

Abstract
Enzyme replacement therapy (ERT) for Pompe disease using recombinant acid alpha-glucosidase (rhGAA) has resulted in increased survival although the clinical response is variable. Cross Reactive Immunological Material (CRIM)-negative status has been recognized as a poor prognostic factor. CRIM-negative patients make no GAA protein and develop sustained high antibody titers to ERT that render the treatment ineffective. Antibody titers are generally low for the majority of CRIM-positive patients and there is typically a better clinical outcome. Because immunomodulation has been found to be most effective in CRIM-negative patients prior to, or shortly after, initiation of ERT, knowledge of CRIM status is important before ERT is begun. We have analyzed 243 patients with infantile Pompe disease using a Western blot method for determining CRIM status and using cultured skin fibroblasts. Sixty-one out of 243 (25.1%) patients tested from various ethnic backgrounds were found to be CRIM-negative. We then correlated the CRIM results with GAA gene mutations where available (52 CRIM-negative and 88 CRIM-positive patients). We found that, in most cases, CRIM status can be predicted from GAA mutations, potentially circumventing the need for invasive skin biopsy and time wasted in culturing cells in the future. Continued studies in this area will help to increase the power of GAA gene mutations in predicting CRIM status as well as possibly identifying CRIM-positive patients who are at risk for developing high antibody titers.
Keywords: Pompe disease, acid alpha-glucosidase, cross reactive immunological material, enzyme replacement therapy, GAA gene
INTRODUCTION
Pompe disease (Glycogen Storage Disease type II; acid maltase deficiency; OMIM# 232300) is an autosomal recessive disorder of glycogen metabolism caused by deficiency of the lysosomal enzyme acid alpha-glucosidase (GAA) [Hirchhorn and Reuser, 2001; Kishnani et al., 2006b]. Although the disease presents as a continuum of clinical spectrum, it can be broadly classified into infantile-onset and late-onset forms, according to the age at presentation [Kishnani et al., 2006b]. The classical infantile form is rapidly progressive and presents with hypertrophic cardiomyopathy by the first few months of life and has a fatal outcome within the first year of life if left untreated, while the non-classical form progresses more slowly and has less severe cardiac involvement [Kishnani et al., 2006b]. Late-onset Pompe disease is variable in age at presentation and extent of organ involvement and includes a juvenile form with onset any time after first year of life through early childhood, and an adult-onset form with symptoms appearing in the second to sixth decade of life [Kishnani et al., 2006b].
Development of recombinant human GAA (rhGAA) enzyme replacement therapy derived from Chinese hamster ovary (CHO) cells and transgenic rabbit milk led to subsequent clinical trials which showed a positive response in infantile Pompe patients [Amalfitano et al., 2001; Angelini and Semplicini, 2011; Kishnani et al., 2006a; Kishnani et al., 2007; Nicolino et al., 2009; van den Hout et al., 2000; van den Hout et al, 2004]. In 2006, CHO-derived rhGAA was approved in the USA, Europe, and Canada with subsequent approvals in many other countries worldwide. However, the clinical response to ERT varies considerably between patients. Various factors, including age and extent of muscle damage at initiation of ERT, muscle fiber type, and defective autophagy, have been associated with varied response to treatment [Hawes et al., 2007; Kishnani et al., 2007; Raben et al., 2007; Angelini and Semplicini, 2011]. In addition, Cross Reactive Immunological Material (CRIM) status has been found to be an important predictor of clinical response [Amalfitano et al., 2001; Kishnani et al., 2006a; Angelini and Semplicini, 2011; Banugaria et al., 2011; Chakrapani et al., 2010; Kishnani et al., 2010]. CRIM-negative patients are unable to make any GAA protein, due to the presence of underlying deleterious null GAA alleles, and as a result their immune system recognizes rhGAA as a foreign protein. Approximately 20% of classical infantile patients are CRIM-negative (personal experience). Despite being on ERT, these patients usually fare poorly due to development of a sustained high titer of neutralizing antibodies to rhGAA that renders the treatment ineffective [Banugaria et al., 2011]. CRIM-positive patients, in contrast, produce some residual GAA protein, although non-functional inactive form. They typically have low antibody titers and a better clinical outcome without the need for immunomodulation. Interestingly, some CRIM-positive infantile patients have been reported to develop high antibody titers and thus reduced overall benefit from ERT similar to CRIM negative patients [Banugaria et al., 2011]. These CRIM-positive patients develop high sustained antibody titers after the first 6 months or so of treatment, just like CRIM negative patients, which is typically followed by clinical decline concomitant with a rise in antibodies [Banugaria et al., 2011; Kishnani et al., 2010].
Determining CRIM status is important so that decisions can be made about immunomodulation therapy prior to or shortly after starting treatment. Recently, immunomodulation therapy has been shown to be effective in preventing an immune response in CRIM-negative patients who are naïve to ERT or who have had a short exposure to ERT [Mendelsohn et al., 2009; Messinger et al., in press]. However, attempts to eliminate anti-rhGAA antibodies in CRIM-negative patients who have been on ERT for an extended period with an entrenched immune response have failed [Amalfitano et al., 2001; Hunley et al., 2004]. A delay in treatment even for a short period of time in a rapidly progressive disease like infantile Pompe is detrimental to clinical outcome, thus methods for determining CRIM status need to be rapid and accurate [Chien et al., 2009; Kemper et al., 2007].
We present data from 10 years of experience of determining CRIM status from skin fibroblasts and correlation with underlying GAA mutation data. We have utilized a Western blot analysis method to determine CRIM status using homogenates from cultured skin fibroblasts in 243 patients with infantile Pompe disease [Bali et al., 2011; Kishnani et al., 2006a; Klinge et al., 2005]. This method is currently the only one available, although blood-based immunological methods are being developed. We then correlated CRIM status, determined by Western blot, with pathogenic GAA gene mutations where available (n=140 patients; 52 CRIM-negative and 88 CRIM-positive). As expected, in many cases, it was possible to predict CRIM status based on GAA mutation data alone. While use of immunoblotting techniques to confirm CRIM status is still recommended, it may be possible to determine CRIM status from GAA mutations alone in the future as more data is gathered, thus circumventing the need for skin biopsies which are invasive and require several weeks before results are available.
