

Abstract
Additions of methylphenylsulfonium methylide onto chiral non-racemic N-sulfinyl imines (R′-SO-N=CH-R, R′=t-butyl, R=protected diol), followed by ring closure, yield terminal aziridines with high diastereoselectivity. Control reactions have established that both N- and C- iminyl substituents impact product preference, and when properly matched, one addition product is selected almost exclusively. Using solution-phase density functional computational methods, minima and transition state searches have been performed to reveal the structural origins of the diastereoselectivity. Our computational findings indicate that ring closure is fast and irreversible, and consequently, the relative energies of the transition states for the competing Re/Si addition steps determine the product diastereomeric ratios. Analysis of addition transition state structures reveals the causes of selectivity as arising from the N- and C- iminyl substituents, and we identify the S (R) configuration of the N-sulfinyl sulfur atom as the dominant director of Si (Re) addition. The control attributed to the sulfur configuration is tied to an important favorable internal interaction between the sulfinyl oxygen and the iminyl hydrogen. The protected diol acts as a secondary director, owing to steric/electrostatic interactions with the approaching ylide.
Keywords: transition states, B3LYP, stereoselectivity, sulfinyl imines, aziridination
Introduction
Terminal aziridines are common intermediates for the synthetic assembly of many molecules of medicinal or biological significance, including several HIV protease inhibitors.[1] Achievement of stereocontrol in such syntheses is both highly desirable and challenging. Sulfur ylides have been used successfully as nucleophilic alkylidene transfer agents to introduce small-ring functionality into organic molecules.[2]
When methylphenylsulfonium methylide (sulfur ylide 1) is generated in the presence of an imine or aldehyde,[3] it attacks the iminyl/carbonyl carbon to form a betaine intermediate with a new chiral center, Scheme 1. Ring closure via an intramolecular nucleophilic attack results in a terminal aziridine/epoxide ring with methylphenyl sulfide as sideproduct.[4] With the particular choice of N-sulfinyl imine 2, Figure 1, as the target of the addition[5], a high level of diastereocontrol (dr > 95:5) is achieved. Imine 2 has chiral substituents on both the iminyl nitrogen (sulfinyl sulfur atom in the S configuration) and on the iminyl carbon (BDA, as derived from D-mannitol). It should be noted that the sulfinyl sulfur atom has local pyramidal geometry and does not invert, making for a rather unexpected stereocenter. Control reactions with variant imines 3-5, Figure 1, all exhibit reduced selectivity, Table 1.[5] Switching from the BDA moiety to one which is achiral (cyclohexyl group (imine 3)) results in the greatest loss of diastereoselectivity (61:39). Reducing the bulkiness of the N-substituent, 4-methylphenyl (imine 4) versus t-butyl, also results in a substantial loss of selectivity (67:33). Moderate loss is observed for imine 5 (79:21), for which the sulfinyl sulfur stereocenter is in the R configuration. Apparently, one of the two factors, either the BDA moiety or the configuration of the sulfinyl sulfur, is dominant in directing diastereocontrol.
Scheme 1.
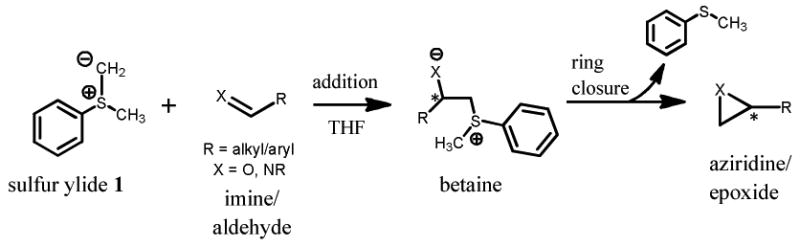
Figure 1.
Imine 2 and Control Imines 3-5. Imine 2 has chiral substituents t-butyl sulfinyl (sulfur in the S configuration) and protected diol (BDA) on the iminyl N and C atoms, respectively. The sulfur of imine 5 is in the R configuration.
Table 1.
