

Abstract
Design of non-nucleoside inhibitors of HIV-1 reverse transcriptase is being pursued with computational guidance. Extension of azine-containing inhibitors into the entrance channel between Lys103 and Glu138 has led to the discovery of potent and structurally novel derivatives including dimeric inhibitors in an NNRTI-linker-NNRTI motif.
Non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs) are a mainstay of combination therapies for the treatment of HIV infection.1 They bind to an allosteric site, which leads to deactivating conformational changes at the proximal polymerase active site.2,3 However, the clinical utility of NNRTIs is challenged by rapid emergence of drug-resistant, variant strains of the virus.4 Thus, much effort has been put into the development of new NNRTIs with improved resistance profiles with simultaneous concern for diminished side-effects and ease of administration.5 The work has mostly featured classical medicinal chemistry with extensive analoging of multiple core structures, which have typically arisen from high-throughput screening.3 As an alternative, our group has emphasized computer-aided structure-based design in an attempt to reduce the number of compounds that need to be synthesized and assayed.6 Significant success has been achieved in several series; for example, 1 – 3 have been reported to inhibit replication of wild-type HIV-1 (IIIB) in infected human T-cells with EC50 values of 2, 11, and 0.3 nM.7–9 These structures illustrate the diversity of NNRTIs; however, crystallographic10 and modeling studies reveal common features with the side chains for inhibitors like 2 – 4 fitting into two channels in the NNRTI binding site.
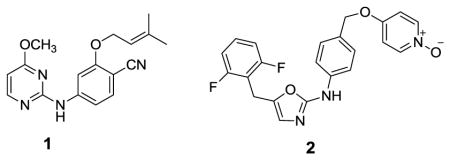
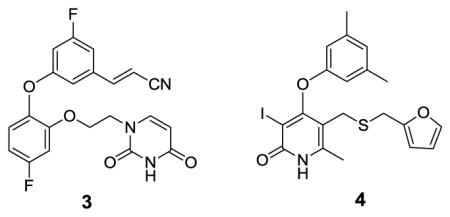
As illustrated for 4 in Figure 1, the benzyl or phenoxy groups of 2 – 4 reside in the tunnel lined by Tyr181, Tyr188, Trp229, and Phe227, which leads towards the polymerase active site, while the heteroaryl containing side chains occupy the groove lined by Phe227, Tyr318, Pro225, and Pro236. Another common feature is a hydrogen bond between the amino groups of 1, 2, and 4 and the carbonyl oxygen of Lys101 (not illustrated). And, for 1, the dimethylallyl group arches into the former channel interacting with Tyr181, Tyr188, and Trp229, as in the 1rt4 crystal structure.11 In addition to the tunnel and groove, the largely open region in front of Lys103, Glu138, and Val179 in Figure 1 is considered to form the entrance channel for the NNRTI binding site. This region appears to have been previously unexplored in the development of NNRTIs. Thus, we set out to consider growing substituents into this area envisioning possible benefits for activity and modulation of molecular properties. In the extreme, we are also interested in pursuing development of chimeric NNRTIs. E.g., an NNRTI1-linker-NNRTI2 construct could be intriguing given different resistance profiles for the two NNRTIs. The next step would be “keychain inhibitors” in which multiple, different NNRTIs or other anti-HIV agents are linked to a core, effectively providing combination therapy in a single compound. Naturally, molecular weight/bioavailability issues would likely become a concern at some point. The present work just addresses growth of substituents into the entrance channel and the viability of dimeric NNRTIs with simultaneous occupancy of the entrance channel and NNRTI binding site.
Figure 1.

Rendering of the 2be2 crystal structure10 of 4 bound to HIV-RT. Carbon atoms of 4 are in green. Some residues are omitted for clarity.
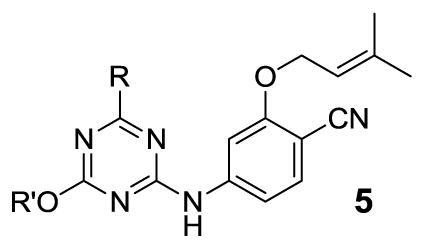
We chose to begin with analogues of the triazene corresponding to 1, which show similar antiviral activity, but diminished cytotoxicity relative to the corresponding pyrimidines.7 We had also found previously that the dimethoxy compound 5b was only two-fold less potent than the monomethoxy 5a (Table 1). Model building was then carried out for analogues in which the methoxy group near the entrance channel was elaborated. The calculations consisted of generation of structures of the complexes and conformational searches with the BOMB program,6 followed by conjugate-gradient optimizations using MCPRO12 and the OPLS/CM1A force field.13 The protein coordinates were based on the 1s9e crystal structure for which the ligand is a diaminotriazine derivative.14 The computations indicated that growth into the entrance channel was sterically allowable using PEG-like extensions (Figure 2).
