

Abstract
Podophyllotoxin (1) has been known to possess anti-tumor activity and is still considered an important lead for research and development of antineoplastic agents. Derivatives of podophyllotoxin, namely etoposide (2), etopophos (3) and teniposide (4) have been developed and are currently used in clinic for the treatment of a variety of malignancies. These agents are also used in combination therapies with other drugs. Due to the drug resistance developed by cancer cells as well as side effects associated with the use of these agents in clinic, the search for new effective anticancer analogues of podophyllotoxin remains an intense area of research. The structural complexity of podophyllotoxin, arising from the presence of four stereogenic carbons in ring C has restricted most of the structural activity relationship (SAR) studied by derivatization of the parent natural product rather than by de novo multi-step chemical synthesis. These issues provide strong impetus to a search for analogues of 1 with simplified structures, which can be accessible via short synthetic sequences from simple starting materials. Even if such initial compounds might have diminished cytotoxic potencies compared with the parent cyclolignan, the ease of preparation of carefully designed libraries of analogues would lead to more informative SAR studies and expeditious structure optimization. In this regard, during the last two decades considerable efforts have been made to synthesize aza- analogs of podophyllotoxin, i. e. aza-podophyllotoxins, with hetero atoms at different positions of the podophyllotoxin skeleton, while keeping the basic podophyllotoxin structure. Recently, there have been significant efforts towards the convenient synthesis of aza-analogs of 1. The use of multicomponent reactions (MCRs) and the synergies of ultrasound and microwave irradiations have increased the synthetic speed and variety of azapodophyllotoxins which are and will be available to be tested against a diverse population of carcinomas and other diseases. It has been reported that several aza-podophyllotoxins retain a great fraction of the cytotoxicity associated with the parent lignan. This review focuses on the strategies towards synthesis of various aza-podophyllotoxin analogues and their biological activities.
Keywords: Antineoplastic, azapodophyllotoxin, podophyllotoxin
1. INTRODUCTION
Podophyllotoxin (1, Fig. 1), is an antimitotic cyclolignan isolated [1, 2] from plants of the genus Podophyllum pelatum L. and P. emodi L, studied extensively as an antitumor agent, but clinical results were disappointing due to severe gastrointestinal side effects [3]. However, continuous efforts towards the synthesis of its analogues led to the discovery of new anticancer drugs. For example its semisynthetic derivatives, etoposide (2) and etoposide phosphate (3) are currently used in the clinic for the treatment of a variety of malignancies including, lung and testicular cancers, lymphoma, nonlymphocytic leukemia, and glioblastoma multiforme [4]. Teniposide (4), also prepared by semisynthesis from 1, is important for the treatment of childhood acute lymphocytic leukemia [4]. In contrast to the parent podophyllotoxin, which binds to tubulin and inhibits microtubule assembly, etoposide is not antimitotic. In fact, molecules such as etoposide, amsacrine, and mitoxantrone are topoisomerase II inhibitors that induce cell death by enhancing topoisomerase II-mediated DNA cleavage through stabilization of the transient DNA/topoisomerase II cleavage complex [5]. Recently, it has been suggested that etoposide- topoisomerase II interactions mediate cleavage complex stabilization, rather than etoposide-DNA interactions, as is the case with amsacrine, another potent topoisomerase II inhibitor [6]. Despite its extensive use, etoposide is not devoid of toxic side effects. Bone marrow depression is a frequent, dose-limiting, and toxic side effect encountered by patients receiving etoposide. The use of effective doses of the drug is also associated with an increased risk of secondary acute myelogenous leukemia [7, 8]. For this reason, the development of more potent analogues with less side toxicity remains a highly valuable objective. Extensive structural activity relationship (SAR) studies with etoposide analogues have suggested that the activity of the epipodophyllotoxin derivatives is related to three structurally distinct pharmacophoric domains: the DNA-intercalating moiety (central part of the molecule, see structure of 1), the binding site (lower part), and a variable substituent region (glycoside moiety) [9–11].
Fig. (1).

Podophyllotoxins.
Most of the SAR studies involving 1 have been restricted to the derivatization of the parent natural product rather than through novel chemical synthesis. This is due to its structural complexity, which arises due to the presence of four stereogenic carbons in ring C [12, 13]. Such approaches, however, are rather limited by the type of chemistry that 1 can undergo. For example, variations of the ring E are not easy to perform because of the three methoxy groups in the starting lignan. No matter how laborious, total synthesis efforts are indispensable in this regard. These issues provide strong impetus to a search for analogues of 1 with simplified structures, which can be prepared via short synthetic sequences from simple starting materials. Even if such initial compounds might have diminished cytotoxic potencies compared with the parent cyclolignan, the ease of preparation of carefully designed libraries of analogues would lead to more informative SAR studies and expeditious structure optimization. In this regard, during the last two decades considerable efforts have been made to synthesize aza- analogues of podophyllotoxin, i.e. azapodophyllotoxins, with heteroatoms at different positions of the podophyllotoxin skeleton, while keeping the basic podophyllotoxin structure [14]. It is worth mentioning that replacing carbon with nitrogen at specific positions, e.g. at the stereocenters at C-2 and C-3, could solve the problem of epimerization at C-2 that has plagued the clinical development of 1 and its stereochemically complex derivatives due to the formation of the significantly less potent cis-lactone metabolite [15]. Recently, significant efforts have been made towards the convenient synthesis of aza-analogues of 1. The use multicomponent reactions (MCRs) with ultrasound and microwave irradiation has greatly increased the speed at which azapodophyllotoxins can be synthesized, making a greater variety available to test against a diverse population of carcinomas and other diseases. It has been reported that several azapodophyllotoxins retain a great fraction of the cytotoxicity associated with the parent lignan. This review focuses on the strategies towards the synthesis of various azapodophyllotoxin analogues and their biological activities. What follows is a description of the various approaches taken towards the synthesis of aza-analogues of podophyllotoxin, i.e. azapodophyllotoxins, and their biological activities.
2. SYNTHESIS OF AZAPODOPHYLLOTOXIN DERIVATIVES
2.1. Classical Synthesis
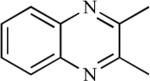
The first synthesis of 2-azapodophyllotoxin was reported by Pearce et al. in 1989 [16]. This report details a stereo-controlled synthesis of 2-azapodophyllotoxin (7), a stable analogue of podophyllotoxin (1). Inversion of C4 stereochemistry in 5 was accomplished by first oxidizing 5 with pyridinium chlorochromate in dichloromethane at 23 °C to give 4-keto-2-azapodophyllotoxin (6), which on reaction with lithium tri-t-butyloxyaluminum hydride in THF at 23 °C afforded 2-azapodophyllotoxin (7) in 65% overall yield [16].
Tomioka and Koga et al. proposed several guidelines for the design of azapodophyllotoxin derivatives: the stereochemical structure should be similar to podophyllotoxin; the carbonyl oxygen should have enough electron density to form a hydrogen-bond; and the compounds should have minimal stereoisomers, shorter synthetic routes, and be available in an optically pure form. Based on these guidelines, synthesis of racemic and optically pure azadeoxypodophyllotoxin derivatives was reported. A cyclic urethane 9, obtained from a known racemic amino acid 8, on condensation with 3,4,5-trimetoxybenzaldehyde in the presence of H2SO4/CH2Cl2 provided a separable mixture of diastereomers 10 and 11. The trans-isomer 10 was formed predominantly, while cis-isomer 11 was the minor product. A 4′ -demethoxy derivative 12 was obtained from 10 or 11 by treating with HBr in 1,2-dichloroethane. (Scheme 1) [17, 18]. Optically pure cyclic urethanes (R)−(−)−9 and (S)−(+)−9 were also prepared starting from the corresponding optically active L- and D-amino acid 8, respectively, which afforded optically pure (−)−10, (−)−11 and (+)−10, (+)−11, respectively [18]. The same group proposed a direct introduction of oxygen functionality at the benzylic position, i.e. at C4 of azadeoxypodophyllotoxin, but the direct oxidation of 10 was unsuccessful [18, 19]. Finally, the desired oxygenated azapodophyllotoxin derivatives 15–18 were synthesized by reacting acetoxy urethane 13 and 3,4,5-trimethoxybenzaldehyde, possibly via intermediate 14 (Scheme 2) [18].
Scheme 1.