Diastereometric Ratios for R′-SO-N=CH-R/SulfurYlide 1 Aziridinations and Computed Relative Energies of Transition States (kcal/mol) for Re versus Si Addition Steps.a
| Imine | R′ | S stereocenterb | Rc | yield | drd | ΔΔE‡e |
|---|---|---|---|---|---|---|
| 2 | t-butyl | S | BDA | 92% | > 95:5 | +1.8 |
| 3 | t-butyl | S | cyclohexyl | 81% | 61:39 | +0.9 |
| 4 | 4-methylphenyl | S | BDA | 80% | 67:33 | +1.1 |
| 5 | t-butyl | R | BDA | 88% | 79:21 | -1.2 |
| ent-2f | t-butyl | R | ADB | 94% | > 95:5 | -1.8 |
| ent-5f | t-butyl | S | ADB | 89% | 79:21 | +1.2 |
Experimental data from Ref. [5].
Configuration of sulfinyl sulfur atom.
BDA denotes the protected diol as shown in Figure 1, as derived from D-mannitol; ADB denotes the mirror image of BDA, as derived from ascorbic acid.
Determined by 1H NMR (crude reaction mixture).
ΔΔE‡ estimated as E(TSRe) − E(TSSi), based on ZPE-corrected B3LYP/6-311+G**(THF) energies. Positive value indicates Si TS favored, negative value indicates Re TS favored.
Enantiomers of imines 2 and 5, respectively. ΔΔE‡ values are given by symmetry.
Crystallizations of the aziridination products have proven unsuccessful, and as of now, the configurations of the major/minor products are still unknown, with one exception. Concurrent with this submission, an x-ray crystal structure was obtained for the sulfone derivative of the dominant product for the imine 2 reaction using dimethylsulfonium methylide; the new chiral center has the S configuration.[5] Quantum computational methods have been employed here to study the complete Re-addition and Si-addition reaction pathways for the imine 2/sulfur ylide 1 system leading to the R and S diastereoisomers of the aziridine product, respectively, Figures 2 and 3. Our goal is to identify the causes of diastereoselectivity in the principal and control reactions represented in Table 1.
Figure 2.
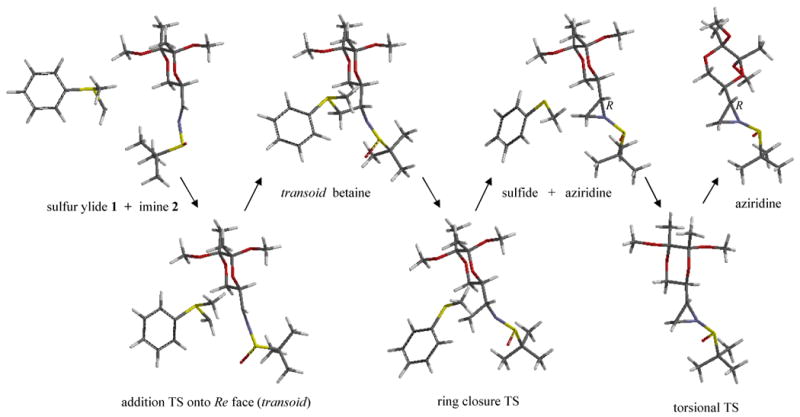
Reaction Pathway for Re Face Addition onto Imine 2 by Sulfur Ylide 1. Re addition onto imine 2 gives rise to the R stereocenter in the aziridine product. B3LYP/6-311+G**(THF) optimized structures.
Figure 3.
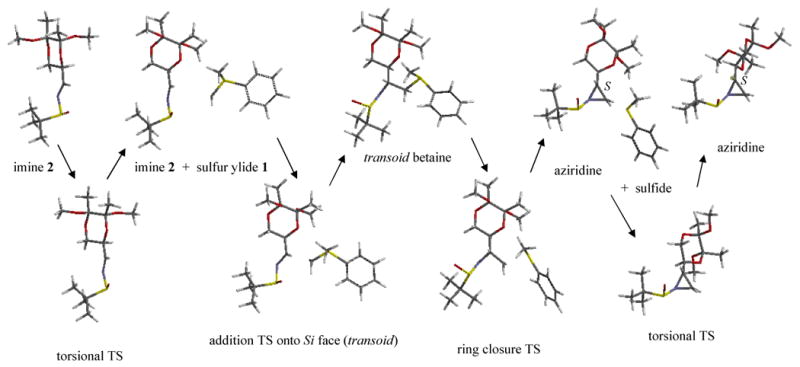
Reaction Pathway for Si Face Addition onto Imine 2 by Sulfur Ylide 1. Si addition onto imine 2 gives rise to the S stereocenter in the aziridine product. B3LYP/6-311+G**(THF) optimized structures.