MATERIALS AND METHODS
Samples
Skin fibroblasts (1st or 2nd pass) were available from 243 children from diverse ethnicities including Caucasian (non-Hispanic and Hispanic), Asian (Indian and East Asian), African-American, and middle-Eastern populations, with a diagnosis of infantile Pompe disease (onset of symptoms <2 years of age) confirmed by deficient GAA enzyme activity in blood and/or skin fibroblasts. Sequencing data from all 19 coding exons and surrounding intron/exon boundaries of the GAA gene were available for 140 of these children (88 CRIM-positive and 52 CRIM-negative). Some of the patients in this study may have been previously published. The Duke University Institutional Review Board approved this retrospective data analysis study.
Western blot analysis
Twenty micrograms of protein homogenate obtained from patient fibroblast cells were loaded per well onto 4-12% gradient pre-cast SDS-PAGE gels (Invitrogen, CA, USA). In addition to samples from patients, fibroblast homogenates from a known CRIM-negative patient (negative quality control), a CRIM-positive quality control (normal human fibroblast), and a molecular weight marker (Magic Mark XP Western Protein Standard, Invitrogen, CA, USA) were loaded on each gel. Beta-actin was used as the loading control. After blotting the gel onto nitrocellulose membrane (BioRad, CA, USA), GAA protein was detected by probing the membrane with polyclonal anti-GAA antibody (Duke Biochemical Genetics Laboratory, NC, USA) produced against placental GAA protein [Kishnani et al., 2007; Kishnani et al., 2010], and/or monoclonal anti-GAA antibody kindly provided by Genzyme corporation (Genzyme Genetics, Framingham, MA, USA). A total of 70 patient cell extract samples were analyzed using both polyclonal and monoclonal anti-GAA antibodies, to determine specificity and compare results. The secondary antibody was horse-radish peroxidase linked anti-rabbit IgG (from donkey). After final washing of membrane with 1 x TBS-T buffer, patient protein bands were visualized using an enhanced chemiluminescence (ECL) Western Blotting Detection Reagents (Amersham, GE Healthcare, NJ, USA) and exposure of the membrane on X-ray film. Protein bands were labeled as 110, 95, 76 or 70 KDa, based on the molecular weight marker.
GAA gene sequencing
GAA mutation analysis was performed through full gene sequencing using genomic DNA isolated from peripheral blood or from skin fibroblasts of Pompe patients. The coding regions of the GAA gene and surrounding exon/intron boundaries were sequenced following PCR amplification, amplicon purification, and loading onto an ABI 3130xl Genetic Analyzer (Perkin Elmer, CA, USA). Sequences were compared to the GAA reference DNA sequence (GenBank Accession: NM_000152) to identify pathogenic mutations. Sequencing results were available for 140 children (88 CRIM-positive and 52 CRIM-negative). The pathogenicity of novel missense mutations was predicted using the PolyPhen-2 program (http://genetics.bwh.harvard.edu/pph/) [Ramensky et al., 2002], and the effect of splice site nucleotide changes was investigated using the Berkeley Drosophila Genome Project splice site prediction program at http://www.fruitfly.org/seq_tools/splice.html
RESULTS
Comparison of CRIM results using monoclonal versus polyclonal antibody
Western blot analysis gave essentially the same result whether monoclonal or polyclonal antibodies were used (n=70, Figure 1). Western blots of normal human fibroblasts showed a band at 95 kDa representing an intermediate form, and 76 kDa and 70 kDa bands representing the fully functional mature enzyme. A band at 110 kDa representing the inactive precursor protein of 952 amino acids is rarely seen in the amounts loaded for normal samples [Bali et al., 2011; Moreland et al., 2005].
Figure 1.
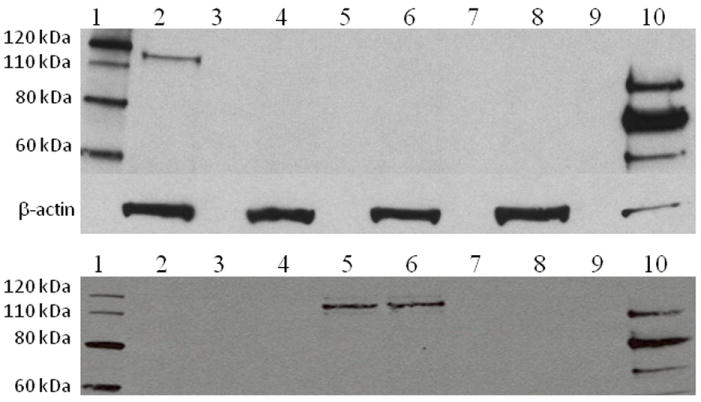
Western blot analysis on protein extracts (20ug loaded) from human skin fibroblast and probed with anti-GAA antibody (Top panel= polyclonal rabbit anti-GAA antibody against human placental GAA; Lower panel= monoclonal anti-GAA antibody, GAA1 provided by Genzyme). β-actin was used as a protein loading control.
Top panel: Lane 1-Protein molecular weight marker; Lane 2 – CRIM-positive Pompe disease patient; Lanes 3, 5, 7 and 9 – Empty; Lanes 4, 6, and 8 – CRIM-negative patients; Lane 10 – Normal human fibroblasts.
Lower panel: Lane 1 – Protein molecular weight marker; Lanes 2 and 3 – Duplicate lanes for CRIM-negative sample; Lanes 4,7 and 9 – Empty; Lanes 5 and 6 – Duplicate lanes for CRIM-positive sample; Lane 8 – CRIM-negative control (known CRIM-negative patient); Lane 10 – Normal human fibroblasts.
Percentage of CRIM negative patients
Out of the 243 patients in this study, 61 (25.1%) were CRIM-negative based on Western blot analysis. CRIM-negative status was found in subjects from various ethnic backgrounds including Caucasian (non-Hispanic and Hispanic), Asian (Indian and East Asian), African-American, and middle-Eastern populations.
