

Letter to the Editor
Regimens of high dose chemotherapy followed by autologous blood and marrow transplantation (ABMT) have been used to treat patients with acute myelogenous leukemia (AML), but leukemia relapse is frequent, likely due to marrow contamination with leukemic hematopoietic stem and progenitor cells (HSPCs) and residual disease in protective niches. Purging autologous hematopoietic grafts of contaminating cancer cells prior to transplant serves as a viable strategy for increasing transplant efficacy but would also require sparing normal HSPCs within the same hematopoietic cell graft. Previous purging strategies using ex vivo incubation with cytotoxic agents and cell culture techniques have generally resulted in the loss of normal HSPC numbers or functionality.(1)
Oncolytic poxviruses, such as vaccinia virus and myxoma virus (MYXV), are promising new instruments in targeting human cancer.(2, 3) MYXV’s natural host tropism is highly restricted to European rabbits (Oryctolagus cuniculus), and it is nonpathogenic for all other vertebrate species tested, including humans.(4, 5) There have been no reports of MYXV infection in humans and no cases of any clinical complications, such as aplastic anemia, despite widespread MYXV exposure in countries such as Australia. MYXV has a large double-strand DNA genome permitting insertion of large numbers (at least 25 kb) of therapeutic eukaryotic genes. Despite its narrow host specificity, we have shown in cultured cells and in preclinical animal model studies that MYXV is capable of infecting and killing a variety of established human cancer cell lines in vitro and in vivo, but its efficacy and safety in targeting primary human cancer cells while sparing normal cells have not been defined.(3) Thus, to determine whether oncolytic viruses can be used as a therapeutic modality for purging leukemia from autologous grafts, we evaluated MYXV in a pre-clinical model of AML and normal HSPC transplantation. We demonstrate that ex vivo incubation with MYXV ablates in vitro colony forming potential of fms-like tyrosine kinase receptor-3 (FLT3) mutant AML cells and dramatically decreases engraftment levels in mice. High risk AMLs, such as those with FLT3 internal tandem duplications (ITD), are notoriously chemotherapy insensitive and prone to relapse. Moreover, using identical ex vivo conditions, MYXV does not affect either in vitro colony forming potential or in vivo engraftment of normal HSPCs derived from healthy human bone marrow (BM). These results show safety and efficacy of using oncolytic MYXV to purge AML HSPCs from autologous HSPC grafts intended for hematopoietic rescue.
Although there have been no case reports of human bone marrow failure despite widespread MYXV exposure, to document MYXV safety in the context of normal human HSPC function, we incubated BM-derived HSPCs from healthy donors (n=3) with a GFP expressing MYXV construct (vMYX-GFP) in vitro over a 3-day period at a high multiplicity of infection (MOI) of 10 per cell. After exposure to vMYX-GFP, BM-derived mononuclear cells (MNCs) were analyzed by flow cytometry to determine the extent of infectivity. Greater than 99.6 ± 0.2 % of BM MNCs were free of vMYX-GFP infection (Figure 1A). A small population of MNCs accounting for 0.5 ± 0.2 % of the total population was GFP-positive by this treatment, which was essentially indistinguishable from mock (vehicle only) treated cells and is typical of a nonpermissive infection by MYXV. No GFP expression was observed in normal CD34+ HSPC cells. On lineage subset analysis, CD15+ myeloid cells and CD19+ B lymphocytes also showed no signs of infection (data not shown).
Figure 1. MYXV treatment of normal and leukemic HSPCs in vitro.