Computational Details
Minima and transition state (TS) searches for the sulfinyl imine/sulfur ylide 1 structures shown in Figures 2 and 3 were performed using the Gaussian 09 program package.[6] Quantum calculations were carried out using the B3LYP density functional method[7] and the valence triple-zeta 6-311+G** basis set[8] augmented with diffuse functions.[9] THF was represented as solvent using the integral equation formalism variant of the Polarizable Continuum Model (PCM)[10] in all optimizations and subsequent frequency calculations. All species were fully optimized under the default parameters of Gaussian. TS structures and minima were confirmed by analytic computation of vibrational frequencies, which also provided solution-phase estimates of zero-point energy (ZPE) corrections. A comparable computational model was used by successfully by Robiette (B3LYP/6-311+G**(CH3CN))//B3LYP/6-31G*(CH3CN)) in the examination of cis/trans aziridination pathways of N-sulfonyl imines with sulfur ylides, but with a warning that although B3LYP model satisfactorily reproduces G3(MP2) energies for such systems, the gas-phase ring-closure barrier is underestimated by 2-3 kcal/mol.[11]
In our searches for transition states for Re and Si face additions, transoid (S and N antiperiplanar) and cisoid (S and N synperiplanar) modes were considered, as well as the three potential rotational orientations of the BDA moiety. Also, relative positioning of the ylidic phenyl and methyl groups were tested. For ring-closure transition states, the position of the sulfinyl S-O bond relative to the inchoate ring was tested as well.
On the basis of our findings for the most stable addition TS structures, targeted TS searches were carried out to find analogous transition states for the additions of sulfur ylide 1 onto imines 3-5.
Finally, the preferences for the Re versus Si face additions were estimated as ΔΔE‡ = E(TSRe) − E(TSSi), based on a reasonable assumption of reversibility of Re and Si dimeric complexation leading to TS formation. We can expect cancellation of errors and high accuracy in computing ΔΔE‡ values because they represent relative energies of isodesmic structures. Moreover, the imine 2 and 5 systems are on the same potential energy surface, and a direct comparison of these addition TS energies is warranted.
Results and Discussion
Imine 2/Sulfur Ylide 1 Reaction Pathways
Optimized structures for the imine 2/sulfur ylide 1 reaction pathways are shown in Figures 2 and 3. The energetics are summarized in Figure 4. Although earlier implementations of implicit solvent together with weak convergence criteria have sometimes produced uncertain imaginary frequencies for very soft vibrational modes, the convergence criteria of Gaussian 09 and its new implementation of the PCM model allowed us to obtain unambiguous confirmation of minima and transition states for this system. The addition steps proceed with barrier heights of about 6 (Si) and 8 (Re) kcal/mol to produce respective betaine intermediates. Small barriers for ring closure (< 3.5 kcal/mol) lead to the formation of the energetically stable aziridines.
Figure 4.
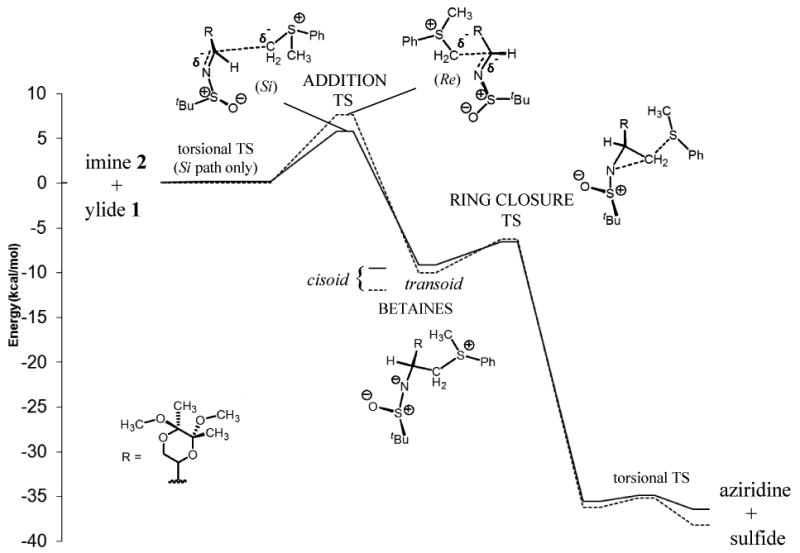
Potential Energy Surface for the Aziridination Reaction of Imine 2 with Sulfur Ylide 1. Solid line indicates the pathway for the Si face addition leading to the S product; dashed line indicates the pathway for Re face addition leading to the R product. Energies are ZPE-corrected and relative to reactants computed at the B3LYP/6-311+G**(THF) level.