Correlation between CRIM-status and GAA gene mutations
GAA gene sequencing results were available on 52 out of 61 patients who tested CRIM-negative by Western blot. Forty-one different mutations were identified in this group, 24 of which had been previously cited and 17 of which are novel (Table I; Figure 2). The most frequently identified mutations were p.Arg854X and c.525delT (32.7% and 4.8% of CRIM-negative alleles respectively). Consistent with the inability to make GAA protein, most CRIM-negative patients (44/52) were homozygous or compound heterozygotes for nonsense and/or frame shift mutations resulting in premature stop codons, or multi-exon deletions (Table II). No missense mutations were identified in the CRIM-negative patients. However, one CRIM-negative patient was homozygous for a point mutation that abolished the initiator methionine (c.1A>G; p.Met1?). In contrast, most of the CRIM-positive patients (81 out of 88) had one or two missense or in frame deletion mutations that would be predicted to produce some GAA protein (Table II). Ninety-two different mutations were found in the CRIM-positive group, 22 of which are novel (Table III). One CRIM-positive patient was homozygous for a previously reported frame shift mutation which created a premature stop codon in the last exon of the GAA gene (p.Gln914ProfsX30) (Reuser et al., 1995; Hermans et al., 1998). Surprisingly another CRIM-positive patient was a compound heterozygote for two previously reported predicted null mutations, a frame shift mutation in exon 7 (p.Glu389ArgfsX3) and a nonsense mutation (p.Arg854X) (Hermans et al., 1993; Kroos et al., 2008). None of the CRIM-positive patients had the same combination of mutations as any of the CRIM-negative patients.
Table I.
GAA mutations found in CRIM-negative infantile Pompe disease patients in this study
| Location | Nucleotide change | Amino acid change | Ethnicitya | Number of allelesb | Citation |
|---|---|---|---|---|---|
| Frame shift | |||||
| Exon 2 | c.340_341insT | p.Lys114IlefsX32 | Jewish (2) | 4 | [Hermans et al., 2004] |
| Exon 2 | c.525delT | p.Glu176ArgfsX45 | Caucasian (2) | 5 | [Hermans et al., 1994] |
| Exon 2 | c.525_526delTG | p.Asn177ProfsX11 | Middle Eastern (2) | 2 | Not cited |
| Exon 3 | c.685_686insCGGC | p.Arg229ProfsX102 | Asian (1) | 1 | [Kishnani et al., 2006a] |
| Exon 4 | c.722_723delTT | p.Phe241CysfsX88 | 2 | [Kroos et al., 2008] | |
| Exon 4 | c.766_785delinsC | p.Tyr256ArgfsX6 | 1 | [Huie et al., 1998] | |
| Exon 7 | c.1128_1129delinsC | p.Trp376CysfsX16 | Caucasian (1) | 1 | [Kroos et al., 2008] |
| Exon 7 | c.1157dupA | p.Val387GlyfsX119 | Turkish (2) | 2 | [Rohrbach et al., 2010] |
| Exon 8 | c.1209del C | p.Asn403LysfsX37 | 2 | Not cited | |
| Exon 11 | c.1591dupG | p.Asp531GlyfsX7 | Indian (2) | 2 | Not cited |
| Exon 12 | c.1650dupG | p.Thr551AspfsX85 | 1 | Not cited | |
| Exon 12 | c.1654delC | p.Leu552SerfsX26 | Caucasian (1) | 1 | Not cited |
| Exon 13 | c.1826dupA | p.Tyr609Xfs | 2 | [Hermans et al., 2004] | |
| Exon 13 | c.1827delC | p.Tyr609Xfs | 1 | [Hermans et al., 2004] | |
| Exon 16 | c.2300delT | p.Phe767SerfsX14 | 2 | [Kroos et al., 2008] | |
| Exon 17 | c.2432delT | p.Leu811ArgfsX37 | 1 | [Amartino et al., 2006] | |
| Exon 17 | c.2439dupC | p.Ile814HisfsX70 | African/N European (1) | 1 | Not cited |
| Exon 19 | c.2706delG | p.Lys903ArgfsX2 | 2 | Not cited | |
| Nonsense | |||||
| Exon 2 | c.352C>T | p.Gln118X | Caucasian (1) | 1 | Not cited |
| Exon 6 | c.1075G>T | p.Gly359X | 2 | Not cited | |
| Exon 10 | c.1442G>A | p.Trp481X | African/N Europe (1) | 1 | Not cited |
| Exon 10 | c.1496G>A | p.Trp499X | 2 | [Kroos et al., 2008] | |
| Exon 10 | c.1497G>A | p.Trp499X | 2 | [Laforet et al., 2000] | |
| Exon 10 | c.1548G>A | p.Trp516X | Caucasian (1) | 1 | [Hermans et al., 2004] |
| Exon 12 | c.1687C>T | p.Gln563X | 1 | [McVie-Wylie, 2001] | |
| Exon 13 | c.1822C>T | p.Arg608X | 1 | [Kroos et al., 2008] | |
| Exon 16 | c.2237G>A | p.Trp746X | Caucasian (1) | 3 | [Beesley et al., 1998] |
| Exon 16 | c.2238G>A | p.Trp746X | 1 | [Kishnani et al., 2006a] | |
| Exon 18 | c.2560C>T | p.Arg854X | Caucasian (2), AA (10), Middle Eastern (2) | 34 | [Hermans et al., 1993] |
| Exon 18 | c.2608C>T | p.Arg870X | 3 | [McCready et al., 2007] | |
| Splice site | |||||
| Intron 2 | c.546+2T>C | 2 | Not cited | ||
| Intron 2 | c.546+2_+5delTGGG | 1 | Not cited | ||
| Intron 4 | c.858+2T>A | Middle Eastern (2) | 2 | Not cited | |
| Intron 12 | c.1754+1G>A | 1 | Not cited | ||
| Intron 12 | c.1754+2T>A | 1 | Not cited | ||
| Intron 13 | c.1888+1G>A | 2 | [Kroos et al., 2008] | ||
| Intron 16 | c.2331+2T>A | 2 | [Kroos et al., 2008] | ||
| Multiple exon deletion | |||||
| Promoter - ?c | Multiple exon deletionc | Caucasian (1) | 1 | Not cited | |
| Exons 2-4 | c.148_859-11del | p.Glu50HisfsX37 | Asian (1) | 1 | [McCready et al., 2007] |
| Exons 8-15 | c.1195-18_2190-20deld | p.Asp399ValfsX6 | Hispanic (2) | 4 | [Huie et al., 2002] |
| Initiator codon | |||||
| Exon 2 | c.1A>G | p.Met1? | 2 | Not cited | |
Citations listed are the first report of a specific mutation. Further citations for each mutation are available at http://www.pompecenter.nl/en/ which is the website for the Pompe Center at Erasmus Medical Center in Rotterdam, the Netherlands.