Scheme 2.

Vandewalle et al. synthesized the 1,3-cis and 1,3-trans isomer in a multistep synthesis [20]. The alkylation of hippuric acid ester 20 with piperonyl bromide led to an acyclic precursor 21, which failed to cyclize under several reaction conditions due to the presence of the ester function. However, reduction of 21 and protection of the alcohol as acetate afforded 22, which was cyclized by the Bischler-Napieralski reaction to give dihydroisoquinoline 23 in high yield. Methanolysis of acetate and aluminium hydride reduction efficiently afforded the 1,3-cis-isomer 24 as the sole product. In contrast, aluminium hydride reduction of the THP ether of 26 gave a 2:1 mixture of the cis/trans isomers 24/27, which were separated by chromatography after the removal of a protecting group. Isomers 24 and 27 finally led to 4-desoxy-2-azapodophyllotxins 25 and 28 (Scheme 3). The same report described an improved synthesis of optically active analogues starting from L- and D-DOPA (Scheme 4). The ester group of 29 was selectively reduced by following the Bischler-Napieralski approach after the formation of an ethylene dioxy unit (ring A), followed by acetylation to get 30, which was then treated with POCl3, giving optically pure dihydroi soquinoline 31, which upon methanolysis and subsequent reduction afforded only the cis isomer, 32. To obtain the trans isomer 36, 31 was quaternized to give 34 followed by sodium borohydride reduction and hydrogenolysis. The same report also briefly described the synthesis of other analogues containing a modified heterocyclic ring D 38–41 [20].
Scheme 3.

Scheme 4.

The 1,2-cis-2,3-trans configuration of ring C in the azapodophyllotoxin structure has been shown to be crucial for the compound's antitumor activity. This structural feature was a serious obstacle for the total synthesis of α and β hydroxyl at C-4 in podophyllotoxin derivatives. Vandewalle et al. reported a multistep synthesis of α- and β-4-hydroxy azapodophyllotoxins by following Schollkopf's method (Schemes 5 and 6) [21]. Ethyl isocyanoacetate, upon reaction with 42 under thermodynamic conditions followed by reduction, led to trans 43, which upon hydrolytic cleavage of oxazoline gave 44;the diol was protected as acetonide. The Bischler-Napieralski reaction on the threo isomer 46 under Lewis acid conditions led to isoquinoline 47, which was reduced to cis-and trans- tetrahydroisoquinolines 48 and 49. Finally, the acetonide groups were hydrolysed and treated with phosgene to obtain 51 and 53 (Scheme 5). The epidophyllotoxin analogue 53 was also trans formed into podophyllotoxin analogue 54 via oxidation-reduction [21]. For the erythro series, β-keto-ester 51 was prepared by reacting 50 with the lithium-enolate anion of ethyl N-(3,4,5-trimethoxybenzoyl)-glycinate. Ketalization of 52, followed by a Bischler-Napieralski reaction, gave 54, which was stereoselectively reduced to 1,3-cis isomer 55, then treated with phosgene, leading to a mixture of the desired product 57 and the side product 56 (Scheme 6) [21].
Scheme 5.

Scheme 6.

Hitotsuyanagi et al. speculated that substitution of the methylene group at the C-4 position by oxygen would not alter the whole stereochemistry of 1 due to similarities between the C-O and C-CH2 bond lengths and the C-O-C and C-CH2-C bond angles. However, it could have an effect on biological activity due to changes in polarity [22]. Based on this hypothesis, they reported the synthesis of 4-oxa-2-azapodophyllotoxin 63. Benzylation and bromination of seasamol afforded the O-benzyl bromide 58, which upon lithiation through a metal-halogen exchange reaction, followed by treatment with 3,4,5-trimethoxybenzaldehyde, led to benzhydrol 59 in good yield. The reaction of 59 with 2,4-oxazolidinedione under Mitsunobu conditions gave 60, which was reduced and then reoxidized using the Dess-Martin reaction to give aldehyde 61. Careful debenzylation of 61 gave labile hemiaminal 62, which was treated with acetic acid to obtain 4-oxa-2-azapodophyllotoxin 63 (Scheme 7) [22]. Using a similar hypothesis, this group has also synthesized 2,4-diaza-4-deoxypodophyllotoxins 69a–d and 70a–d, where the carbons at the C2 and C4 positions were substituted by nitrogen atoms (Scheme 8) [23, 24]. The objective here was to determine the effect of changes in the whole stereochemical structure on biological properties. The synthesis started with N-benzylanilines 64 and 65, which were reacted with boron trichloride and triethylamine followed by methoxybenzaldehydes to give boracyclic intermediates, which upon hydrolysis led to benzhydryl alcohols 66a–d. The alcohols 66a–d were converted to urethanes 67a–d, which upon reaction with glyoxylic acid, followed by reduction, produced alcohols 68a–d. The cyclization of 68a–d was done by using sodium methoxide to give oxazolones, which were debenzylated to yield the cis analogues 69a–d. The cis analogues, upon treatment with trifluoroacetic acid, were converted into their corresponding trans isomers 70a–d. The N-amino analogues 71 and 72 were also prepared by reacting 69a and 70a, respectively, with O-(2,4-dinitrophenyl)hydroxylamine. Oxidation of 70a was also carried out by reacting it with lead tetraacetate to give the dehydro derivative 73 (Scheme 8) [23, 24]. Later, to evaluate the effect of replacing the methylene group of 1 with nitrogen on biological activity, the same group reported the synthesis of (−)-4-aza-4-deoxypodophyllotoxin (74) in an optically pure form from natural (−)-podophyllotoxin (1) through C-ring cleavage followed by a Curtius rearrangement and intramolecular N-alkylation [25].
In 1997, the first convenient and simple synthetic method for the synthesis of azapodophyllotoxins was reported based on condensation reactions of three components: anilines, tetronic acid and benzaldehydes was reported [26]. In this approach, substituted anilines were treated with an equimolar amount of tetronic acid in dioxane at room temperature, leading to the production of anilinolactones, which were reacted with substituted aldehydes in the presence of p-chloranil in trifluoroacetic acid at room temperature to produce 4-aza-analogues of 1-arylnaphthalene lignans 75a–o in high yield. These lignans were later reduced by an excess amount of sodium cyanoborohydride in acetic acid to the corresponding 4-aza-2,3-dehydro-4-deoxypodophyllotoxins 76a–o (Scheme 9) [14, 26]. Although this method [26] was convenient, its scope was limited, as it required three steps to afford the required azapodophyllotoxins. This method also failed to give N-substituted derivatives, as the alkylation of 76 was unsuccessful. To address this issue, Giorgi-Renault et al. reported the first one-pot MCR for the synthesis of azapodophyllotoxins and their N-alkylated derivatives (77) (Scheme 10) [27].
Scheme 9.

Scheme 10.