We were unable to find a true cisoid (S and N synperiplanar) mode of addition for either Re or Si face approaches; attempts to find such transition states led to TS structures with C and N antiperiplanar and with higher energies than the transoid modes shown in Figures 2 and 3. The absence of cisoid additions contrasts our system with the findings for previous studies of similar systems.[11, 12] In all, 14 addition TS structures were identified. The most stable betaine intermediate arises from Re addition and is near cisoid with d(S-C-C-N) = 39°. Only transoid conformations are suited for immediate ring closure; closure occurs by way of a classic backside nucleophilic attack by the nitrogen on the methylene carbon, with the sulfide as leaving group. A total of 8 ring-closure transition states were found.
Our computational model indicates that the Re addition aziridine is more stable than the Si addition diastereoisomer by about 1.8 kcal/mol, Figure 4. Based on the energetics of the two pathways shown in Figure 4, however, we conclude that the expected major product is the kinetically-favored Si addition aziridine. Our reasoning is as follows: 1) the initial Si addition step is kinetically favored versus the Re addition step, with a smaller barrier of 5.8 versus 7.7 kcal/mol and 2) ring closure is apparently rapid (ring-closure barriers < 3.5 kcal/mol) and nonreversible (ring-opening barriers > 29 kcal/mol). We expect the latter to hold true for all our imine/sulfur ylide systems. Even if the ring-closure barriers are underestimated by ∼3 kcal/mol, the rates of ring closure should be much faster than rates of betaine decomposition (barriers for reverse addition >15 kcal/mol). Thus, the origin of the diastereoselectivity resides in the competition between the Re and Si addition TS structures. The computed ΔΔE‡ values for the additions onto imines 2-5 are shown in Table 1; the sign of ΔΔE‡ permits assignment of the major product for each imine, and the magnitudes roughly reflect the relative experimental dr values.
Transition States for Re and Si Face Additions onto Imines 2 and 5
The TS structures for additions onto the Re and Si faces of imine 2 are shown in detail in Figure 5. As implied earlier, both TS structures are transoid. Inspection of Figure 5 reveals the structural reasons for the greater relative stability of the Si addition TS (5.8 versus 7.7 kcal/mol). The near planarity of the H-C-N-S-O system, as imposed by an important electrostatic interaction between the sulfinyl oxygen and iminyl hydrogen (r(H…O) = 2.45 Å and d(H-C-S-O) = 9.9 °), clearly establishes the Si face as open to addition by the sulfur ylide. This feature is consistent with the synperiplanar (s-cis) arrangement of the C-N-S-O unit observed in crystal structures of sulfinyl imines[13] and in gas-phase calculations for simple sulfinyl imines.[14, 15] The t-butyl group is positioned in front of the Re face by virtue of the sulfinyl sulfur stereocenter (S). A favorable electrostatic interaction between the BDA moiety and the sulfur ylide is present, r(OBDA…H′) = 2.65 Å, and it occurs without steric hindrance. The Re addition TS, in contrast, accommodates the ylide approach by rotation about the N-S bond, disrupting the sulfinyl oxygen/iminyl hydrogen interaction. While a favorable BDA/sulfur ylide interaction is present in the Re addition TS, r(OBDA…H′) = 2.59 Å, an unfavorable van der Waals contact is also present between the methylene moieties of the ylide and of BDA, r(HBDA…H″) = 2.37 Å. In short, the two chiral substituents are matched – both favor Si approach of the ylide.
Figure 5.