Where available, ethnicity of the patient(s) in whom a specific allele was found is stated. The number in parentheses gives the number of alleles found in patients of that ethnicity. AA, African American.
Number of alleles identified in this study.
The end points of this deletion are not defined.
Human Genome Variation Society nomenclature (http://www.hgvs.org/).
Figure 2.
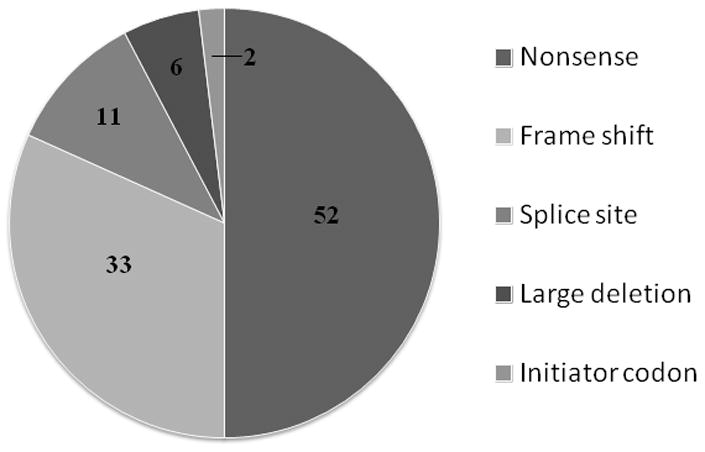
Pie chart showing number of alleles identified with different mutations in CRIM-negative patients (see Table I for details).
Table II.
Number of CRIM-negative and CRIM-positive patients with different types of GAA gene mutations.
| Type of mutations | Number of CRIM-negative patients | Number of CRIM-positive patients |
|---|---|---|
| Two predicted null allelesa | 44 | 1b |
| Homozygous premature stop codon in last exon | 0 | 1c |
| At least one missense or in frame deletion | 0 | 81d |
| Homozygous initiator methionine mutation | 1e | 0 |
| Homozygous splice site mutation | 4 | 3 |
| Compound heterozygote splice site/null mutation | 3 | 2 |
| Total patients | 52 | 88 |
Null allele = nonsense or frame shift mutation resulting in premature termination codon in any exon but the last, or multi-exon deletion.
Compound heterozygote for c.1165delG (p.Glu389ArgfsX3) and c.2560C>T (p.Arg854X)
Homozygous for p.Gln914ProfsX30 which results in a premature stop codon in the last exon of the GAA gene
Two missense alleles (n=39), one missense and one null allele (n=31), one missense and one splice site mutation (n=4), one missense and one in frame exon 18 deletion (n=3), exon 18 deletion and null allele (n=2), homozygous exon 18 deletion (n=1), and small in frame deletion and null allele (n=1).
Homozygous for c.1A>G (p.Met1?).
Table III.
Additional novel GAA mutant alleles identified in this study
| Location | Nucleotide change | Amino acid change | Ethnicitya | Number of allelesb |
|---|---|---|---|---|
| Missensec | ||||
| Exon 7 | c.1118T>G | p.Leu373Arg | 1 | |
| Exon 10 | c.1466A>G | p.Asp489Gly | 1 | |
| Exon 12 | c.1719C>A | p.Asn573Lys | Mexico (2) | 2 |
| Exon 12 | c.1726G>C | p.Gly576Arg | 1 | |
| Exon 13 | c.1802C>T | p.Ser601Leu | Hispanic (1) | 1 |
| Exon 13 | c.1832G>A | p.Gly611Asp | 1 | |
| Exon 15 | c.2105G>T | p.Arg702Leu | African American (1) | 1 |
| Exon 17 | c.2456 G>C | p.Arg819Pro | Middle Eastern (4) | 4 |
| Exon18 | c.2528T>C | p.Leu843Pro | 1 | |
| Exon 19 | c.2783A>G | p.Tyr928Cys | Indian (2) | 2 |
| Frame shift | ||||
| Exon 8 | c.1293_1312del20 | p.Gln433AspfsX66 | 1 | |
| Exon 9 | c.1356delC | p.Ser454AlafsX23 | Asian (1) | 1 |
| Exon 13 | c.1848dupC | p.Val617Argfs19X | 1 | |
| Exon 14 | c.1936_1937insGCCGACG | p.Val646GlyfsX93 | Indian (1) | 1 |
| Exon 17 | c.2408_2426del19 | p.Gln803ProfsX39 | Caucasian (1) | 1 |
| Splice site | ||||
| Intron 9 | c.1437+1G>A | Hispanic (2) | 2 | |
| Intron 10 | c.1551+1G>T | African American (1) | 1 | |
| Intron 12 | c.1754+1G>A | African American (1) | 1 | |
| Intron 15 | c.2189+3G>C | Indian (1) | 1 | |
| Intron 17 | c.2481+2T>C | 1 | ||
| Deletion | ||||
| Exon 2-3 | Deletion of at least exon 2 and proximal exon 3d | Asian (1) | 1 | |
| Exon 2 | c.460_465del6 | p.Arg154_Thr155del | 1 | |
| Predicted benign | ||||
| Exon 5 | c.917C>T | p.Ser306Leu | African American (1) | 1 |
Where available, ethnicity of the patient(s) in whom a specific allele was found is stated. The number in parentheses gives the number of alleles found in patients of that ethnicity.
Number of alleles identified in this study.
All missense mutations listed were predicted to be “probably damaging” by the Polyphen-2 program, except for p.Ser306Leu, which was predicted to benign.
The end points of this deletion are not defined.
DISCUSSION
Based on 10 years of laboratory experience, we report CRIM status determined by Western blot in a large group of patients with infantile Pompe disease and correlation of the results with GAA gene mutation data. Firstly, our results show that Western blot analysis of skin fibroblast homogenate is a reliable method for determining CRIM status in infantile patients with Pompe disease. We observed essentially the same results regardless of whether an affinity purified polyclonal antibody, raised in rabbits against human placental GAA, or a monoclonal antibody (provided by Genzyme; mixture of various protein epitopes) was used in the 70 patients that were tested by both methods. However, as results with the monoclonal antibody were cleaner, without any non-specific bands, and the monoclonal antibody is easier to produce, it is now the method recommended by our laboratory. Based on our experience, loading 20 ug protein for the Western blot is optimal. It is possible that loading less protein may lead to lack of detection of protein in a CRIM-positive patient [Bali et al., 2011].
