Department of Developmental Biology Vice Provost for Undergraduate Education Summer Research Grant and a Stanford Major Grant for undergraduate summer research. We thank Jennifer Kanter for help with lipid handling techniques, Maria Fabian for help with tissue culture, Jonah Chan for helpful suggestions, and Lynette Foo and Junryo Watanabe for constructive comments on this manuscript.
References
- Alberghina M, Viola M, Moschella F, Giuffrida AM. Myelination of regenerating sciatic nerve of the rat: lipid components and synthesis of myelin lipids. Neurochem Res. 1983;8:133–150. doi: 10.1007/BF00963914. [DOI] [PubMed] [Google Scholar]
- Aspenström P. Effectors for the Rho GTPases. Curr Opin Cell Biol. 1999;11:95–102. doi: 10.1016/s0955-0674(99)80011-8. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science. 2008;322:967–970. doi: 10.1126/science.1161151. [DOI] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Bedi KS, Winter J, Berry M, Cohen J. Adult rat dorsal root ganglion neurons extend neurites on predegenerated but not on normal peripheral nerves in vitro. Eur J Neurosci. 1992;4:193–200. doi: 10.1111/j.1460-9568.1992.tb00867.x. [DOI] [PubMed] [Google Scholar]
- Benson MD, Romero MI, Lush ME, Lu QR, Henkemeyer M, Parada LF. Ephrin-B3 is a myelin-based inhibitor of neurite outgrowth. Proc Natl Acad Sci U S A. 2005;102:10694–10699. doi: 10.1073/pnas.0504021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand J, Winton MJ, Rodriguez-Hernandez N, Campenot RB, McKerracher L. Application of Rho antagonist to neuronal cell bodies promotes neurite growth in compartmented cultures and regeneration of retinal ganglion cell axons in the optic nerve of adult rats. J Neurosci. 2005;25:1113–1121. doi: 10.1523/JNEUROSCI.3931-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Furuyashiki T, Ishihara H, Shibasaki Y, Ohashi K, Mizuno K, Maekawa M, Ishizaki T, Narumiya S. A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron. 2000;26:431–441. doi: 10.1016/s0896-6273(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Borthakur G, Cruz MA, Dong JF, McIntire L, Li F, López JA, Thiagarajan P. Sulfatides inhibit platelet adhesion to von Willebrand factor in flowing blood. J Thromb Haemost. 2003;1:1288–1295. doi: 10.1046/j.1538-7836.2003.00156.x. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Buschard K, Blomqvist M, Månsson JE, Fredman P, Juhl K, Gromada J. C16:0 sulfatide inhibits insulin secretion in rat beta-cells by reducing the sensitivity of KATP channels to ATP inhibition. Diabetes. 2006;55:2826–2834. doi: 10.2337/db05-1355. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, Duffy P, Huebner E, Strittmatter SM. MAG and OMgp synergize with Nogo-A to restrict axonal growth and neurological recovery after spinal cord trauma. J Neurosci. 2010;30:6825–6837. doi: 10.1523/JNEUROSCI.6239-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Chen X, Yin X, Yuan R, Wang B, Ye J. NgR RNA interference, combined with zymosan intravitreal injection, enhances optic nerve regeneration. J Neurochem. 2009;110:1628–1634. doi: 10.1111/j.1471-4159.2009.06264.x. [DOI] [PubMed] [Google Scholar]
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- Christophe O, Obert B, Meyer D, Girma JP. The binding domain of von Willebrand factor to sulfatides is distinct from those interacting with glycoprotein Ib, heparin, and collagen and resides between amino acid residues Leu 512 and Lys 673. Blood. 1991;78:2310–2317. [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Colello RJ, Schwab ME. A role for oligodendrocytes in the stabilization of optic axon numbers. J Neurosci. 1994;14:6446–6452. doi: 10.1523/JNEUROSCI.14-11-06446.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Cho KS, So KF, Yip HK. Synergistic effect of Nogo-neutralizing antibody IN-1 and ciliary neurotrophic factor on axonal regeneration in adult rodent visual systems. J Neurotrauma. 2004;21:617–625. doi: 10.1089/089771504774129946. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J. Regeneration of adult axons in white matter tracts of the central nervous system. Nature. 1997;390:680–683. doi: 10.1038/37776. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Suzuki K, Popko B. Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol. 1998a;27:649–659. doi: 10.1023/a:1006908013972. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B. Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci. 1998b;18:1642–1649. doi: 10.1523/JNEUROSCI.18-05-01642.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M. The role and metabolism of sulfatide in the nervous system. Mol Neurobiol. 2008;37:93–103. doi: 10.1007/s12035-008-8022-3. [DOI] [PubMed] [Google Scholar]