Table 1.
Anti-HIV-1 Activity (EC50) and cytotoxicity (CC50), μMa
| ||||
|---|---|---|---|---|
| Compound | R | OR′ | EC50 | CC50 |
| 5ab | OCH3 | H | 0.011 | 42 |
| 5bb | OCH3 | OCH3 | 0.022 | >100 |
| 5c | OCH3 | OCH2CH2OCH3 | 0.097 | 8.6 |
| 5d | OCH3 | (OCH2CH2)2OCH3 | 0.150 | 13 |
| 5e | OCH3 | (OCH2CH2)3OCH3 | 0.380 | 4.2 |
| 5f | CH2CH3 | OCH2CH2OCH3 | 0.057 | 2.1 |
| 5g | CH2CH3 | (OCH2CH2)2OCH3 | 0.540 | 9.8 |
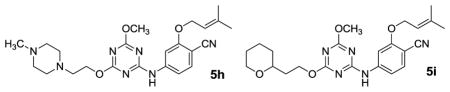
| 5h | OCH3 | OCH2CH2-4-MePip | 0.320 | 7.0 |
| 5i | OCH3 | OCH2CH2-2-THP | 1.2 | 4.2 |
| 5jb | OCH3 | NH2 | 0.009 | 0.11 |
| nevirapine | 0.110 | >100 | ||
4-MePip = N-methylpiperazinyl; 2-THP = 2-tetrahydropyranyl (racemic).
Ref. 7b.
Figure 2.

Computed structure of 5d bound to HIV-RT illustrating extension of the polyether side chain into the entrance channel. Carbon atoms of 5d are in green. Some residues are omitted for clarity.
The desired compounds 5c – 5i were synthesized via SNAr reaction between previously reported chlorotriazines7b and appropriate alkoxides, as summarized in Scheme 1. The alcohol for 5i was prepared by reductive rearrangement of 2-vinyloxytetrahydropyran. Activities against the IIIB strain of HIV-1 were measured using MT-2 human T-cells; EC50 values are obtained as the dose required to achieve 50% protection of the infected cells by the MTT colorimetric method. CC50 values for inhibition of MT-2 cell growth by 50% are obtained simultaneously.7,15.16 The identity of all assayed compounds was confirmed by 1H and 13C NMR and high-resolution mass spectrometry; purity was >95% as judged by high-performance liquid chromatography.
Scheme 1.

Synthesis of substituted triazenes.
It was encouraging that the methoxyethoxy analogue 5c retained good potency at 97 nM and the penalties for additional ethyleneoxy groups were modest for 5d and 5e. 5f, the R = ethyl analogue of 5c, was also prepared and yielded improved activity with an EC50 of 57 nM. Curiously, 5g, the ethyl analogue of 5d, exhibited the opposite trend. To test if more branched alternatives could be tolerated, the N-methylpiperazinyl and 2-tetrahydropyranyl derivatives, 5h and 5i, were considered; they were found to be less active with EC50 values of 0.32 and 1.2 μM. Since the MT-2 assay is cell-based, expectations for 5h were unclear in view of the potential impact of protonation of the piperazine nitrogens on the cell permeability.
Prior results for the amino analogue 5j are also noted in Table 1. It is a 9-nM NNRTI, and the corresponding methylamino analogue with the cyano group replaced by chlorine was found to have good potency, 31 nM, as well.7b Thus, amino connectors might also be viable. Clearly, numerous additional model compounds could be explored with alternative side chains including anionic ones, and further optimization of the group R. Nevertheless, at this point, it was established that substantial molecular growth was possible in the entrance channel with retention of antiviral activity at nanomolar levels.
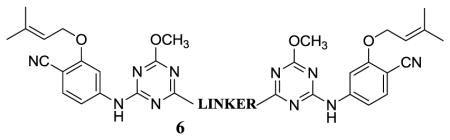
Thus, attention turned to more ambitious Janus dimers, NNRTI-linker-NNRTI. Computational modeling was performed to assess the viability of dimers of analogues of 5a tethered as in 6. Use of the BOMB program readily found low-energy structures with relatively short linkers, e.g., for 6b in Figure 3. The second copy of the NNRTI can be accommodated in a cleft passing between Lys32B and Lys172A. The illustrated conformer of 6b is well extended in the entrance channel. Six compounds were synthesized from the chloromethoxytriazene,7b as summarized in Scheme 2. The corresponding assay results are listed in Table 2.