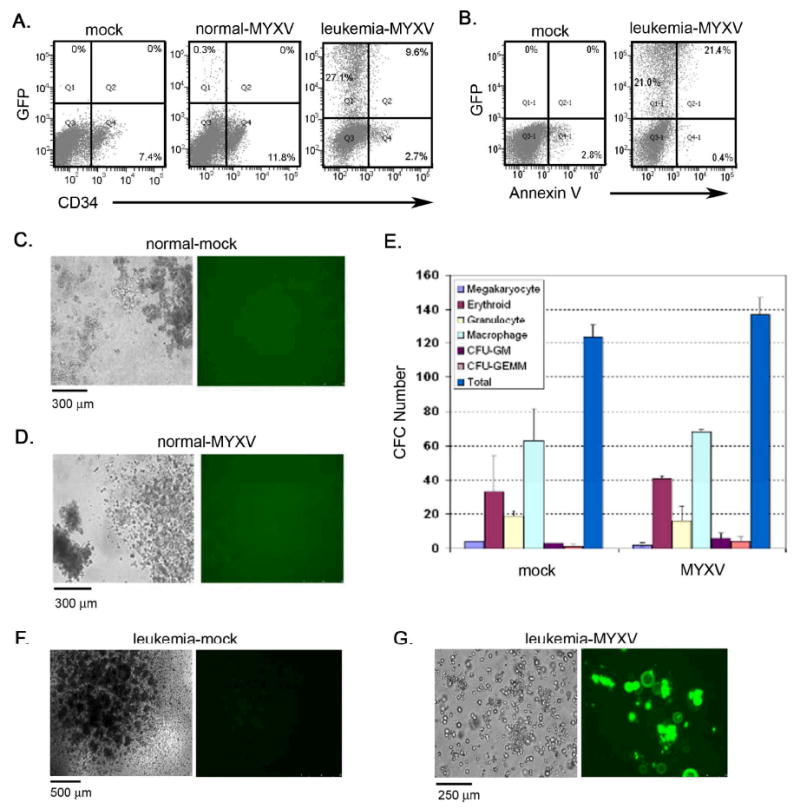
(A) Normal and leukemic cells (FLT ITD+) were incubated with MYXV expressing GFP at 10 MOI. Flow cytometric analysis demonstrated that a small population of normal MNCs was infected while a significant proportion of AML cells were susceptible to MYXV infection. (B) Analysis of apoptotic events was examined and showed that cell death was mainly observed in the GFP+ MYXV infected cell population as shown by Annexin V staining. All gating was established using appropriate isotype controls. (C, D) Normal BM cells were incubated with MYXV expressing GFP at 10 MOI and assessed for colony forming potential. Various types of CFC colonies were formed and showed no GFP expression in either MYXV-treated (D) or mock-treated samples (C) indicating that MYXV was unable to infect normal CFCs. (E) The frequency of each colony type was enumerated following MYXV or mock treatment. Similar frequencies were observed between both cohorts suggesting that MYXV does not prevent normal CFC colony differentiation in vitro. Data are representative of repeat experiments and values represent mean ± standard deviation. (F) Mock-treated AML cells formed AML-CFU colonies. (G) MYXV-treated AML cells showed signs of infection (GFP+) and did not form colonies.
To test whether primary AML cells permitted intracellular MYXV replication and oncolysis in vitro, leukemia cells were obtained from a 71 year-old white male presenting with de novo AML (normal male karyotype, FLT3 ITD+ mutation, NPM1c+ mutation). The myeloblasts were collected via peripheral blood leukapheresis and made up > 98% of the collected product. These AML cells were incubated with vMYX-GFP at 10 MOI for 3 days, and analyzed for infectivity and apoptotic events via Annexin-V staining. Experiments were performed in triplicate. In these samples, 40.3 ± 5.0 % of AML cells were infected with the virus, which was significantly higher in comparison to normal BM-derived MNCs (p<0.05; Figure 1A). Of the total AML CD34+ population, we found that 65 ± 6% were infected with MYXV, which was significantly higher in comparison to normal BM CD34+ cells (p<0.05). Cellular infection correlated with higher levels of apoptotic cell death, as assessed with Annexin V staining, and was restricted to the GFP+ cell population (Figure 1B). Kinetic analysis of cells incubated with vMYX-GFP at either 1.5 or 3.5 days post-infection showed that there was a time dependant increase in the number of HSPCs infected with vMYX-GFP (data not shown). These data indicate that MYXV efficiently infects AML HSPCs in vitro and induces apoptotic cell death through a classic permissive oncolytic poxvirus infection.
To further examine MYXV safety for HSPCs, experiments were performed to determine if MYXV alters normal HSPC differentiation function in vitro. MNCs derived from normal BM (n=3) were incubated with vMYX-GFP at 10 MOI for 3 hours at 37°C, after which cells were assayed for normal HSPC differentiation using an in vitro methylcellulose based colony forming cell (CFC) assay. We observed differentiated colonies forming normally after 14 days using either mock (vehicle only) or vMYX-GFP treated cells, indicating that the CFC potential of HSPCs was not adversely affected by MYXV (Figures 1C and 1D). Resulting colonies were analyzed for viral infection using fluorescent microscopy and showed a uniform lack of GFP+ colonies, further demonstrating that vMYX-GFP did not infect or perturb the viability of normal CFCs in vitro.