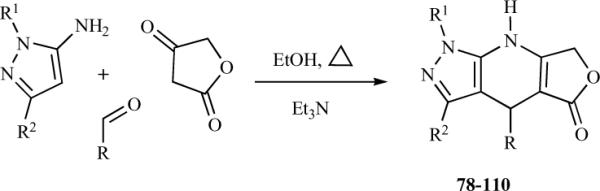
Magedov et al. implemented a bioisosteric replacement of the methylenedioxybenzene subunit of azapodophyllotoxin with a pyrazole moiety to produce tetracyclic dihydropyridopyrazoles 78–110. Libraries of these analogues were prepared using a simple one-step MCR approach involving the condensation of substituted 5-amino-pyrazoles, tetronic acid, and substituted aldehydes (Scheme 11) [28]. The resulting dihydropyridopyrazoles precipitate as the reaction mixtures are allowed to cool to room temperature, are isolated by simple filtration and, in most cases, no further purification is required. Shi et al. reported the synthesis of similar compounds 110–126 using the MCR approach in the presence of organocatalysts in ethanol [29]. The reaction of 5-amino-1-phenyl-3-methylpyrazole, 4-methylbenzaldehyde, and tetronic acid was studied in EtOH in the presence of 10 mol% L-proline at 80 °C (Scheme 12). L-proline is soluble in EtOH and the solubilities of the desired products in EtOH are low. Therefore the products were easily be separated by cooling the reaction to room temperature and filtering it after the reaction was complete. A possible mechanism for the L-proline-catalyzed synthesis of dihydropyridopyrazole derivatives was also proposed in the same report [29].
Scheme 11.
Scheme 12.
Madec et al. reported the synthesis of 4-aza-2,3-didehydropodophyllotoxins, starting with a reaction between N-benzyl tetronamide or N-phenyl tetronamide with the functionalized benzhydrilic alcohol 127 to produce the benzhydrylated N-benzyl tetronamide intermediate 128. The latter was arylated using a copper-mediated Ullmann-type N-arylation. Thus, treatment of 128 with CuI and CsCO3 in dimethylformamide (DMF) (according to Fukuyama's protocol [26]) afforded the expected 4-aza-2,3-didehydropodophyllotoxins (129) in a quantitative yield (Scheme 13) [30].
Scheme 13.

Giorgi-Renault et al. reported a rigid aminologue of 4-aza-2,3-didehydropodophyllotoxins, where the rotation between the tetracycle and the aryl ring was blocked by the creation of an additional pseudocycle [31]. The quinoline-lactone 130 was prepared using the MCR route, followed by oxidation of the dihydroquinoline intermediate in hot DMSO. The 4-chloroquinoline-lactone 131 was then prepared by reacting POCl3 with the N-oxide intermediate obtained by m-CPBA oxidation of 130. This was followed by a nucleophilic substitution by aniline to give 132, which was quaternized with trifluoromethyl sulfonate, leading to the quinolinium salt 133. Deprotonation of 133, followed by reduction or hydrogenation of imine 134, led to the desired aminologue 135 (Scheme 14) [31].
Scheme 14.

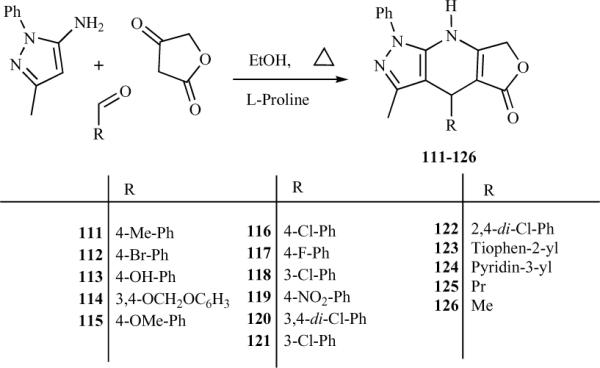
Recently, Shi et al. extended their earlier reported MCR synthesis of dihydropyridopyrazoles [21] using L-proline as an organo-catalyst for the synthesis of 4-azapodophyllotoxin derivatives 136a–x (Scheme 15) [32]. This method has the advantages of high yields, high regioselectivity, extensive adaptability, easy operation, and environmental friendliness. The reaction was carried out with 10 mol% L-proline in refluxing ethanol, resulting in the isolation of product in over 90% yields. The products could be separated by cooling the reaction mixture to room temperature followed by simple filtration. The filtrate containing L-proline can be directly recovered and recycled. Shi et al. found that, even after seven uses, the catalytic efficiency of the L-proline reaction solution was unchanged. The cytotoxic activities of these compounds were also investigated, and some appeared to have good cytotoxic activity against tumor cells [32].
Scheme 15.
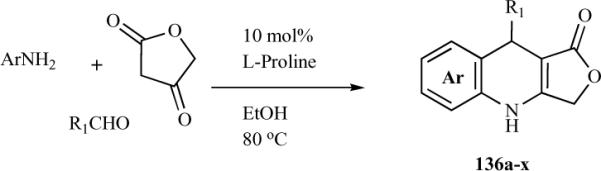
The synthesis of a new series of azapodophyllotoxin analogues and their antivascular activity was recently reported by Giorgi-Renault et al. [33] In this study, a linker was inserted between the trimethoxyphenyl ring E and the tetracyclic ABCD moiety of the 4-aza-1,2-didehydropodophyllotoxins (Scheme 16). Most of these derivatives were N-methylated for synthetic and stability purposes, as N-substitution prevents aromatization of the 1,4-dihydro-quinoline moiety. This linker enables free rotation between the two moieties. The synthesis strategy for 137–140 was straightforward, involving the MCR approach, where substituted aniline, tetronic acid, and substituted aldehyde were reacted to obtain the desired products. The two-carbon homologated analogue 141 was obtained in good yield by catalytic hydrogenation of the exocylic double bond of vinylogue 139 without reduction of the lactone. For the synthesis of ethynologue 143, direct dehydrogenation of 140 using manganese dioxide, selenium dioxide, or dichlorodicyanoquinone was unsuccessful.
Scheme 16.
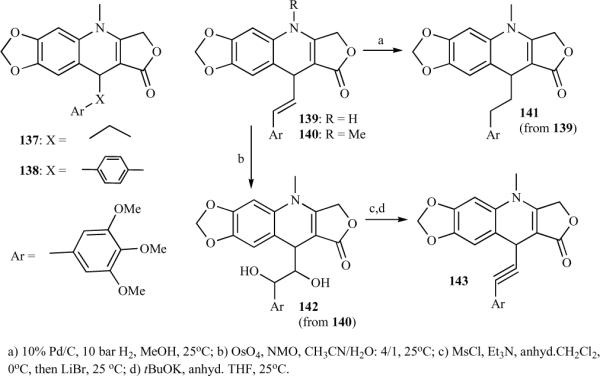
An addition-elimination approach was applied for the synthesis of 143, where 140 was first treated with osmium tetroxide followed by mesylation, bromination, and elimination using potassium tertbutoxide (Scheme 16) [33].
Recently, we reported the synthesis of novel arylamino alcohols 144–146, which we used to prepare novel N-(2-hydroxy-ethyl)-2,3-didehydroazapodophyllotoxins 147–149 by simple reflux in ethanol [34]. The 4-aza-2,3-didehydropodophyllotoxin core was functionalized at C4 of the C ring in a fashion similar to that which occurs in podophyllotoxin derivatives (Scheme 17). These N-(2-hydroxyethyl)-2,3-didehydro-azapodophyllotoxins have the potential to be conjugated to tissue-targeting carriers. This series was recently expanded to include three different types of hydroxyazapodophyllotoxin derivatives: those containing (i) a five-membered methylenedioxy ring (147a–f), (ii) a five-membered ring with no heteroatom (148a–f), and (iii) a six-membered ethylenedioxy ring (149a–f) as ring A [34]. Each series has a different substitution pattern of methoxy groups on ring E. The screening of these azapodophyllotoxin derivatives for their antitumor activity is in progress.
Scheme 17.