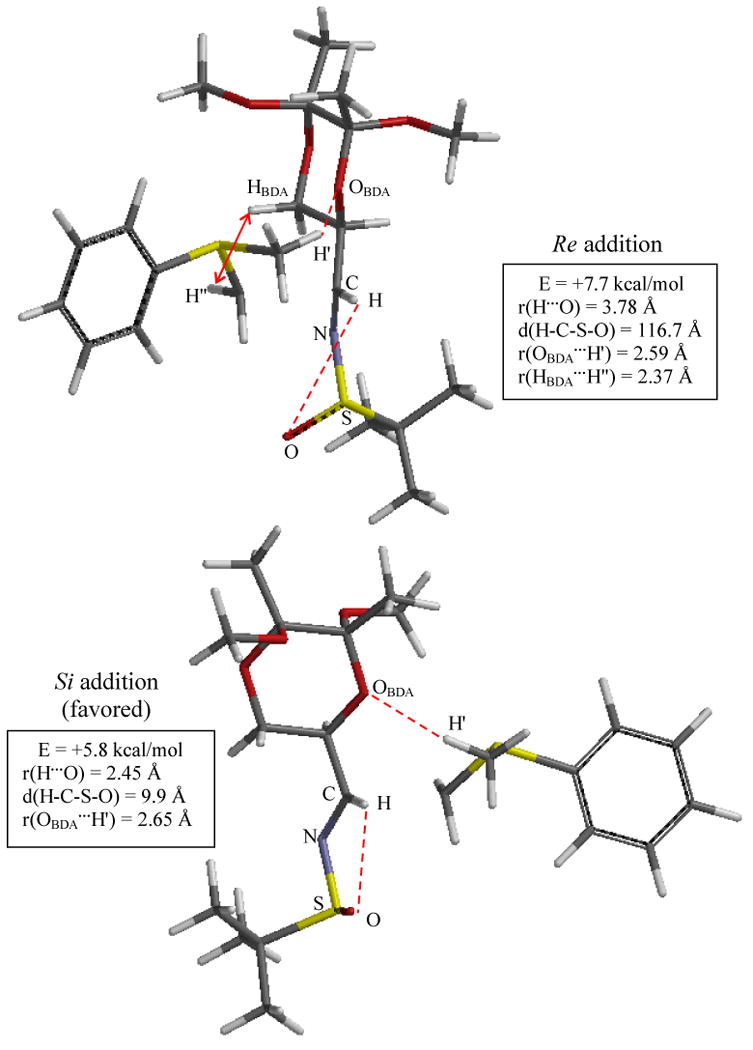
Optimized TS Structures for Additions onto Imine 2 using Sulfur Ylide 1. Structures and energies determined at the B3LYP/6-311+G**(THF) level of theory; energies are ZPE-corrected and relative to reactants. Significant interaction distances are indicated. Bottom: An important intramolecular interaction between the sulfinyl oxygen and the iminyl hydrogen permits ylide approach to the Si face because of the S configuration of the sulfinyl sulfur stereocenter. Top: This interaction is disrupted to allow for Re approach; also, a steric clash with BDA, indicated by the double arrow, occurs with Re face approach. Imine 2's N- and S-substituents are matched, as both favor Si addition.
The TS structures for the less stereodiscriminating addition onto imine 5 are illustrated in Figure 6. In accord with experiment, the magnitude of ΔΔE‡ is only 1.2 kcal/mol for imine 5 versus 1.8 kcal/mol for imine 2. Analogous to the Si TS of imine 2, the Re addition TS of Figure 4 shows that the inverted sulfinyl sulfur stereocenter (R) of imine 5 leaves the Re face open to addition without interference, as determined by the important sulfinyl oxygen/iminyl hydrogen interaction. However, a steric clash with the BDA moiety, r(HBDA…H″) = 2.35 Å, disfavors Re face approach, and so the Re TS suffers from a small energy penalty of about 0.6 kcal/mol, relative to the baseline set by the Si TS of imine 2. In comparison, the imine 5 Si TS accommodates the ylide approach by disruption of the sulfinyl oxygen/iminyl hydrogen interaction with a larger energy penalty of about 1.8 kcal/mol. For imine 5, Re addition is selected because the R sulfinyl sulfur stereocenter is apparently the dominant factor, but the selectivity is weakened because BDA is a mismatch for Re addition. Finally, the dr values reported for the enantiomers of imine 2 (ent-2) and imine 5 (ent-5) are independent confirmations of the observed selectivities afforded by their partner imines, Table 1. Our analysis implies that ent-5 is selective for Si addition but suffers from a mismatch, whereas ent-2, with a proper match of substituents, both favoring Re addition, is strongly selective for Re addition.