- Ellezam B, Bertrand J, Dergham P, McKerracher L. Vaccination stimulates retinal ganglion cell regeneration in the adult optic nerve. Neurobiol Dis. 2003;12:1–10. doi: 10.1016/s0969-9961(02)00013-x. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Farooqui T, Yates AJ, Horrocks LA. Regulation of protein kinase C activity by various lipids. Neurochem Res. 1988;13:499–511. doi: 10.1007/BF00973288. [DOI] [PubMed] [Google Scholar]
- Fidler PS, Schuette K, Asher RA, Dobbertin A, Thornton SR, Calle-Patino Y, Muir E, Levine JM, Geller HM, Rogers JH, Faissner A, Fawcett JW. Comparing astrocytic cell lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. J Neurosci. 1999;19:8778–8788. doi: 10.1523/JNEUROSCI.19-20-08778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, He Z, Benowitz LI. Counteracting the Nogo receptor enhances optic nerve regeneration if retinal ganglion cells are in an active growth state. J Neurosci. 2004a;24:1646–1651. doi: 10.1523/JNEUROSCI.5119-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Petkova V, Thanos S, Benowitz LI. Switching mature retinal ganglion cells to a robust growth state in vivo: gene expression and synergy with RhoA inactivation. J Neurosci. 2004b;24:8726–8740. doi: 10.1523/JNEUROSCI.2774-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman P, Mânsson JE, Rynmark BM, Josefsen K, Ekblond A, Halldner L, Osterbye T, Horn T, Buschard K. The glycosphingolipid sulfatide in the islets of Langerhans in rat pancreas is processed through recycling: possible involvement in insulin trafficking. Glycobiology. 2000;10:39–50. doi: 10.1093/glycob/10.1.39. [DOI] [PubMed] [Google Scholar]
- Garcia J, Callewaert N, Borsig L. P-selectin mediates metastatic progression through binding to sulfatides on tumor cells. Glycobiology. 2007;17:185–196. doi: 10.1093/glycob/cwl059. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–1864. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Vargas ME, Wang JT, Mandemakers W, Oster SF, Sretavan DW, Barres BA. An oligodendrocyte lineage-specific semaphorin, Sema5A, inhibits axon growth by retinal ganglion cells. J Neurosci. 2004;24:4989–4999. doi: 10.1523/JNEUROSCI.4390-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPré T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- Halder RC, Jahng A, Maricic I, Kumar V. Mini review: immune response to myelin-derived sulfatide and CNS-demyelination. Neurochem Res. 2007;32:257–262. doi: 10.1007/s11064-006-9145-4. [DOI] [PubMed] [Google Scholar]
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci U S A. 2002;99:4227–4232. doi: 10.1073/pnas.032068299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, McKerracher L, Braun PE, David S. A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron. 1999;24:639–647. doi: 10.1016/s0896-6273(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Isaac G, Pernber Z, Gieselmann V, Hansson E, Bergquist J, Månsson JE. Sulfatide with short fatty acid dominates in astrocytes and neurons. FEBS J. 2006;273:1782–1790. doi: 10.1111/j.1742-4658.2006.05195.x. [DOI] [PubMed] [Google Scholar]
- Jain A, Brady-Kalnay SM, Bellamkonda RV. Modulation of Rho GTPase activity alleviates chondroitin sulfate proteoglycan-dependent inhibition of neurite extension. J Neurosci Res. 2004;77:299–307. doi: 10.1002/jnr.20161. [DOI] [PubMed] [Google Scholar]
- Kadoya K, Tsukada S, Lu P, Coppola G, Geschwind D, Filbin MT, Blesch A, Tuszynski MH. Combined intrinsic and extrinsic neuronal mechanisms facilitate bridging axonal regeneration one year after spinal cord injury. Neuron. 2009;64:165–172. doi: 10.1016/j.neuron.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- Keirstead HS, Hasan SJ, Muir GD, Steeves JD. Suppression of the onset of myelination extends the permissive period for the functional repair of embryonic spinal cord. Proc Natl Acad Sci U S A. 1992;89:11664–11668. doi: 10.1073/pnas.89.24.11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto T, Yin Y, Omura K, Gilbert HY, Kim D, Cen LP, Moko L, Kügler S, Benowitz LI. Long-distance axon regeneration in the mature optic nerve: contributions of oncomodulin, cAMP, and pten gene deletion. J Neurosci. 2010;30:15654–15663. doi: 10.1523/JNEUROSCI.4340-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci. 2004;24:6209–6217. doi: 10.1523/JNEUROSCI.1643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Geoffroy CG, Chan AF, Tolentino KE, Crawford MJ, Leal MA, Kang B, Zheng B. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron. 2010;66:663–670. doi: 10.1016/j.neuron.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19:7537–7547. doi: 10.1523/JNEUROSCI.19-17-07537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI. Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci. 2000;20:4615–4626. doi: 10.1523/JNEUROSCI.20-12-04615.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, Yurchenco PD. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol. 2005;169:179–189. doi: 10.1083/jcb.200501098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacBrinn MC, O'Brien JS. Lipid composition of optic nerve myelin. J Neurochem. 