Role of GAA gene mutations in determining CRIM status
Although our Western blot method is reliable, it is invasive and the turn-around time is several weeks owing to culture time for fibroblasts. We therefore sought to correlate the results of the Western blot with GAA mutations to investigate the possibility of a quicker, mutation-based method for determining CRIM-status. While we identified a wide range of different mutations in both the CRIM-negative and CRIM-positive groups, there is a dramatic difference in the frequency of mutation types in the two groups (Table II). As expected, most CRIM-negative patients were homozygous or compound heterozygotes for alleles that would not be expected to produce any GAA protein including nonsense, frame shift, and multi-exon deletions (44 out of 52 CRIM-negative patients had two of these alleles).
Nonsense, frame shift, and multiple exon deletion mutations
With the exception of two CRIM-positive patients, all individuals who had two of these types of mutations were CRIM-negative. One exception was a CRIM-positive patient who was homozygous for p.Gln914ProfsX30 [Reuser et al., 1995; Hermans et al., 1998; Banugaria et al, 2011]. This mutation is expected to result in a premature termination codon in the last exon of the GAA gene. Although it is known that premature termination codons result in nonsense-mediated decay of mRNA and production of no protein, however if the premature termination codon occurs in the last exon of a gene or up to about 50 nucleotides from the 3’ end of the penultimate exon, the nonsense mediated decay machinery appears to miss it, and some protein is made [Silva and Romao, 2009]. This mechanism may explain the CRIM-positive status of this patient. Another patient, determined to be CRIM-positive by the presence of a clear but weak band on Western blot, surprisingly was a compound heterozygote for a frame shift mutation (p.Glu389ArgfsX3) and a nonsense mutation (p.Arg854X), both of which are predicted to be null alleles (Hermans et al., 1993; Kroos et al, 2008). As the p.Arg854X mutation is known to be a CRIM-negative mutation [Kishnani et al., 2010], we considered the possibility that the c.1165delG (p.Glu389ArgfsX3) allele in exon 7 could somehow result in protein production. Using a splice site prediction program (http://www.fruitfly.org/seq_tools/splice.html), we found that the c.1165delG mutation creates a new donor splice site within exon 7. If used, this splice site would cause an in frame deletion removing the last 33 base pairs of exon 7 and result in a protein missing 11 amino acids but maintaining the normal reading frame. While we have not performed cDNA sequencing studies to determine if this occurs, it does provide a possible explanation for the CRIM-positive status of this patient. In summary, the presence of two nonsense or frame shift mutations is a good predictor of CRIM status, typically resulting in CRIM-negative status unless the premature stop codon is in the last exon of the gene. However, until further data are gathered on specific mutations, confirmation of CRIM status using immunological methods is recommended to identify unusual cases where a predicted null allele may actually produce some protein, as is in the case described where splicing was unexpectedly affected by a frame shift mutation. While splice site prediction programs could be used to look for possible effects of splicing for any sequence variant, we understand that they should be viewed as a prediction tool and cannot always fully and accurately predict what occurs in vivo.
Missense mutations
It is predicted that if a patient has at least one missense mutation, some GAA protein will be produced and therefore the patient would be CRIM-positive, although the amount, level of processing, and activity of the protein would vary considerably depending on the underlying mutation [Banugaria et al., 2011; Kishnani et al., 2010; Kroos et al., 2008]. Overall, 77 of the 88 CRIM-positive patients had at least one missense mutation (Table II). In fact, all patients with at least one missense mutation were CRIM-positive. Possessing an in frame deletion was also predicted to result in CRIM-positive status. For example, a previously identified deletion of exon 18 was found in six CRIM-positive patients (Table II) but not in any CRIM-negative patients. This mutation has been previously reported and shown to produce GAA protein [Ausems et al., 1996]. Our results therefore suggest that missense mutations and in frame deletions result in CRIM-positive status. However, it remains important to confirm CRIM-status by Western blot to ensure that no unusual cases are missed.
Interestingly, one of the CRIM-negative subjects had a point mutation that abolished the initiator methionine codon (c.1A>G; p.Met1?). Nucleotide substitutions affecting the initiator methionine have been reported for many different genetic disorders. When the initiator methionine codon is altered, a downstream methionine is used in some cases. Of note, the effect of initiator methionine codon alteration on protein production appears to depend on the specific gene and ranges from normal protein expression, to N-terminal deletions (resulting from use of a downstream methionine), to reduced or no detectable protein. The effect on mRNA levels is also variable [Boneh et al., 2005]. After the initiator methionine, the next in frame ATG codon in GAA is located at amino acid position 122. An initiation codon prediction program (ATGpr, http://atgpr.dbcls.jp/) predicts that the likelihood of this codon being used as an initiation codon is low [Nishikawa et al., 2000]. Even if this codon were to be used as an alternative initiation codon, the amino-terminus of GAA including the signal sequence required for targeting to the endoplasmic reticulum would be deleted [Moreland et al., 2005]. If this does occur, we would assume that the protein is so unstable that it cannot be detected by traditional Western blot analysis. Another mutation of the initiator codon (c.3G>A) has also been reported in a patient with Pompe disease [Kroos et al., 2008], and was found in compound heterozygosity with a missense mutation (p.His308Pro) in one of our CRIM-positive patients. No protein studies have been performed but the effect of c.3G>A was predicted to be very severe [Kroos et al., 2008].