2.2. Microwave-irradiated Synthesis
It is well documented in the literature that microwave irradiation accelerates organic reactions and has been used as a tool for the efficient synthesis of a variety of organic compounds. Also, as described earlier, MCRs offer a wide range of possibilities for the efficient, single-step construction of azapodophyllotoxins. Tu et al. have utilized the combination of these two tools extensively in their study of azapodophyllotoxin synthesis, providing diversified libraries of these compounds. They first reported a microwave-irradiated MCR consisting of aldehyde, aromatic amine, and tetronic acid in water, without a catalyst, yielding a new series of 4-azapodophyllotoxin derivatives (150) (Scheme 18) [35]. Several solvents were examined viz.: water, glycol, DMF, glacial acetic acid, and ethanol at 80 to 120 °C, and the microwave irradiation power was optimized. The highest yield was obtained when water was used as the solvent in the reaction of equimolar amounts of 4-bromophenyl aldehyde, p-toluidine, and tetronic acid at 100 °C with a microwave power of 150 W. For comparison, the synthesis was performed under both microwave irradiation and classical heating conditions at 100 °C. Microwave irradiation promoted the reaction more efficiently:The reaction time was shorted from six hours (under the traditional heating conditions) to only four minutes. Additionally, the yield was increased from 72% to 97%. [35]. The same group also reported the synthesis of a new series of azapodophyllotoxin analogues containing a furo[3,4-b]quinoline motif (151) using a facile MCR consisting of an aldehyde, tetronic acid, and an enaminone in glacial acetic acid, using microwave irradiation. Optimization of reaction conditions by screening different solvents, reaction temperatures, and microwave power levels revealed that the best yields were obtained at 300 W and 100 °C using acetic acid as a solvent and no catalyst (Scheme 19). Comparing microwave conditions with conventional heating showed that microwave-irradiated reactions proceed much faster and have higher yields [36].
Scheme 18.

Scheme 19.

Using a similar approach, Tu et al. later reported the synthesis of N-substituted furo[3,4-b]indeno[2,1-e]pyridine analogues of azapodophyllotoxin 152 through an MCR of aldehyde, 2H-indene-1,3-dione, and 4-(arylamino)-furan-2(5H)-one using ethylene glycol as solvent at 100 °C under microwave irradiation at 200 W without a catalyst (Scheme 20). Although the detailed mechanism of the reaction remains to be fully clarified, the authors speculate that the formation of N-substituted furo[3,4-b]indeno[2,1-e]pyridine analogues of aza-podophyllotoxin is due to a reaction sequence of condensation, addition, cyclization, and dehydration [37].
Scheme 20.

A series of new polycyclic-fused isoxazolo[5,4-b]pyridines was obtained by a one-pot tandem reaction under microwave irradiation in water, without the use of additional reagents or catalysts [38]. The three-component equimolar reaction of 4-fluorobenzaldehyde, 3-methylisoxazol-5-amine, and tetronic acid was investigated to establish the feasibility of the strategy and optimize the reaction conditions. Reaction temperatures and different solvents (including ethylene glycol, ethanol, acetic acid and water) were screened using the model reaction and the optimum yields were obtained in water at 120 °C with 200 W of microwave power (Scheme 21). A range of novel structures (153) was reported, with good to excellent yields. The reaction of aromatic aldehydes bearing electron-withdrawing or electron-donating groups proceeded smoothly; however, aliphatic aldehydes were unable to react under similar conditions. Another active methylenic compound, 1,3-indanedione, was examined as a replacement for tetronic acid in the reaction with aromatic aldehydes and 3-methylisoxazol-5-amine and a new series of isoxazolo[5,4-b]pyridines (154) was successfully synthesized (Scheme 21) [38].
Scheme 21.

Most of the reported modifications on azapodophyllotoxins have been performed on rings B and C; the modifications on ring A were not well documented. Recently, Tu et al. reported a microwave-assisted, four-component reaction of dimedone, aromatic aldehydes, and tetronic acid with excess ammonia water. Instead of the suspected product, 156, the presence of aromatic aldehydes with electron-withdrawing groups led to product 155 in high yields. In contrast, the same reaction with electron-donating aldehydes gave the product 157, which suggested that tetronic acid did not take part in the reaction (Scheme 22) [39].
Scheme 22.

2.3. Ultrasound-irradiated Synthesis
The application of ultrasound in organic synthesis has increased over the last couple of decades. Ultrasound-mediated synthesis is usually more convenient and easily controlled compared to traditional methods and has often resulted in shorter reaction times, utilizing milder reaction conditions. Tu et al. reported the synthesis of furo[3′,4′:5,6]pyrido[2,3d]pyrimidine 158 and indeno[2′,1′:5,6] pyrido[2,3-d]pyrimidine 159 derivatives via a three-component condensation of aldehydes, 2,6- diaminopyrimidine-4(3H)-one, and tetronic acid or 1,3-indanedione in ethylene glycol, using ultrasonic irradiation without a catalyst (Scheme 23). The reactions using ethylene glycol as the solvent resulted in higher yields and shorter reaction times than those using AcOH, DMF, or EtOH [40].
Scheme 23.