Figure 6.
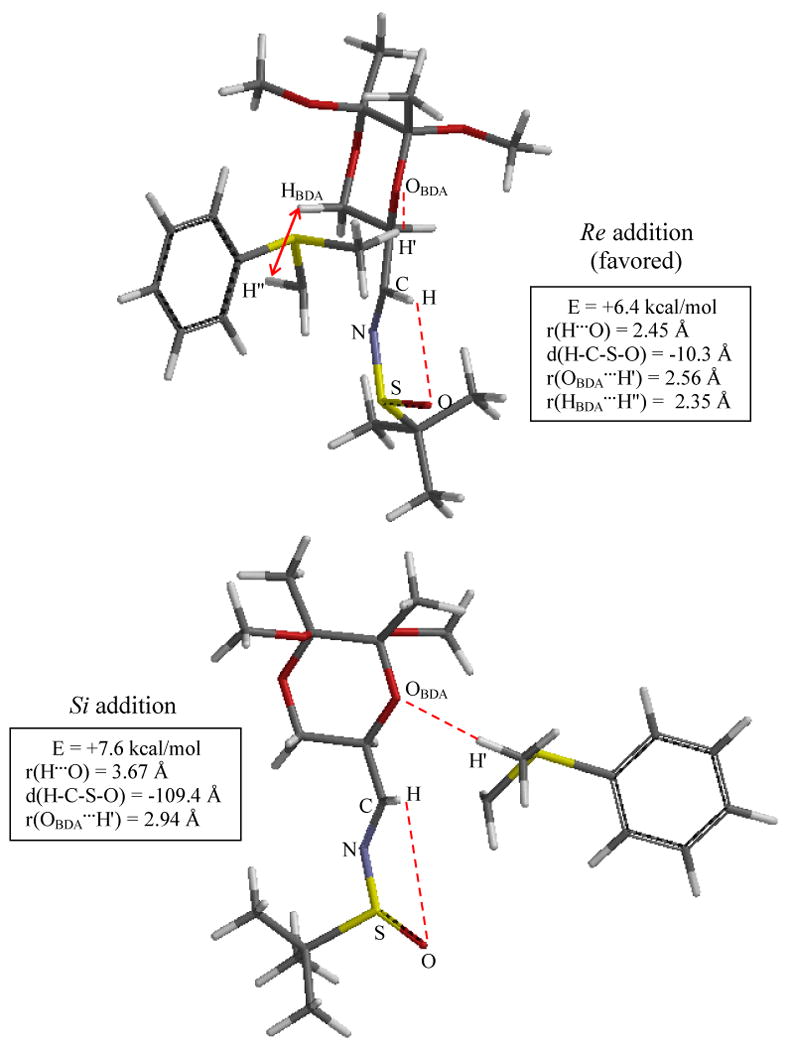
Optimized TS structures for Additions onto Imine 5 using Sulfur Ylide 1. Structures and energies determined at the B3LYP/6-311+G**(THF) level of theory; energies are ZPE-corrected and relative to reactants. Significant interaction distances are indicated. Top: The R configuration of the sulfinyl sulfur permits ylide approach to the Re face; Re face approach is favored in spite of a steric clash with BDA. Bottom: The sulfinyl oxygen/iminyl hydrogen interaction is disrupted to permit Si approach, but Si approach occurs without steric clash with BDA. Imine 5's N- and S- substituents are not matched, with the former favoring Re addition and dominant, while the latter favors Si addition.
Transition States for Re and Si Face Additions onto Imines 3 and 4
As in the case of imine 2, our computations show that imines 3 and 4, with the sulfinyl sulfur in the S configuration, also favor Si addition (ΔΔE‡ > 0), Table 1. The TS structures of imine 3 are analogous to those of imine 2 shown in Figure 5, as would be expected; however, replacement of BDA with the achiral cyclohexyl group diminishes the magnitude of ΔΔE‡ by about half, consistent with the observed loss of selectivity in the experimental dr. Upon switching from t-butyl to the less bulky 4-methylphenyl, imine 4, the level of facial discrimination is simply not as high, as indicated by both the experimental dr and the computational model. Indeed, inspection of the imine 4 Re addition TS shows that Re approach of the ylide does maintain the sulfinyl oxygen/iminyl hydrogen interaction despite steric crowding. The steric crowding, of course, is sufficient to make the Re approach less favorable than the Si approach for imine 4.