Splicing mutations
About 15% of GAA sequence changes causing Pompe disease are predicted to affect splicing [Zampieri et al., 2011]. However, the effect of splice site mutations on CRIM status remains difficult to predict. By virtue of having being found in CRIM-negative patients, the splice site mutations found in our CRIM-negative patients are expected to be null alleles, producing no protein (Table I). In CRIM-positive patients, if a splice site mutation exists in homozygosity, or in compound heterozygosity with a known null mutation, then it is expected to produce protein. Most of the splice site mutations found in CRIM-positive patients in this study were found in compound heterozygosity with missense changes, and thus no conclusion could be made regarding whether these alleles can produce any protein based on this alone. However, three CRIM-positive patients were homozygous for different splice site mutations (c.1195-2A>G, c.1327-2A>G, and c.1437+1G>A), and two had a predicted null allele and a splice site mutation (c.1551+1G>T and c.2189+3G>C). None of these splice site mutations were seen in homozygosity in any of our CRIM-negative patients. Based on these results, these splice site alleles are expected to produce protein. Two of these mutations, c.1195-2A>G and c.1327-2A>G, have been cited before [Hamdan et al., 2008; Kroos et al., 2008; Oba-Shinjo et al., 2009]. A Brazilian patient with severe infantile Pompe disease, who died at 8 months of age, was a compound heterozygote for c.1195-2A>G and a second unidentified mutation [Oba-Shinjo et al., 2009]. The patient homozygous for c.1327-2A>G was treated with ERT from 18 hours of age and reportedly had a favorable outcome at 10 months. CRIM status and antibody titers were not reported [Hamdan et al., 2008]. This patient is included in our study and was found to be CRIM-positive by Western blot analysis of skin fibroblast homogenate. For the other three mutations (c.1437+1G>A, c.1551+1G>T, and c.2189+3G>C) different substitutions in the same splice site have been reported (c.1437+2T>C, c.1551+1G>C, c.1551+1G>A, and c.2189+1G>A) [Huie et al., 1994; Kroos et al, 2008; Montalvo et al., 2006; Orlikowski et al.; Pittis et al., 2008; Stroppiano et al., 2001]. Studies of cDNA have shown that c.1437+2T>C and c.1551+1G>C, cause in frame exon skipping [Huie et al., 1994; Stroppiano et al., 2001; Zampieri et al., 2011]. A patient who is homozygous for c.1551+1G>A, as well as heterozygous for p.Arg725Trp, has been described with onset of symptoms at 14 years [Orlikowski et al., 2011] suggesting that c.1551+1G>A allows for production of some protein, possibly by in frame exon skipping. Although we cannot be sure without performing cDNA studies, it is possible that in frame exon skipping in our patients could explain their CRIM positive status. Further studies of specific splice site mutations are needed to determine their effect on CRIM status.
SUMMARY
Based on the above predictions, CRIM status could be predicted from GAA gene mutation analysis for 126/140 patients (90%). The remaining patients, for whom we could not easily predict CRIM status, had splice site mutations (7 CRIM-negative and 5 CRIM-positive), and an initiator methionine mutation (one CRIM-negative patient). In addition, we predicted that a patient who was a compound heterozygote for a frame shift and a nonsense mutation would be CRIM-negative when, in fact, the patient was CRIM-positive, possibly because the mutation creates a novel splice site. Continued studies in this area will help to increase the power of predicting CRIM status from GAA gene mutations, and may circumvent the need for invasive biopsies. Future studies are needed to correlate Western blot banding pattern and GAA mutations with development of high antibody titers in CRIM-positive patients.
Acknowledgments
We gratefully acknowledge the patients with Pompe disease and their families for participating in this study. We thank Joan Keutzer from Genzyme Corporation for helpful discussions throughout the study and for providing the GAA monoclonal antibody. Funding for this study was provided in part by Genzyme Corporation and by the Lysosomal Disease Network (LDN). The LDN is a part of NIH Rare Diseases Clinical Research Network (RDCRN). This publication was also made possible by Grant Number UL1RR024128 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Priya S. Kishnani and Deeksha Bali have received research/grant support and honoraria from Genzyme Corporation. Priya S. Kishnani is a member of the Pompe and Gaucher Disease Registry Advisory Board for Genzyme Corporation.
Biographies
Deeksha Bali is director of the Duke Glycogen Storage Disease laboratory and has been highly involved in laboratory diagnosis and research on Pompe disease and other glycogen storage diseases for the past 15 years. New methods for non-invasive diagnosis of lysosomal storage diseases have been developed so that patients can benefit from early diagnosis and initiation of treatment.
Jennifer L. Goldstein is a clinical research coordinator at Duke Medicine with a strong interest in laboratory diagnosis of glycogen storage diseases. Other interests are creatine deficiency syndrome, molybdenum cofactor disorders, newborn screening, and autism research.
Suhrad Banugaria is actively involved in clinical and pre-clinical research involving Pompe disease at Duke Medicine. He is involved in projects working towards improvement of GAA enzyme delivery into muscle tissue of patients with Pompe disease using mouse models. CRIM status and associated immune modulation in CRIM-negative patients is one of his keen interests.
Jian Dai is a laboratory technician at Duke Medicine who has vast experience in performing Western blots to determine CRIM status in patients with Pompe disease. She has been involved in pre-clinical and clinical work involving Pompe disease using GAA knock out and GAA-muscle M6PR double knock out mouse models.
Joanne Mackey is a nurse practitioner who has worked in the Duke Medical Genetics Program and involved with patients with Pompe disease and their families for the past 15 years. She has been involved in enzyme replacement therapy clinical trials for Pompe disease since the first trial was performed at Duke. Her other interests are Down syndrome and various treatment modalities for metabolic diseases.
Catherine Rehder is director of the Molecular Diagnostics and Cytogenetics Laboratories at Duke. She has been involved with genotyping patients with Pompe disease and other metabolic diseases for the past 5 years through gene sequencing.
Priya S. Kishnani is Chief of Medical Genetics, Professor of Pediatrics, and a clinician scientist who is dedicated to the care and treatment of individuals with Pompe disease and other metabolic disorders. She has been involved in numerous clinical trials for the treatment of Pompe disease, Down syndrome, and many other conditions. She continues to research new therapies for Pompe disease, Down syndrome, and other glycogen storage diseases.