1969:16, 7–12. doi: 10.1111/j.1471-4159.1969.tb10337.x. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE. Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron. 1994;13:805–811. doi: 10.1016/0896-6273(94)90247-x. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, Crowell T, Cate RL, McCoy JM, Pepinsky RB. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Mirsky R, Winter J, Abney ER, Pruss RM, Gavrilovic J, Raff MC. Myelin-specific proteins and glycolipids in rat Schwann cells and oligodendrocytes in culture. J Cell Biol. 1980;84:483–494. doi: 10.1083/jcb.84.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier PP, Sierra A, Schwab JM, Henke-Fahle S, Mueller BK. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci. 2003;22:319–330. doi: 10.1016/s1044-7431(02)00035-0. [DOI] [PubMed] [Google Scholar]
- Moon LD, Asher RA, Rhodes KE, Fawcett JW. Regeneration of CNS axons back to their target following treatment of adult rat brain with chondroitinase ABC. Nat Neurosci. 2001;4:465–466. doi: 10.1038/87415. [DOI] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, Love C, Jones EY, Kikutani H, Lubetzki C, Dusart I, Chédotal A. The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci. 2003;23:9229–9239. doi: 10.1523/JNEUROSCI.23-27-09229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973a;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: changes in myelin composition during brain maturation. J Neurochem. 1973b;21:759–773. doi: 10.1111/j.1471-4159.1973.tb07520.x. [DOI] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, Garcia KC, He Z. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbielska M, Hogan EL. Molecular and immunogenic features of myelin lipids: incitants or modulators of multiple sclerosis? Mult Scler. 2009;15:1011–1029. doi: 10.1177/1352458509106708. [DOI] [PubMed] [Google Scholar]
- Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, Michalovich D, Simmons DL, Walsh FS. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383–384. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- Roberts DD, Rao CN, Liotta LA, Gralnick HR, Ginsburg V. Comparison of the specificities of laminin, thrombospondin, and von Willebrand factor for binding to sulfated glycolipids. J Biol Chem. 1986;261:6872–6877. [PubMed] [Google Scholar]
- Saadat L, Dupree JL, Kilkus J, Han X, Traka M, Proia RL, Dawson G, Popko B. Absence of oligodendroglial glucosylceramide synthesis does not result in CNS myelin abnormalities or alter the dysmyelinating phenotype of CGT-deficient mice. Glia. 2010;58:391–398. doi: 10.1002/glia.20930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio T, Schwab ME. Lesioned corticospinal tract axons regenerate in myelin-free rat spinal cord. Proc Natl Acad Sci U S A. 1990;87:4130–4133. doi: 10.1073/pnas.87.11.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, Murray B, Jung V, Pepinsky RB, Mi S. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Sivasankaran R, Pei J, Wang KC, Zhang YP, Shields CB, Xu XM, He Z. PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat Neurosci. 2004;7:261–268. doi: 10.1038/nn1193. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol. 1981;83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- Sommer I, Schachner M. Cell that are O4 antigen-positive and O1 antigen-negative differentiate into O1 antigen-positive oligodendrocytes. Neurosci Lett. 1982;29:183–188. doi: 10.1016/0304-3940(82)90351-2. [DOI] [PubMed] [Google Scholar]
- Vance DE, Vance JE. Biochemistry of lipids, lipoproteins, and membranes. Ed 4. Amsterdam: Elsevier; 2002. [Google Scholar]
- Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A. 2010;107:11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos JP, Lopes-Cardozo M, Gadella BM. Metabolic and functional aspects of sulfogalactolipids. Biochim Biophys Acta. 1994;1211:125–149. doi: 10.1016/0005-2760(94)90262-3. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002a;417:941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002b;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Weibel D, Cadelli D, Schwab ME. Regeneration of lesioned rat optic nerve fibers is improved after neutralization of myelin-associated neurite growth inhibitors. Brain Res. 1994;642:259–266. doi: 10.1016/0006-8993(94)90930-x. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Dubreuil CI, Lasko D, Leclerc N, McKerracher L. Characterization of new cell permeable C3-like proteins that inactivate Rho and stimulate neurite outgrowth on inhibitory substrates. J Biol Chem. 2002;277:32820–32829. doi: 10.1074/jbc.M201195200. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Neubauer D, Dyess K, Ferguson TA, Muir D. Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Exp Neurol. 1998;154:654–662. doi: 10.1006/exnr.1998.6951. [DOI] [PubMed] [Google Scholar]