3. BIOLOGICAL ACTIVITY OF AZAPODOPHYLLOTOXINS
Podophyllotoxin (1) has been shown to isomerize into inactive picropodophyllotoxin via epimerization at the C2 center under physiological conditions. Tomioka et al. replaced the sp3 C2 center with sp2 nitrogen, which is incapable of epimerizing at the C2 center, and studied the antitumor activity of the resulting racemic and optically pure azapodophyllotoxin derivatives [17–19]. In vitro growth inhibition data of these compounds for KB cells is provided in Table 1, and indicate promising growth inhibition by the racemic compounds. Some of these compounds also exhibited promising in vivo activity against P-388 mouse (T/C 145 (10) and 170 (11)). It is also important to note that the data indicate that the cytotoxicity of azadeoxypodophyllotoxin relies primarily on the absolute configuration at the Cl position, not that at the C3 position [18].
Table 1.
Growth Inhibition of KB Cells by Azapodophyllotoxin Derivatives
| Entry | Compound | ED50 (μg/mL) |
|---|---|---|
| 1 | rac 10 | <0.3 |
| 2 | rac 12 | <0.3 |
| 3 | rac 28 | 4.55 |
| 4 | rac 29 | <0.3 |
| 5 | rac 26 | 0.62 |
| 6 | rac 27 | 2.75 |
| 7 | (−)-10 | <0.3 |
| 8 | (+)-11 | <0.3 |
| 10 | (−)-11 | 38.5 |
Hitotsuyanagi et al. replaced the C4 methylene group of 1 with oxygen, which would not alter the stereochemistry of the molecule, but could change its polarity. The resulting compound 63 showed significant activity (IC50 0.031 μg/mL) against adriamycin-resistant P-388 leukemia cells [22]. Later, the same group sythesized diazapodophyllotixin analogues and studied their cytotoxicity and antitumor activity. All analogues of 69–73 were assayed for in vitro cytotoxicity against L1210 murine leukemia cells and for antitumor activity against P-388 leukemia cells in mice; the results were compared with podophyllotoxin (1) as control. Podophyllotoxin (1) was found to be the most cytotoxic in vitro against L1210 leukemia cells; however, it showed no in vivo activity against P-388 leukemia cells. In contrast, both the cis analogue 69a and the trans analogue 70a showed potent antitumor activity. Analogues 69b–d and 70b–d also showed significant activity (Table 2). These results suggest that the ethylenedioxy group on ring B and the 3,5-dimethoxy group on ring E are important for activity. The N-amino analogues 71 and 72 retained activity both in vitro and in vivo. In general, the trans analogues were more cytotoxic than the corresponding cis analogues. This could be attributed to the former being topologically more similar to podophyllotoxin (1) than the latter, which would also explain the reduced activity of the dehydrated analogue 73. However, it is still not clear whether the improved antitumor activity of analogues in vivo is due to pharmacokinetics or other factors.
Table 2.
Antitumor and Cytotoxicity Activity of Diazapodophyllotoxin Analogues 69–73
| Compds | Antitumor activity against P-388 leukemia T/C (%) @ different doses (mg/kg/injection)a | Cytotoxicity L1210 IC50 (μg/mL) | ||||||
|---|---|---|---|---|---|---|---|---|
| 2.5 | 5 | 10 | 25 | 50 | 100 | 250 | ||
| 1 | 111 | 109 | 111 | <85 | 0.0036 | |||
| 69a | 117 | 144 | 161 | 239 | 0.42 | |||
| 69b | 123 | 137 | 159 | 214 | 0.22 | |||
| 69c | 119 | 131 | 153 | 165 | 233 | <85 | 0.052 | |
| 69d | 117 | 133 | 150 | 172 | 94 | <85 | 0.080 | |
| 70a | 111 | 124 | 128 | 150 | 161 | 172 | >326 | 0.050 |
| 70b | 110 | 139 | 156 | 206 | 206 | 0.16 | ||
| 70c | 139 | 144 | 156 | 177 | <85 | 0.055 | ||
| 70d | 114 | 131 | 147 | 199 | <85 | 0.028 | ||
| 71 | 111 | 122 | 147 | 147 | 161 | 0.75 | ||
| 72 | 104 | 109 | 114 | 134 | 153 | <85 | 0.090 | |
| 73 | 104 | 109 | 109 | 111 | 109 | 144 | 0.54 | |
single i.p. treatment on day 1. T/C, median survival time of test animals/median survival time of control animals; 125% or above considered active; <85% considered toxic.
The activities of analogues 69a and 70a, which showed the most promising activity in terms of their T/C values, were further investigated using vincristine-resistant P388 leukemia (P388/VCR) and B-16 melanoma cells (Table 3). Interestingly, 1 showed only marginal (T/C = 125%) activity against the P388/VCR cells and was inactive against the B16 melanoma cells, while 69a and 70a showed significant activity against both P388/VCR and B16 melanoma cells. The mode of action of 69a and 70a was examined by assessing the effects of these compounds on an assembly of microtubules prepared from bovine brain. The concentrations of 1, 69a, and 70a necessary to inhibit microtubule assembly by 50% were 0.13, 1.7, and 0.42 μg/mL, respectively, which correlated to their in vitro cytotoxicity against L1210 leukemia cells (Table 2). These results suggest that incorporation of heteroatoms within the podophyllotoxin core structure could be a possible approach for producing more promising analogues [22].
Table 3.
Antitumor Activity of Podophyllotoxin and its Diaza-analogues Against P-388/VCR Leukemia and B16 Melanoma Cells
| Compds | P-388/VCR T/C (%)a @ different doses (mg/kg/injection) | B16 melanoma T/C (%)b @ different doses (mg/kg/injection) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2.5 | 5 | 10 | 25 | 25 | 50 | 100 | |
| 1 | 122 | 125 | <85% | 110 | ||||
| 69a | 137 | <85 | 163 | 190 | ||||
| 70a | 113 | 129 | 152 | 152 | 153 | 197 | ||
Dose given i.p on days 1–5. T/C, median survival time of test animals/median survival time of control animals; 125% or above considered active; <85% considered toxic.
Dose given i.p. on days 1, 5, and 9.
The screening results for in vivo antitumor activity against vincristine-resistant P388 leukemia cells showed that 2,4-diaza-4-deoxypdophyllotoxin exhibit better activity than podophyllotoxin (1) and 2-aza-4-deoxypodophyllotoxin. This suggests that substitution of the carbon atom at position 4 by a nitrogen atom is important for antitumor activity. Hitotsuyanagi et al. synthesized (−)-4-aza-4-deoxypodophyllotoxin (74) in an optically pure form from natural (−)-podophyllotoxin (1) and the biological activity evaluation revealed that both 1 and its 4-aza analogue 74 possess same the antitumor activity against P-388 leukemia cells (IC50 = 0.0050 μg/mL for both) [25]. The same group synthesized the 4-aza-2,3-dehydro-4-deoxypodophyllotoxin analogues 76a–n through quinolines 75a–n and studied their cytotoxicity using P-388 leukemia cells (Table 4) [14]. In general, the quinoline analogues 75a–n showed very weak or no activity. However, 4-aza-justicidin B (75e) showed moderate activity (IC50 = 2.0 μg/mL), which is greater than the reported data for justicidin B, a cytotoxic principal from several plants (IC50 = 3.3 μg/mL against same cell line). In contrast, dihydroquinoline analogues 75a–n showed significant antitumor activity. The screening results revealed the substituent effects of the ring B on the activity of these compounds. Analogues 76a (which has the same 1,3-benzodioxole structure as 1), 76b, and 76g (both of which have a cyclic ring A structure consisting of 1,4-benzo-ioxane and indan, respectively) showed higher activity than the analogues that contain a dimethoxyphenyl (76c), a trimethoxyphenyl (76f), or a fluorene (76h) unit. Analogue 76c, with its two free rotatable methoxy groups, was more than 2700 times less toxic than analogues 76a and 76b with a cyclic ether structure, although the electronic effects of the methylenedioxy, ethylenedioxy, and o-dimethoxy groups are quite similar. Also, while sterically very similar but electronically dissimilar, the indan analogue 76g was only 2.3 times less toxic than analogue 76a. These results suggest that the steric effects are more influential than the electronic effects. The reduced activity of fluorene analogue 76h compared to that of indan analogue 76g also supports this possibility. The same tendency was also seen between 76d and 76i, and between 76e and 76m. A comparison of the 76a–m analogues illustrates some of the substituent effects of ring E. Interestingly, 76l, with an unsubstituted phenyl group as the ring E, was only 2.9 times less toxic than analogue 76a. The results also suggest that an m-methoxy group may either enhance or not influence the activity, while a p-methoxy group reduces the activity. The potency of the analogues was found to be in the order of 76a>76j=76l>76i>76k. The cyclic ketone analogue 76n was about 15 times less toxic than the lactone analogue 76a, indicating that an oxygen atom is more effective than a methylene group at position 12 (Table 4) [14].