Figure 3.
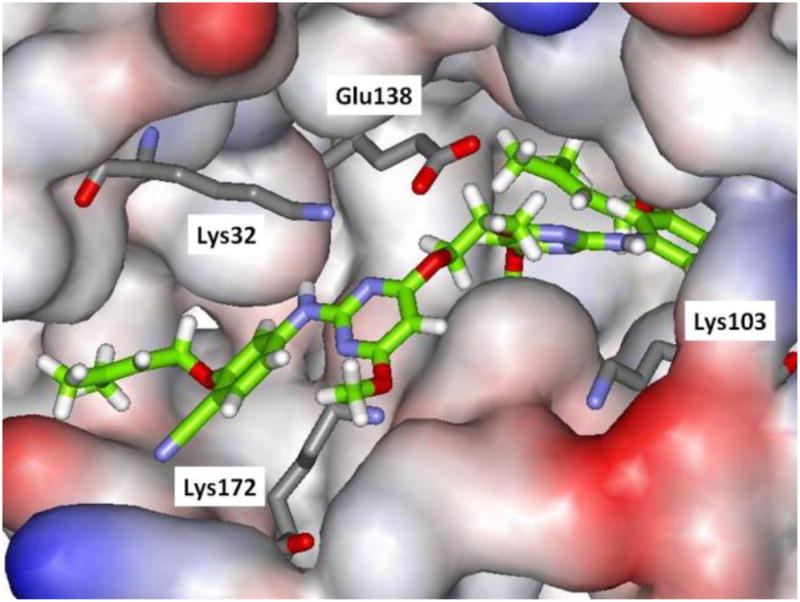
Computed structure of 6b bound to HIV-RT illustrating extension into the entrance channel. Carbon atoms of the inhibitor are in green. Some residues are omitted for clarity.
Scheme 2.

Synthesis of dimeric inhibitors.
Table 2.
Anti-HIV-1 Activity (EC50) and cytotoxicity (CC50), μMa
| |||
|---|---|---|---|
| Compound | Linker | EC50 | CC50 |
| 6a | OCH2CH2O | 0.390 | 42 |
| 6b | OCH2CH2CH2O | 0.170 | 21 |
| 6c | OCH2CH2CH2CH2O | NA | >100 |
| 6d | OCH2CH2OCH2CH2O | NA | >100 |
| 6e | NHCH2CH2NH | NA | 6.0 |
| 6f | NHCH2CH2CH2NH | NA | 50 |
NA = not active.
The diethers with the shortest linkers, 6a and 6b, do show good activity with EC50 values of 390 and 170 nM, and low cytotoxicity, 20 – 40 μM. The dimeric constructs with longer linkers or amino connectors (6e, 6f) did not show anti-viral activity below their CC50 levels. As illustrated in Figure 3, 6b may fill the entrance groove well. Longer linkers could cause the protruding NNRTI to be pushed farther from the protein’s surface. The similar activities for 5d and 6b indicate the addition of some favorable contacts between 6b and the protein to offset the increased loss of conformational freedom. The proof-of-concept success with 6a and 6b is striking and provides a foundation for investigations of heterodimers, NNRTI1-linker-NNRTI2.
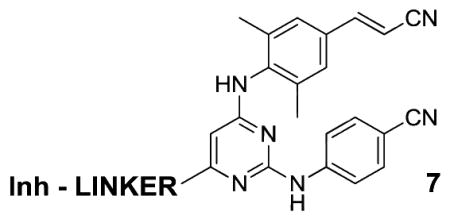
The stage is also set for construction of bifunctional inhibitors, NNRTI-linker-Inh, where Inh is a member of a different class of anti-HIV agent, and the necessary attachment point to the NNRTI is evident. Bifunctional inhibitors have previously been explored with NNRTIs. Early work on NNRTI-(CH2)n-NRTI constructs provided compounds where the anti-viral activity seemed to arise solely from the NNRTI component.17 The structural situation is unclear, though it is possible that the NRTI resides in the entrance channel. More recent efforts designed NNRTI-linker-NRTI inhibitors such that the linker is in the tunnel passing Trp229; however, there was no evidence that synergistic binding to the NNRTI and NRTI sites was achieved.18 NNRTIs, particularly in the HEPT class, have also been linked to a characteristic diketoacid fragment of HIV integrase inhibitors.19 The linking to the NNRTI in these cases was at the terminus that would reside in the Pro225-Pro236 groove (Figure 1). Though these constructs retained strong inhibition of HIV-RT, the inhibition of HIV integrase has only been in the micromolar range.19 The present results open up the possibility of exploring an alternative topology via connections to NNRTIs such that the second inhibitor resides in the NNRTI-entrance channel. Aside from the present triazines and related pyrimidines (1) such connections should be possible for the oxazole (2) and catechol diether (3) series as well as for some other known NNRTIs such as rilpivirine, which has an excellent resistance profile;20 e.g., the appropriate chimera would be 7.