References
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. [PubMed] [Google Scholar]
- Amartino H, Painceira D, Pomponio RJ, Niizawa G, Sabio Paz V, Blanco M, Chamoles N. Two clinical forms of glycogen-storage disease type II in two generations of the same family. Clin Genet. 2006;69:187–188. doi: 10.1111/j.1399-0004.2005.00557.x. [DOI] [PubMed] [Google Scholar]
- Angelini C, Semplicini C. Enzyme Replacement Therapy for Pompe Disease. Curr Neurol Neurosci Rep. 2011 Oct 15; doi: 10.1007/s11910-011-0236-5. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Ausems MG, Kroos MA, Van der Kraan M, Smeitink JA, Kleijer WJ, Ploos van Amstel HK, Reuser AJ. Homozygous deletion of exon 18 leads to degradation of the lysosomal alpha-glucosidase precursor and to the infantile form of glycogen storage disease type II. Clin Genet. 1996;49:325–328. doi: 10.1111/j.1399-0004.1996.tb03801.x. [DOI] [PubMed] [Google Scholar]
- Bali DS, Tolun AA, Goldstein JL, Dai J, Kishnani PS. Molecular analysis and protein processing in late-onset Pompe disease patients with low levels of acid alpha-glucosidase activity. Muscle Nerve. 2011;43:665–670. doi: 10.1002/mus.21933. [DOI] [PubMed] [Google Scholar]
- Banugaria SG, Prater SN, Ng YK, Kobori JA, Finkel RS, Ladda RL, Chen YT, Rosenberg AS, Kishnani PS. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: Lessons learned from infantile Pompe disease. Genet Med. 2011;13:729–736. doi: 10.1097/GIM.0b013e3182174703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesley CE, Child AH, Yacoub MH. The identification of five novel mutations in the lysosomal acid a-(1-4) glucosidase gene from patients with glycogen storage disease type II. Hum Mutat. 1998;11:413. doi: 10.1002/(SICI)1098-1004(1998)11:5<413::AID-HUMU16>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Boneh A, Korman SH, Sato K, Kanno J, Matsubara Y, Lerer I, Ben-Neriah Z, Kure S. A single nucleotide substitution that abolishes the initiator methionine codon of the GLDC gene is prevalent among patients with glycine encephalopathy in Jerusalem. J Hum Genet. 2005;50:230–234. doi: 10.1007/s10038-005-0243-y. [DOI] [PubMed] [Google Scholar]
- Chakrapani A, Vellodi A, Robinson P, Jones S, Wraith JE. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis. 2010;33:747–750. doi: 10.1007/s10545-010-9206-3. [DOI] [PubMed] [Google Scholar]
- Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, Huang AC, Wu MH, Huang PH, Tsai FJ, Chen YT, Hwu WL. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics. 2009;124:e1116–1125. doi: 10.1542/peds.2008-3667. [DOI] [PubMed] [Google Scholar]
- Hamdan MA, Almalik MH, Mirghani HM. Early administration of enzyme replacement therapy for Pompe disease: Short-term follow-up results. J Inherit Metab Dis. 2008;(Suppl 2):S431–436. doi: 10.1007/s10545-008-1000-0. [DOI] [PubMed] [Google Scholar]
- Hawes ML, Kennedy W, O’Callaghan MW, Thurberg BL. Differential muscular glycogen clearance after enzyme replacement therapy in a mouse model of Pompe disease. Mol Genet Metab. 2007;91:343–51. doi: 10.1016/j.ymgme.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Hermans MM, De Graaff E, Kroos MA, Mohkamsing S, Eussen BJ, Joosse M, Willemsen R, Kleijer WJ, Oostra BA, Reuser AJ. The effect of a single base pair deletion (delta T525) and a C1634T missense mutation (pro545leu) on the expression of lysosomal alpha-glucosidase in patients with glycogen storage disease type II. Hum Mol Genet. 1994;3:2213–2218. doi: 10.1093/hmg/3.12.2213. [DOI] [PubMed] [Google Scholar]
- Hermans MM, de Graaff E, Kroos MA, Wisselaar HA, Willemsen R, Oostra BA, Reuser AJ. The conservative substitution Asp-645-->Glu in lysosomal alpha-glucosidase affects transport and phosphorylation of the enzyme in an adult patient with glycogen-storage disease type II. Biochem J. 1993;289(Pt 3):687–693. doi: 10.1042/bj2890687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans MMP, Kroos MA, Smeitink JAM, van der Ploeg AT, Kleijer WJ, Reuser AJJ. Glycogen storage disease type II: Genetic and biochemical analysis of novel mutations in infantile patients from Turkish ancestry. Hum Mutat. 1998;11:209–215. doi: 10.1002/(SICI)1098-1004(1998)11:3<209::AID-HUMU5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Hermans MM, van Leenen D, Kroos MA, Beesley CE, Van Der Ploeg AT, Sakuraba H, Wevers R, Kleijer W, Michelakakis H, Kirk EP, Fletcher J, Bosshard N, Basel-Vanagaite L, Besley G, Reuser AJJ. Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II. Hum Mutat. 2004;23:47–56. doi: 10.1002/humu.10286. [DOI] [PubMed] [Google Scholar]
- Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid alha-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AC, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2001. pp. 3389–3420. [Google Scholar]
- Huie ML, Anyane-Yeboa K, Guzman E, Hirschhorn R. Homozygosity for multiple contiguous single-nucleotide polymorphisms as an indicator of large heterozygous deletions: identification of a novel heterozygous 8-kb intragenic deletion (IVS7-19 to IVS15-17) in a patient with glycogen storage disease type II. Am J Hum Genet. 2002;70:1054–1057. doi: 10.1086/339691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie ML, Chen AS, Tsujino S, Shanske S, DiMauro S, Engel AG, Hirschhorn R. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (-13T-->G) mutation in a majority of patients and a novel IVS10 (+1GT-->CT) mutation. Hum Mol Genet. 1994;3:2231–2236. doi: 10.1093/hmg/3.12.2231. [DOI] [PubMed] [Google Scholar]
- Huie ML, Tsujino S, Sklower Brooks S, Engel A, Elias E, Bonthron DT, Bessley C, Shanske S, DiMauro S, Goto YI, Hirschhorn R. Glycogen storage disease type II: identification of four novel missense mutations (D645N, G648S, R672W, R672Q) and two insertions/deletions in the acid alpha-glucosidase locus of patients of differing phenotype. Biochem Biophys Res Commun. 1998;244:921–927. doi: 10.1006/bbrc.1998.8255. [DOI] [PubMed] [Google Scholar]
- Hunley TE, Corzo D, Dudek M, Kishnani P, Amalfitano A, Chen YT, Richards SM, Phillips JA, 3rd, Fogo AB, Tiller GE. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics. 2004;114:e532–535. doi: 10.1542/peds.2003-0988-L. [DOI] [PubMed] [Google Scholar]