Table 4.
Cytotoxicity of Aza-Lignans 75a-n and 76a-n Against P-388 Leukemia Cells
| Compd | IC50 (μg/mL) | Compd | IC50 (μg/mL) |
|---|---|---|---|
| 1 | 0.0043 | ||
| 75a | >100 | 76a | 0.0018 |
| 75b | 80 | 76b | 0.0017 |
| 75c | >100 | 76c | 4.9000 |
| 75d | 39 | 76d | 0.7600 |
| 75e | 2 | 76e | 0.7700 |
| 75f | 29 | 76f | 2.6000 |
| 75g | >100 | 76g | 0.0041 |
| 75h | 63 | 76h | 0.9200 |
| 75i | 40 | 76i | 0.0480 |
| 75j | >100 | 76j | 0.0053 |
| 75k | >100 | 76k | 0.1300 |
| 75l | 60 | 76l | 0.0053 |
| 75m | >100 | 76m | 0.0300 |
| 75n | 71 | 76n | 0.0280 |
It is importat to note that podophyllotoxin (1) possesses four chiral centers on the ring C carbon atoms, its 1,2-cis-2,3-trans system provides the two aromatic rings with an appropriate topology, and that the absolute configuration at the C-1 center is critical for activity expression [18]. On the contrary, analogues 76a and 76b, with one chiral center, are more than twice as cytotoxic as 1, despite being racemic. A comparison of the crystal structure of podophyllotoxin (1) and the MM2*10 energy-minimized structure of analogue 76a also revealed that the two aromatic rings in analogue 76a well mimic the topology of those in 1, which could account for its potent activity [14].
Magedov et al. implemented a bioisosteric replacement of the methylenedioxybenzene subunit of podophyllotoxin with a pyrazole moiety to synthesize tetracylcic dihydropyridopyrazoles 78–110 and evaluated their antiproliferative activity against three cancer cell lines: HeLa (human cervical), MCF-7/AZ (breast adenocarcinomas), and Jurkat (T-cell leukemia) (Table 5) [28]. Podophyllotoxin 1 and its 4-aza-2,3-didehydro analogues 5 and 7 were also tested for comparision. The pyrazole moiety of the dihydropyridopyrazole was optimized by putting different substitutents on this ring (compounds 78–83). Screening results showed that 83 exhibited potency and activity profiles similar to 76a and 76c, indicating that the alkoxybenzene ring can be replaced by the pyrazole moiety without loss of potency. However, the potency of podophyllotoxin (1) remained superior to all of them. The analogues with larger substituents on their pyrazole rings (78–81) and the methylated pyrazole 82 exhibited more limited activity than 83. Optimization of ring E revealed that, in addition to the significant antiproliferative and apoptosis-inducing activity of 83 (which possesses a trimethoxy ring E just like podophyllotoxin), analogues possessing the m-bromo substitution (91–96) were the most potent. Comparing the profiles of analogues 91–96 reveals that the potency remains unchanged, regardless of whether there are other substituents at the 4' or 5' position. However, m-chloro, m-fluoro, and o- or p-bromo–substituted analogues were comparatively less active. Also, the heterocyclic analogues 104–110 were found to be either inactive or much less potent (Table 5). In general, the level of apoptosis induction in Jurkat cells showed a trend similar to that of the analogue's antiproliferative potency, with the meta-bromo analogues 91–96 having the highest level, at 50–58%. The GI50 (concentration required for 50% of the effect) values of selected analogues obtained by dose-dependent studies against a HeLa cell line revealed that dihydropyridopyrazoles (83, 91–96) and Itokawa/Takeya's analogues (76a and 76c) have similar potencies, possessing GI50 values in the low micromolar range, while analogue 96 showed submicromolar activity (Table 6). All meta-bromo analogues exhibited activity 35–400 times higher than that of podophyllotoxin (1) [28].
Table 5.
Antiproliferative Activities of Dihydropyridopyrazoles 78–110
| Compd | 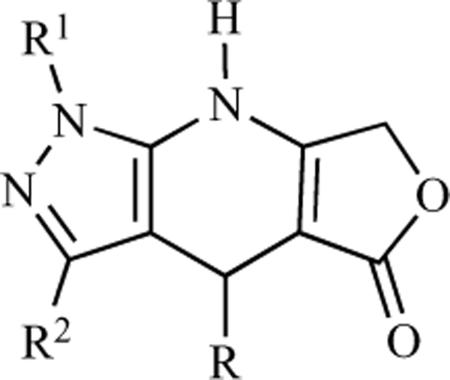 |
% cell viability | % apoptosis Jurkat | ||||
|---|---|---|---|---|---|---|---|
| HeLa | MCF-7/AZ | Jurkat | |||||
| R1 | R2 | R | |||||
| 1 | 19 ± 5 | 55 ± 3 | 18 ± 5 | 54 ± 2 | |||
| 2 | 91 ± 2 | 76 ± 2 | 75 ± 5 | 4 ± 3 | |||
| 76a | 53 ± 5 | 58 ± 4 | 35 ± 6 | 49 ± 4 | |||
| 76c | 54 ± 6 | 52 ± 3 | 54 ± 1 | 55 ± 4 | |||
| 78 | Ph | 4-Br-Ph | 3,4,5-tri-OMe-Ph | 73 ± 5 | 90 ± 3 | 99 ± 1 | 2 ± 1 |
| 79 | 4-Me-Ph | Me | 3,4,5-tri-OMe-Ph | 77 ± 4 | 71 ± 5 | 77 ± 2 | 23 ± 1 |
| 80 | H | 4-Br-Ph | 3,4,5-tri-OMe-Ph | 67 ± 4 | 98 ± 3 | 78 ± 7 | 5 ± 1 |
| 81 | iPr | Me | 3,4,5-tri-OMe-Ph | 83 ± 5 | 99 ± 0 | 77 ± 9 | 4 ± 0 |
| 82 | Me | Me | 3,4,5-tri-OMe-Ph | 55 ± 3 | 98 ± 1 | 79 ± 4 | 5 ± 1 |
| 83 | H | Me | 3,4,5-tri-OMe-Ph | 50 ± 2 | 58 ± 5 | 47 ± 3 | 55 ± 4 |
| 84 | H | Me | 3,4-di-OMe-Ph | 73 ± 3 | 95 ± 4 | 88 ± 2 | 5 ± 1 |
| 85 | H | Me | 4-OMe-Ph | 73 ± 7 | 82 ± 3 | 94 ± 4 | 11 ± 1 |
| 86 | H | Me | 4-OCF3-Ph | 97 ± 2 | 84 ± 3 | 97 ± 6 | 9 ± 2 |
| 87 | H | Me | 4-SMe-Ph | 45 ± 2 | 95 ± 4 | 87 ± 4 | 14 ± 6 |
| 88 | H | Me | Ph | 74 ± 4 | 84 ± 3 | 87 ± 5 | 10 ± 2 |
| 89 | H | Me | 2-NO2-Ph | 71 ± 4 | 99 ± 2 | 64 ± 1 | 6 ± 1 |
| 90 | H | Me | 4-OH-3-OMe-5-NO2-Ph | 78 ± 3 | 89 ± 5 | 90 ± 2 | 2 ± 1 |
| 91 | H | Me | 3-Br-Ph | 51 ± 3 | 63 ± 4 | 68 ± 3 | 58 ± 2 |
| 92 | H | Me | 3,5-di-Br-4-OH-Ph | 58 ± 7 | 60 ± 4 | 58 ± 3 | 53 ± 0 |
| 93 | H | Me | 3-Br-4-OEt-5-OMe-Ph | 52 ± 2 | 49 ± 4 | 28 ± 5 | 49 ± 1 |
| 94 | H | Me | 4-OAc-3-Br-5-OMe-Ph | 47 ± 2 | 46 ± 2 | 36 ± 6 | 58 ± 1 |
| 95 | H | Me | 3-Br-4-NMe2-Ph | 55 ± 3 | 59 ± 3 | 34 ± 5 | 41 ± 1 |
| 96 | H | Me | 3-Br-4,5-di-OMe-Ph | 16 ± 3 | 47 ± 1 | 29 ± 3 | 56 ± 1 |
| 97 | H | Me | 3-Cl-Ph | 43 ± 4 | 54 ± 3 | 76 ± 4 | 28 ± 4 |
| 98 | H | Me | 3,4-di-Cl-Ph | 88 ± 1 | 56 ± 3 | 71 ± 4 | 33 ± 4 |
| 99 | H | Me | 3-F-Ph | 99 ± 1 | 76 ± 2 | 84 ± 2 | 28 ± 1 |
| 100 | H | Me | 2-Br-Ph | 57 ± 4 | 100 ± 3 | 90 ± 1 | 8 ± 2 |
| 101 | H | Me | 4-Br-Ph | 62 ± 3 | 92 ± 2 | 90 ± 1 | 10 ± 2 |
| 102 | H | Me | Me | 70 ± 4 | 97 ± 2 | 78 ± 5 | 5 ± 1 |
| 103 | H | Me |
|
95 ± 5 | 91 ± 4 | 89 ± 6 | 14 ± 2 |
| 104 | H | Me |
|
58 ± 2 | 100 ± 1 | 94 ± 2 | 6 ± 1 |
| 105 | H | Me |
|
82 ± 4 | 98 ± 3 | 74 ± 4 | 18 ± 1 |
| 106 | H | Me |
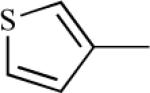
|
98 ± 2 | 100 ± 2 | 82 ± 5 | 6 ± 4 |
| 107 | H | Me |
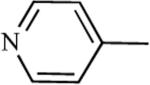
|
94 ± 1 | 100 ± 1 | 92 ± 5 | 4 ± 2 |
| 108 | H | Me |
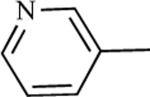
|
69 ± 4 | 96 ± 2 | 88 ± 9 | 11 ± 3 |
| 109 | H | Me |
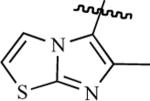
|
84 ± 4 | 100 ± 3 | 85 ± 6 | 7 ± 2 |
| 110 | H | Me |
|
72 ± 6 | 100 ± 4 | 97 ± 3 | 11 ± 2 |
Table 6.
Comparative Antiproliferative Potencies Expressed as GI50
| Compd | GI50 (μM)a HeLa | Compd | GI50 (μM)a HeLa | Compd | GI50 (μM)a HeLa |
|---|---|---|---|---|---|
| 1 | 0.02 | 83 | 5 ± 1 | 94 | 4 ± 1 |
| 2 | 8 ± 2 | 91 | 5 ± 1 | 95 | 6 ± 1 |
| 76a | 6 ± 1 | 92 | 8 ± 2 | 96 | 0.75 ± 0.1 |
| 76c | 6 ± 1 | 93 | 5 ± 1 | 97 | 10 ± 2 |
Concentration required to reduce the viability of HeLa cells by 50%.
Shi et al. recently screened 4-azapodohyllotoxin analogues 136 for anticancer activity using the sulforhodamine B (SRB) method against the human liver cancer cell line HepG2 (Table 7) and found a strong inhibitory effect for most of the analogues [32].
Table 7.
Inhibition Rates of 4-Azapodophyllotoxins 136a-w on HepG2 Cells
| Compd | 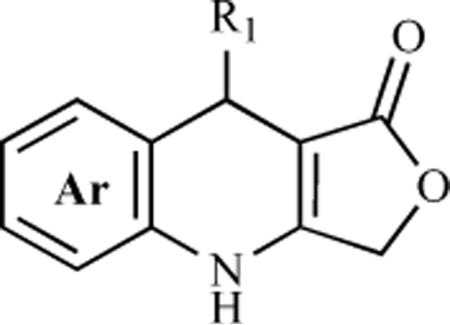 |
Inhibition rate (%) | |