We next examined whether MYXV could alter the efficiency of normal HSPC differentiation by calculating the frequency of different colonies derived from either mock or vMYX-GFP treated cells. Following infection, cells were plated in the CFC assay (n=3) and the number and type of colonies determined after 16 to 18 days. The frequency of colonies formed was similar between mock and vMYX-GFP treated groups (Figure 1E). Since the number and type of hematopoietic colonies generated were similar between groups, it suggested that vMYX-GFP did not alter CFC viability or frequency in normal HSPC specimens. The data showed that MYXV does not adversely affect normal HSPC developmental potential following in vitro incubation.
In order to determine whether MYXV infects AML cells and perturbs their growth and differentiation in vitro, primary AML cells were incubated with vMYX-GFP at MOI of 10 for 3 hours, and then grown in methylcellulose based media (n=3). Mock treated AML cells formed pleomorphic colonies consistent with AML-colony forming units (CFUs) (Figure 1F). When exposed to vMYX-GFP, AML cells did not form recognizable AML-CFU colonies and instead remained heterodispersed (Figure 1G). Prior to plating in methylcellulose, AML infection was confirmed using flow cytometric analysis. Together, these data indicated that MYXV effectively infects AML in vitro, and impairs clonal proliferation.
Based on the in vitro safety results showing normal colony forming potential from MYXV treated normal HSPCs, we next tested safety by in vivo repopulation assays. Normal BM MNCs (n=3) or CD34+ selected cells (n=6) were exposed to 10 MOI MYXV for 3 hours ex vivo and then xenotransplanted into sublethally irradiated (325 cGy) NOG mice. After 6 to 8 weeks, BM was harvested from the mice and analyzed by flow cytometry for human cell engraftment using antibodies to human CD45 and HLA-abc. Mock-treated BM MNCs (n=4) and CD34+ cells (n=6) were also transplanted into NOG mice and served as controls. Typical human engraftment with CD45+/HLA-abc+ cells was observed in both treatment groups (Figures 2A). Quantitatively, similar numbers of mice engrafted with human hematopoietic cells in mock and MYXV-treated cohorts (70% vs. 78%; p=0.72; Figure 2B). The percent engraftment varied within individual mice as determined by double staining for CD45 and HLA-abc, but overall levels of engraftment were indistinguishable between mice receiving mock or MYXV treated normal HSPCs (1% vs. 2%, p=0.41; Figure 2B). Interestingly, some MYXV treated mice generated higher engraftment levels in comparison to mock treated mice, but this trend was not statistically significant. Histological analysis of various tissues from mice transplanted with MYXV-treated cells showed normal tissue morphology (data not shown). None of the immunocompromised NOG mice developed any evidence of viral replication in their skin or in any internal tissues, indicating that even severely immunodeficient hosts are non-permissive for MYXV infection. The data indicate that ex vivo treatment with MYXV does not reduce in vivo engraftment potential of normal BM-derived MNCs or CD34+ HSPCs. This critical safety data supports the notion that MYXV would be a safe candidate for purging leukemic cells from autologous hematopoietic grafts, even in immunocompromised recipients.
Figure 2. Normal and leukemic human HSPC engraftment in vivo following MYXV treatment.