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551) for support. Receipt of reagents through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is also greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flexner C. Nature Rev Drug Disc. 2007;6:959. doi: 10.1038/nrd2336. [DOI] [PubMed] [Google Scholar]
- 2.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Science. 1992;256:1783. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 3.Prajapati DG, Ramajayam R, Yadav MR, Giridhar R. Bioorg Med Chem. 2009;17:5744. doi: 10.1016/j.bmc.2009.06.060. [DOI] [PubMed] [Google Scholar]
- 4.De Clerq E. Nature Rev Drug Disc. 2007;6:1001. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 5.Adams J, Patel N, Mankaryous N, Tadros M, Miller CD. Ann Pharmacotherapy. 2010;44:157. doi: 10.1345/aph.1M359. [DOI] [PubMed] [Google Scholar]
- 6.Jorgensen WL. Acc Chem Res. 2009;42:724. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ruiz-Caro J, Basavapathruni A, Kim JT, Wang L, Bailey CM, Anderson KS, Hamilton AD, Jorgensen WL. Bioorg Med Chem Lett. 2006;16:668. doi: 10.1016/j.bmcl.2005.10.037. [DOI] [PubMed] [Google Scholar]; (b) Thakur VV, Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2006;16:5664. doi: 10.1016/j.bmcl.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 8.Leung CS, Zeevaart JG, Domaoal RA, Bollini M, Thakur VV, Spasov K, Anderson KS, Jorgensen WL. Bioorg Med Chem Lett. 2010;20:2485. doi: 10.1016/j.bmcl.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KS, Jorgensen WL. J Med Chem. 2011;54:8582. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Himmel DM, Das K, Clark AD, Hughes SH, Benjahad A, Oumouch S, Guillemont J, Coupa S, Poncelet A, Csoka I, Meyer C, Andries K, Nguyen CH, Grierson DS, Arnold E. J Med Chem. 2005;48:7582. doi: 10.1021/jm0500323. [DOI] [PubMed] [Google Scholar]
- 11.Ren J, Esnouf RM, Hopkins AL, Warren J, Balzarini J, Stuart DI, Stammers DK. Biochem. 1998;37:14394. doi: 10.1021/bi981309m. [DOI] [PubMed] [Google Scholar]
- 12.Jorgensen WL, Tirado-Rives J. J Comput Chem. 2005;26:1689. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen WL, Tirado-Rives J. Proc Natl Acad Sci USA. 2005;102:6665. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das K, Clark AD, Jr, Lewi PJ, Heeres J, de Jonge MR, Koymans LMH, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, de Béthune MP, Boyer PL, Clark P, Hughes SH, Janssen PAJ, Arnold E. J Med Chem. 2004;47:2550. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 15.Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Biochem Pharmacol. 1994;47:171. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 16.Ray AS, Yang Z, Chu CK, Anderson KS. Antimicrob Agents Chemother. 2002;46:887. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Velázquez S, Alvarez R, San-Félix A, Jimeno ML, De Clercq E, Balzarini J, Camarasa MJ. J Med Chem. 1995;38:1641. doi: 10.1021/jm00010a008. [DOI] [PubMed] [Google Scholar]
- 18.Younis Y, Hunter R, Muhanji CI, Hale I, Singh R, Bailey CM, Sullivan TJ, Anderson KS. Bioorg Med Chem. 2010;18:4661. doi: 10.1016/j.bmc.2010.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. J Med Chem. 2007;50:3416. doi: 10.1021/jm070512p. [DOI] [PubMed] [Google Scholar]; (b) Wang Z, Vince R. Bioorg Med Chem Lett. 2008;18:3587. doi: 10.1016/j.bmcl.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 20.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD, Jr, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. J Med Chem. 2005;48:1901. doi: 10.1021/jm049534r. [DOI] [PubMed] [Google Scholar]