|---|---|---|---|
| Ar | R1 | ||
| 136a | 4-Me-Ph | 4-Br-Ph | 74.63 |
| 136b | 4-Me-Ph | 4-Me-Ph | 80.22 |
| 136c | 4-Me-Ph | 4-Cl-Ph | 72.39 |
| 136d | 4-Me-Ph | 4_NO2-Ph | 77.61 |
| 136e | 4-Me-Ph | 3,4-di-Cl-Ph | 79.97 |
| 136f | 3-Me-Ph | 4-Me-Ph | 82.64 |
| 136g | 3,4-OCH2OC6C3 | 3,4,5-tri-OMe-Ph | 78.73 |
| 136h | 3,4-OCH2OC6C3 | 4-F-Ph | 76.49 |
| 136i | 3,4-OCH2OC6C3 | 4-OMe-Ph | 80.97 |
| 136j | 3,4-OCH2OC6C3 | 4-Br-Ph | 80.60 |
| 136k | 3,4-OCH2OC6C3 | 4-Cl-Ph | 80.22 |
| 136l | naphthalene -1-yl | 4-OMe-Ph | 64.06 |
| 136m | naphthalene -1-yl | 4-Br-Ph | 73.74 |
| 136n | naphthalene -1-yl | 4-Cl-Ph | 78.63 |
| 136o | naphthalene -1-yl | 3,4-OCH2OC6C3 | 76.41 |
| 136p | naphthalene -1-yl | 4-Me-Ph | 82.19 |
| 136q | naphthalene -1-yl | 2,4-di-Cl-Ph | 76.41 |
| 136r | naphthalene -1-yl | thiophen-2-yl | 71.51 |
| 136t | quinolin-6-yl | 4-Cl-Ph | 65.30 |
| 136u | 3-Cl-4-MeC6H3 | 4-Cl-Ph | 74.63 |
| 136v | 3-Cl-4-MeC6H3 | 4-Br-Ph | 85.76 |
| 136w | 3-Cl-4-MeC6H3 | 3,4-di-Cl-Ph | 87.98 |
Podophyllotoxin (1) works by acting on the colchicine binding site of tubulin, resulting in microtubule depolymerization. Colchicine and podophyllotoxin bind to β-tubulin at its interface with α-tubulin and the trimethoxyphenyl nucleus being hidden into the β-subunit. Dynamic NMR and X-ray studies show that, in solution, 1 adopts a single conformation, with its trimethoxyphenyl ring in a quasi-axial position nearly perpendicular to the tricyclic moiety. Giorgi-Renault et al. investigated this point for 4-aza-2,3-didehydropodophyllotoxins by synthesizing two series of novel azapodophyllotoxin derivatives and studying their tubulin polymerization inhibition activity. In the first series, a linker was inserted between the tetracyclic moiety and ring E of 4-aza-1,2-didehydropodophyllotoxins to allow free rotation between the two parts (compounds 137–141 and 143) [33]. The second series involved a conformational restriction of the ring E nucleus via a rigid analogue in which the rotation between the tetracycle and the aryl ring was blocked by the creation of an additional pseudocycle (compounds 132, 134, 135) (Table 8) [31, 33]. The activities of the parent compound 76a and reference compound combretastatin A-4 (159) were also tested for comparison. The reference compounds 76a (ITP = 0.47) and 159 (ITP = 0.72) both showed strong anti-microtubule activity, with IC50 values lower than that of colchicine.
Table 8.
Inhibition of Tubulin Polymerization, Cytotoxicity, and Morphological Effects on EA.hy 926 Endothelial Cells Caused by Azapodophyllotoxin Analogues
| Compd | ITPa | Cytotoxicity: IC50 [uM]b | ||
|---|---|---|---|---|
| B16 | NIH 3T3 | EA.hy926 | ||
| 76a | 0.47 | 0.016 ± 0.001 | 0.15 ± 0.07 | 0.15 ± 0.05 |
| 132 | 1.17 | 0.40 ± 0.01 | 0.5 ± 0.1 | 2.8 ± 1.5 |
| 134 | >40 | >30 | >30 | >30 |
| 135 | 2.89 | 38.4 ± 10.1 | 2.1 ± 0.8 | 8.6 ± 2.4 |
| 137 | >40 | >30 | >30 | >30 |
| 138 | >40 | >10 | >10 | >10 |
| 139 | 0.75 | 0.41 ± 0.21 | 0.08 ± 0.02 | 0.77 ± 0.05 |
| 140 | >40 | 14.1 ± 2.9 | 7.5 ± 2.1 | 19.1 ± 1.8 |
| 141 | >40 | >30 | >30 | >30 |
| 143 | >40 | >30 | >30 | >30 |
| 159 | 0.72 | 0.010 ± 0.003 | 0.020 ± 0.005 | 0.03 ± 0.01 |
Inhibition of tubulin polymerization (ITP) is expressed as the ratio of IC50 compd/IC50 colchicine. IC50 compd is the concentration of compound required to inhibit 50% of the rate of microtubule assembly, and the average IC50 value for colchicine was 0.36 mM under the given conditions.
Concentration of compound corresponding to 50% growth inhibition after a 48-hour incubation.
Among the carbon homologue series, only the N-unsubstituted vinylogue 139 was found to be significantly active (ITP = 0.75). This shows that the insertion of a double bond between the tetraline moiety and the trimethoxyphenyl ring of 76a is tolerable. As the trimethoxyphenyl group of podophyllotoxin is buried within a hydrophobic pocket of β-tubulin, the additional double bond may reinforce the Van der Waals interactions between vinylazapodophyllotoxin 139 and tubulin.The compounds 137, 138, 140, 141, and 143, containing both a spacer group and N-methylation, were inactive for tubulin polymerization inhibition as indicated by their higher ITP values (>40). In contrast, N-methylation was well tolerated with an amino spacer, as aminologue 135 showed significant ITP activity (ITP = 2.89). The imine 134 was found to be inactive compared to aminologue 135 and quinoline 132, which suggests that the hydrogen bond between the amino linker and the lactone carbonyl plays a crucial role for tubulin inhibition activity (Table 8) [33]. The cytotoxicity of these compounds has also been evaluated against a solid-tumor-derived murine B16 melanoma cell line and the EA.hy 926 cell line (an immortalized HUVE line). The results were compared to the compounds' cytotoxicity against a murine fibroblast NIH 3T3 cell line as a normal cell control. The reference compounds 76a and 159 showed significant toxicity, with IC50's in the nanomolar range compared to linker analogues 139, 140, 132, and 135. Similar activity profiles were observed in all three cell lines. Vinylogue 139 and quinoline 132 showed 40-fold lower anti-proliferative effects against B16 cells than those of 159, whereas their anti-microtubule activities were similar. Aminologue 135 displayed high ITP activity associated with poor B16 cytotoxicity, which is considered a good indicator for antivascular activity [41, 42].
4. CONCLUSIONS AND FUTURE PERSPECTIVES
Even after drastically reducing the natural structural complexity found in podophyllotoxin (1), many of the azapodophyllotoxin analogues described above expressed more pronounced antitumor activity than that of 1. Thus, the incorporation of heteroatoms within the podophyllotoxin core structure should constitute a convenient approach for the production of even more promising analogues. Moreover, some of the new compounds are racemic, and thus the separation and testing of individual enantiomers will undoubtedly lead to the discovery of more potent compounds.
The use of MCRs in combination with ultrasound and microwave irradiation has increased the speed of synthesis and the variety of azapodophyllotoxins available to test against a diverse population of carcinomas and other diseases.
Dihydropyridopyrazolyl azapodophyllotoxins are able to induce apoptosis in Jurkat cells on a scale similar to that of podophyllotoxin, while showing virtually no apoptotic activity against noncancerous, nucleated white blood cells. This opens new avenues for the synthesis of cancer-selective azapodophyllotoxins. How azapodophyllotoxin compounds exert their cytotoxic effect, whether through a podophyllotoxin-like antitubulin mechanism, by targeting topoisomerase II, or have a totally independent mode of action, constitutes the subject of further investigation.
The structural complexity of 1, arising from the presence of four stereogenic carbons in ring C has restricted most of the structural activity relationship (SAR) studies to those using derivatives of the parent natural product rather than compounds obtained through de novo chemical synthesis. Such approaches, however, are rather limited by the type of chemistry that 1 can undergo. These issues provide a strong impetus to search for analogues of 1 with simplified structures, which can be prepared via short, synthetic sequences from simple starting materials, such as many of the azapodophyllotoxin derivatives described here. Even if such initial compounds might have diminished cytotoxic potencies compared with the parent cyclolignan, having an easy method to produce carefully designed analogue libraries would lead to more informative SAR studies and expeditious structure optimization.
Fig. (2).
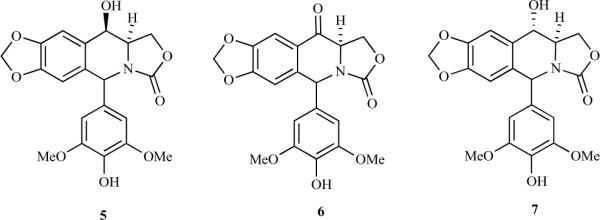
Chiral analogues of podophyllotoxins.
Fig. (3).