- Kemper AR, Hwu WL, Lloyd-Puryear M, Kishnani PS. Newborn screening for Pompe disease: synthesis of the evidence and development of screening recommendations. Pediatrics. 2007;120:e1327–1334. doi: 10.1542/peds.2007-0388. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, Bali D, Smith SA, Li JS, Mandel H, Koeberl D, Rosenberg A, Chen YT. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006a;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O’Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006b;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Gorlinger K, Wallot M, Richards S, Voit T. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord. 2005;15:24–31. doi: 10.1016/j.nmd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Kroos M, Pomponio RJ, van Vliet L, Palmer RE, Phipps M, Van der Helm R, Halley D, Reuser A. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat. 2008;29:E13–26. doi: 10.1002/humu.20745. [DOI] [PubMed] [Google Scholar]
- Laforet P, Nicolino M, Eymard PB, Puech JP, Caillaud C, Poenaru L, Fardeau M. Juvenile and adult-onset acid maltase deficiency in France: genotype-phenotype correlation. Neurology. 2000;55:1122–1128. doi: 10.1212/wnl.55.8.1122. [DOI] [PubMed] [Google Scholar]
- McCready ME, Carson NL, Chakraborty P, Clarke JT, Callahan JW, Skomorowski MA, Chan AK, Bamforth F, Casey R, Rupar CA, Geraghty MT. Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II. Mol Genet Metab. 2007;92:325–335. doi: 10.1016/j.ymgme.2007.07.006. [DOI] [PubMed] [Google Scholar]
- McVie-Wylie A, Lowery M, Faulkner E, Lamson D, Chen YT. GSD type II: Description of four novel mutations causing acid alpha-glucosidase deficiency. ASHG Annual Meeting abstracts, Program # 2706 2001 [Google Scholar]
- Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med. 2009;360:194–195. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]
- Messinger YH, Mendelsohn NJ, Rhead W, Hershkovitz E, Champion M, Jones S, Olson R, White A, Wells C, Bali D, Case LE, Young S, Rosenberg AS, Kishnani PS. Successful Immune Tolerance Induction to Enzyme Replacement Therapy in CRIM-Negative Infantile Pompe Disease. Genetics in Medicine. doi: 10.1038/gim.2011.4. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalvo AL, Bembi B, Donnarumma M, Filocamo M, Parenti G, Rossi M, Merlini L, Buratti E, De Filippi P, Dardis A, Stroppiano M, Ciana G, Pittis MG. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum Mutat. 2006;27:999–1006. doi: 10.1002/humu.20374. [DOI] [PubMed] [Google Scholar]
- Moreland RJ, Jin X, Zhang XK, Decker RW, Albee KL, Lee KL, Cauthron RD, Brewer K, Edmunds T, Canfield WM. Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J Biol Chem. 2005;280:6780–6791. doi: 10.1074/jbc.M404008200. [DOI] [PubMed] [Google Scholar]
- Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, Arnold GL, Pivnick EK, Ottinger CJ, Robinson PH, Loo JC, Smitka M, Jardine P, Tatò L, Chabrol B, McCandless S, Kimura S, Mehta L, Bali D, Skrinar A, Morgan C, Rangachari L, Corzo D, Kishnani PS. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Ota T, Isogai T. Prediction whether a human cDNA sequence contains initiation codon by combining statistical information and similarity with protein sequences. Bioinformatics. 2000;16:960–967. doi: 10.1093/bioinformatics/16.11.960. [DOI] [PubMed] [Google Scholar]
- Oba-Shinjo SM, da Silva R, Andrade FG, Palmer RE, Pomponio RJ, Ciociola KM, S Carvalho M, Gutierrez PS, Porta G, Marrone CD, Munoz V, Grzesiuk AK, Llerena JC, Jr, Berditchevsky CR, Sobreira C, Horovitz D, Hatem TP, Frota ER, Pecchini R, Kouyoumdjian JA, Werneck L, Amado VM, Camelo JS, Jr, Mattaliano RJ, Marie SK. Pompe disease in a Brazilian series: clinical and molecular analyses with identification of nine new mutations. J Neurol. 2009;256:1881–1890. doi: 10.1007/s00415-009-5219-y. [DOI] [PubMed] [Google Scholar]
- Orlikowski D, Pellegrini N, Prigent H, Laforet P, Carlier R, Carlier P, Eymard B, Lofaso F, Annane D. Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord. 2011;21:477–482. doi: 10.1016/j.nmd.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Pittis MG, Donnarumma M, Montalvo AL, Dominissini S, Kroos M, Rosano C, Stroppiano M, Bianco MG, Donati MA, Parenti G, D’Amico A, Ciana G, Di Rocco M, Reuser A, Bembi B, Filocamo M. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum Mutat. 2008;29:E27–36. doi: 10.1002/humu.20753. [DOI] [PubMed] [Google Scholar]
- Raben N, Takikita S, Pittis MG, Bembi B, Marie SK, Roberts A, Page L, Kishnani PS, Schoser BG, Chien YH, Ralston E, Nagaraju K, Plotz PH. Deconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sand. Autophagy. 2007;3:546–52. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuser AJJ, Kroos MA, Hermans MMP, Bijvoet AGA, Verbeet MP, van Diggelen OP, Kleijer WJ, van der Ploeg AT. Glycogenosis type II (acid maltase deficiency) Muscle and Nerve. 1995;3:S61–S69. doi: 10.1002/mus.880181414. [DOI] [PubMed] [Google Scholar]
- Rohrbach M, Klein A, Kohli-Wiesner A, Veraguth D, Scheer I, Balmer C, Lauener R, Baumgartner MR. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis. 2010;33:751–757. doi: 10.1007/s10545-010-9209-0. [DOI] [PubMed] [Google Scholar]
- Silva AL, Romao L. The mammalian nonsense-mediated mRNA decay pathway: to decay or not to decay! Which players make the decision? FEBS letters. 2009;583:499–505. doi: 10.1016/j.febslet.2008.12.058. [DOI] [PubMed] [Google Scholar]
- Stroppiano M, Bonuccelli G, Corsolini F, Filocamo M. Aberrant splicing at catalytic site as cause of infantile onset glycogen storage disease type II (GSDII): molecular identification of a novel IVS9 (+2GT-->GC) in combination with rare IVS10 (+1GT-->CT) Am J Med Genet. 2001;101:55–58. doi: 10.1002/ajmg.1310. [DOI] [PubMed] [Google Scholar]
- Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–57. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet. 2000;356:397–8. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- Zampieri S, Buratti E, Dominissini S, Montalvo AL, Pittis MG, Bembi B, Dardis A. Splicing mutations in glycogen-storage disease type II: evaluation of the full spectrum of mutations and their relation to patients’ phenotypes. Eur J Hum Genet. 2011;19:422–431. doi: 10.1038/ejhg.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]