Conclusions
Computational studies have been performed on the addition of sulfur ylide 1 onto a series of chiral non-racemic N-sulfinyl imines, Figure 1, which afford varying diastereoselectivity in the aziridine product, Table 1. Examination of the Re and Si addition TS structures have revealed the causes of selectivity arising from the N- and C- iminyl substituents. Based on our computational findings, we have assigned the configurations of the major/minor aziridine products in the principal and control reactions in the absence of x-ray crystallographic data. Specifically, we have assigned the new chiral center for the major aziridine product for the reaction with imine 2 on the basis that it arises from Si face addition; the assignment is consistent with the S configuration now determined for its sulfone adduct.[5]
Our computational model for the principal sulfur ylide 1/imine 2 reaction pathways indicates that ring closure of the betaine intermediates is rapid and irreversible; consequently, we have concluded that the relative energies of the transition states for the competing Re/Si addition steps (ΔΔE‡) govern the experimental dr values, Table 1. We have identified the configuration of the N-sulfinyl sulfur atom as the dominant director of addition: S directs Si addition, while R directs Re addition. This stereocontrol is tied to the presence of a favorable internal interaction between the sulfinyl oxygen and iminyl hydrogen of the imine, Figures 5 and 6. The iminyl C- substituent, BDA, acts as a secondary director: if derived from D-mannitol, Si addition is favored; if from ascorbic acid, Re addition is favored. A match of the N- and C- substituents in imine 2 results in almost exclusive Si addition. Likewise, its enantiomer (ent-2) is highly selective for Re addition.
A synperiplanar (s-cis) arrangement of the C-N-S-O unit in sulfinyl imines has also been observed in x-ray crystal structures[16] and has been shown to be a favored conformation in gas-phase calculations for simple sulfinyl imines.[14,15] This arrangement has been used to explain observed selectivities in Diels-Alder additions[14] and has served as a basis for proposed transition states in mediated allylations.[16] To the best of our knowledge, however, no computational results regarding actual transition state structures for reactions of sulfinyl imines have been presented until now. As our systems involve methylene transfer onto sulfinyl imines resulting in terminal aziridines – not the formation of cis/trans aziridines,[11, 12] a completely new examination of facial diastereoselective additions has been conducted.
Supplementary Material
Acknowledgments
This work was supported in part by NIGMS (NIH NIGMS 1R15GM085936), NSF (CHE 0514004), the Camille and Henry Dreyfus Foundation (TH-06-008), and by a grant of high performance computing resources and technical support from the Alabama Supercomputing Authority.
Footnotes
Supporting Information Available: Tables of selected geometric parameters, energies, and coordinates for all discussed TS species.