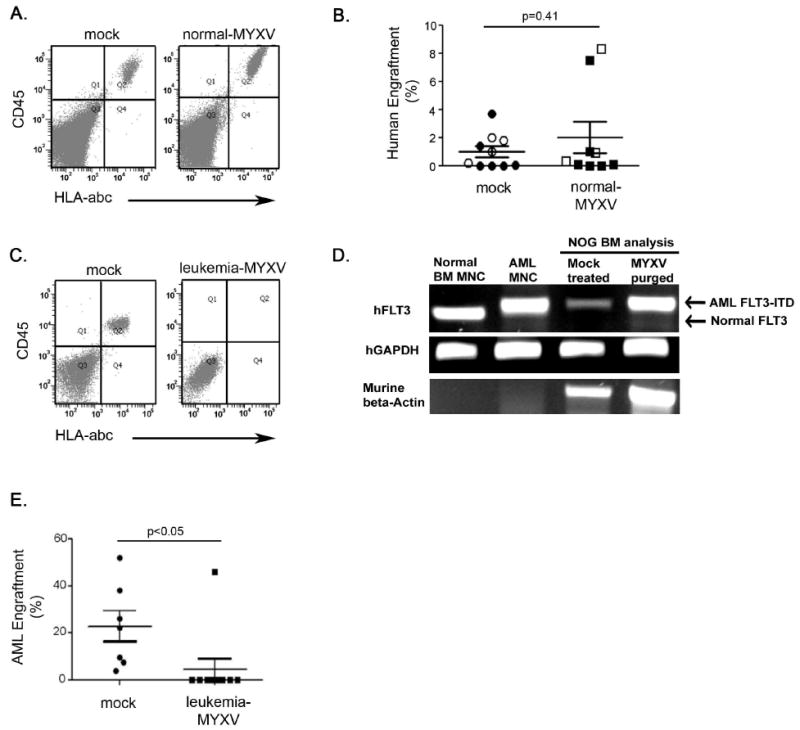
(A) Normal BM-derived MNCs or CD34+ selected cells were exposed to MYXV at 10 MOI for 3-hours ex vivo and transplanted into irradiated NOG mice. Analysis of mouse BM after 6 to 8-weeks demonstrated normal human cell engraftment with CD45+/HLA-abc+ cells observed in both MYXV treated (n=9) and mock treated (n=10) groups. Shown are representative flow cytometry plots. (B) Similar engraftment frequencies were observed between mock and MYXV treated cells. As expected, the percent engraftment values varied between mice; however, the overall levels of engraftment were similar between mice receiving mock or MYXV treated cells (p=0.41). Symbols representing mice transplanted with MNCs or CD34+ cells are shown in open and filled shapes (circle: Mock treated, square: MYXV purged), respectively. (C) AML cells were mock treated or exposed to MYXV at 10 MOI for 3-hours ex vivo and subsequently transplanted into irradiated NOG mice. Mock treated AML cells demonstrated leukemic engraftment in all mice tested (n=7). MYXV treated AML cells resulted in leukemic engraftment in only 10% of transplanted mice as analyzed by flow cytometry (n=10). Shown are representative flow cytometry plots. (D) Leukemic engraftment was also demonstrated using PCR for the FLT3 ITD mutation. Leukemic human cells in mouse bone marrow all exhibited the AML FLT3 ITD genotype. The FLT3 ITD mutation in AML cells created the larger FLT3 band, compared to normal controls. Only engrafted samples are shown: one representative of 7 from mock-treated and the single FACS-positive engrafted sample from the 10 MYXV-purged mice. Normal BM cells and AML MNCs were used as PCR controls. (E) MYXV treatment resulted in significantly lower percent engraftment levels in comparison to mock treated controls (p<0.05). Values represent mean±SEM. Gating was established using appropriate isotype controls.
Finally, we set out to test MYXV oncolytic effects on primary AML HSPCs in vivo. An AML specimen harboring a FLT3 ITD mutation was used in this study because this high risk AML represents a hematologic malignancy that is notoriously chemotherapy insensitive and can be tracked at the molecular level with PCR. Primary AML FLT3 ITD+ MNCs were treated with MYXV at 10 MOI for 3 hours and transplanted into sublethally irradiated NOG mice. Experiments were performed in duplicate and mouse numbers maximized due to the rarity of obtaining AML cells. At 6 to 8 weeks post-transplantation, mouse BM was harvested and examined for AML engraftment. By flow cytometric analysis, mock-treated cells resulted in CD45+/HLA-abc+ human engraftment in 100% of transplanted mice (n=7; Figure 2C and 2E). Conversely, MYXV-treated AML cells only engrafted in 10% of mice, whereas the remaining 90% of the recipients were phenotypically free of any detectable AML cells (n=10; Figure 2C and 2E). In both cohorts, PCR analysis for human AML FLT3 ITD+ cells in mouse BM was used to confirm leukemic engraftment (Figure 2D). Comparing results, MYXV treatment resulted in significantly lower mean levels of engraftment in comparison to mock treated controls (4.5% vs. 24%, p<0.05; Figure 2E). In the primary, high risk AML specimen tested, normal HSPC levels were extremely low, thus normal HSPC engraftment in immunocompromised mice did not occur, as expected. Taken together, the data indicate that MYXV incubation ex vivo significantly inhibited subsequent AML HSPC engraftment in vivo, resulting in phenotypic and molecular remissions of FLT3 ITD AML.