References
- 1.(a) Izawa K, Onishi T. Chem Rev. 2006;106:2811. doi: 10.1021/cr050997u. [DOI] [PubMed] [Google Scholar]; (b) Honda Y, Katayama S, Kojima M, Suzuki T, Kishibata N, Izawa K. Org Biomol Chem. 2004;2:2061. doi: 10.1039/b404071f. [DOI] [PubMed] [Google Scholar]; (c) Wang D, Schwinden MD, Radesca L, Patel B, Kronenthal D, Huang MH, Nugent W. J Org Chem. 2004;69:1629. doi: 10.1021/jo035733r. [DOI] [PubMed] [Google Scholar]; (d) Evans BE, Rittle KE, Homnick CF, Springer JP, Hirshfield J, Veber DF. J Org Chem. 1985;50:4615. [Google Scholar]; (e) Beaulieu PL, Wernic D, Duceppe JS, Guindon Y. Tetrahedron Lett. 1995;36:3317. [Google Scholar]; (f) Reetz MT, Binder J. Tetrahedron Lett. 1989;30:5425. [Google Scholar]; (g) Rotella DP. Tetrahedron Lett. 1995;36:5453. [Google Scholar]
- 2.(a) McGarrigle EM, Myers EL, Illa O, Shaw MA, Riches SL, Aggarwal VK. Chem Rev. 2007;107:5841. doi: 10.1021/cr068402y. [DOI] [PubMed] [Google Scholar]; (b) Dalko PI, Moisan L. Angew Chem, Int Ed. 2004;43:5138. doi: 10.1002/anie.200400650. [DOI] [PubMed] [Google Scholar]; (c) Dai LX, Hou XL, Zhou YG. Pure and Applied Chemistry. 1999;71:369. [Google Scholar]
- 3.Forbes DC, Standen MC, Lewis DL. Org Lett. 2003;5:2283. doi: 10.1021/ol034612a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Forbes DC, Bettigeri SV, Amin SR, Bean CJ, Law AM, Stockman RA. Synth Commun. 2009;39:2405. doi: 10.1080/00397910802654898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Forbes DC, Bettigeri SV, Patrawala SA, Pischek SC, Standen MC. Tetrahedron. 2009;65:70. doi: 10.1016/j.tet.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Forbes DC, Amin SR, Bean CJ, Standen MC. J Org Chem. 2006;71:8287. doi: 10.1021/jo061370u. [DOI] [PubMed] [Google Scholar]
- 5.(a) Forbes DC, Bettigeri SV, Pischek SC. Chem Commun. 2009:1882. doi: 10.1039/b822779a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moragas-Solà T, Lewis W, Bettigeri SV, Stockman RA, Forbes DC. Acta Cryst E. 2010 doi: 10.1107/S1600536810048816. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts RE, Stratmann O, Yazyev AJ, Austin R, Cammi C, Pomelli JW, Ochterski R, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision A.1. Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 7.(a) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (b) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (c) Vosko SH, Wilk L, Nusair M. Can J Phys. 1980;58:1200. [Google Scholar]; (d) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623. [Google Scholar]
- 8.(a) McLean AD, Chandler GS. J Chem Phys. 1980;72:5639. [Google Scholar]; (b) Raghavachari K, Binkley JS, Seeger R, Pople JA. J Chem Phys. 1980;72:650. [Google Scholar]
- 9.Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PvR. J Comp Chem. 1983;4:294. [Google Scholar]
- 10.(a) Miertuš S, Scrocco E, Tomasi J. J Chem Phys. 1981;55:117. [Google Scholar]; (b) Miertuš S, Tomasi J. J Chem Phys. 1982;65:239. [Google Scholar]; (c) Pascual-Ahuir JL, Silla E, Tuñón I. J Comp Chem. 1994;15:1127. [Google Scholar]; (d) Cancès E, Mennucci B, Tomasi J. J Chem Phys. 1997;107:3032. [Google Scholar]
- 11.Robiette R. J Org Chem. 2006;71:2726. doi: 10.1021/jo052559t. [DOI] [PubMed] [Google Scholar]
- 12.(a) Aggarwal VK, Charmant JPH, Ciampi C, Hornby JM, O'Brien CJ, Hynd G, Parsons R. J Chem Soc, Perkin Trans. 2001;1:3159. [Google Scholar]; (b) Aggarwal VK, Harvey JN, Richardson J. J Am Chem Soc. 2002;124:5747. doi: 10.1021/ja025633n. [DOI] [PubMed] [Google Scholar]
- 13.(a) Owens TD, Souers AJ, Ellman JA. J Org Chem. 2003;68:3. doi: 10.1021/jo020524c. [DOI] [PubMed] [Google Scholar]; (b) Davis FA, Reddy RE, Szewczyk JM, Reddy GV, Portonova PS, Zhang H, Fanelli D, Reddy RT, Zhou P, Carrol PJ. J Org Chem. 1997;62:2555. doi: 10.1021/jo970077e. [DOI] [PubMed] [Google Scholar]
- 14.Tietze LF, Schuffenhauer A. Eur J Org Chem. 1998:1629. [Google Scholar]
- 15.Bharatam PV, Uppal P, Kaur A, Kaur D. J Chem Soc, Perkin Trans 2. 2000:43. [Google Scholar]
- 16.Sun XW, Xu MH, Lin GQ. Org Lett. 2006;8:4979. doi: 10.1021/ol062216x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.