Various autograft purging strategies have been attempted, however their major limitations are a lack of specificity for AML HSPCs versus normal HSPCs and inadequate elimination of cancer cells upon purging. In this report, we show for the first time that MYXV specifically targets and eliminates primary AML HSPCs using a novel ex vivo purging technique. We also show that MYXV does not affect normal HSPCs, as assessed by in vitro colony forming potential and in vivo engraftment in immunodeficient mice. Interestingly, we show susceptibility of a primary AML specimen harboring an activating mutation in FLT3, which represents an aggressive malignancy well-known for insensitivity to conventional chemotherapy. Efficacy against this leukemia signifies the effectiveness of this strategy and an opportunity for disease eradication in an otherwise grim clinical setting. Together, these results show that oncolytic virotherapy with MYXV represents a promising therapeutic approach to eradicate malignant hematopoietic cells, including residual cancer stem cells, in autografts from patients treated with myeloablative immunosuppression and that require hematopoietic cell rescue.
Subsequent studies will test additional leukemic and myelodysplastic specimens to determine the breadth of applicability of this novel approach, and will focus on understanding the mechanisms involved in the successful purging of AML using MYXV. Additionally, more specific studies examining effects of MYXV on cancer stem cells are ongoing. Given that 65% of AML CD34+ HSPCs showed evidence of MYXV infection after incubation and leukemic engraftment was prevented in 90% of recipients, it is possible that leukemic stem cells may be infected and marked by MYXV incubation. Further investigation of this finding is warranted. At present, the precise method of MYXV specificity for human cancer cells is only partially understood. It is known that poxviruses like MYXV can bind and initiate entry into most mammalian cells, but then discriminates permissive versus nonpermissive cells by virtue of the cell signaling circuitry of the infected cell.(4) For example, we have previously shown that hyperactive AKT signaling, either as constitutive phosphorylation or as induced by the virus infection, dictates MYXV permissiveness in a wide variety of solid tumor cell lines.(6) However, considering the complexity of cancer cell populations, this may not be the only leukemia-specific signaling pathway by which MYXV discriminates normal versus cancerous HSPCs. For example, when normal human macrophages are infected with MYXV, the cells rapidly co-induce two anti-viral cytokines (TNF and type I interferon) via a RIG-I dependant signaling mechanism, which then aborts the MYXV infection in normal human somatic cells in a paracrine-like fashion.(7) Thus, another model would be that normal HSPCs are competent for this synergy whereas cancerous HSPCs, such as AML cells, would be defective in some aspect of the TNF/interferon synergistic pathway. The aforementioned studies will provide insights into leukemia tropism and anti-neoplastic mechanisms of MYXV-mediated oncolytic purging.
References
- 1.Yang H, Eaves C, de Lima M, Lee MS, Champlin RE, McMannis JD, et al. A novel triple purge strategy for eliminating chronic myelogenous leukemia (CML) cells from autografts. Bone Marrow Transplant. 2006 Mar;37(6):575–582. doi: 10.1038/sj.bmt.1705284. [DOI] [PubMed] [Google Scholar]
- 2.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009 Jan;9(1):64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 3.Stanford MM, McFadden G. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert opinion on biological therapy. 2007 Sep;7(9):1415–1425. doi: 10.1517/14712598.7.9.1415. [DOI] [PubMed] [Google Scholar]
- 4.McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005 Mar;3(3):201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanford MM, Werden SJ, McFadden G. Myxoma virus in the European rabbit: interactions between the virus and its susceptible host. Veterinary research. 2007 Mar-Apr;38(2):299–318. doi: 10.1051/vetres:2006054. [DOI] [PubMed] [Google Scholar]
- 6.Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, Gao X, et al. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci U S A. 2006 Mar 21;103(12):4640–4645. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang F, Gao X, Barrett JW, Shao Q, Bartee E, Mohamed MR, et al. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 2008 Jul;4(7):e1000099. doi: 10.1371/journal.ppat.1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]