(−)-4-aza-4-deoxypodophyllotoxin 74.
Fig. (4).
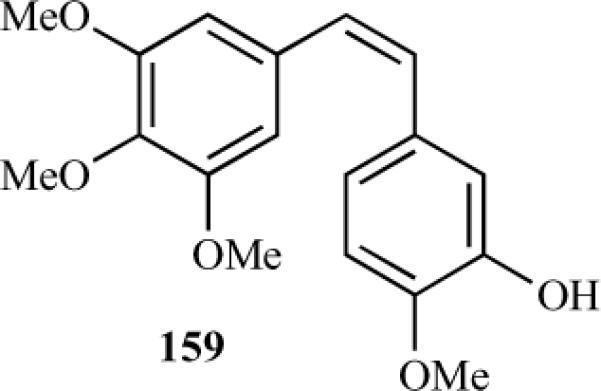
Combretastatin A-4.
ACKNOWLEDGMENTS
The authors are grateful to the NCI Developmental Therapeutics Program for the 60-cell line screening of the azapodophyllotoxin derivatives described in this paper, and for the support of this research from the National Cancer Institute, under contract number HSN261200800001E; the National Institute of General Medical Sciences, under grant number S06-GM008216; and the National Center for Research Resources, under grant number P20 RR-016470 (from the National Institutes of Health). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
REFERENCES
- [1].Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- [2].Feher M, Schmidt JM. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- [3].Imbert TF. Discovery of podophyllotoxins. Biochimie. 1998;80:207–222. doi: 10.1016/s0300-9084(98)80004-7. [DOI] [PubMed] [Google Scholar]
- [4].Bohlin L, Rosen B. Podophyllotoxin derivatives: Drug discovery and development. Drug Discov. Today. 1996;1:343–351. [Google Scholar]
- [5].Wang JC. DNA topoisomerases. Ann. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- [6].Leroy D, Kajava AV, Frei C, Gasser SM. Analysis of etoposide binding to subdomains of human DNA topoisomerase II alpha in the absence of DNA. Biochemistry. 2001;40:1624–1634. doi: 10.1021/bi0019141. [DOI] [PubMed] [Google Scholar]
- [7].Pedersen-Bjergaard J. Radiotherapy- and chemotherapy-induced myelodysplasia and acute myeloid-leukemia. A review. Leukemia Res. 1992;16:61–65. doi: 10.1016/0145-2126(92)90102-d. [DOI] [PubMed] [Google Scholar]
- [8].Felix CA. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim. Biophys. Acta Gene Struct. Expression. 1998;1400:233–255. doi: 10.1016/s0167-4781(98)00139-0. [DOI] [PubMed] [Google Scholar]
- [9].Lazo JS, Li TH, Woo ES, Settineri CE, Allan WP, Yalowich JC. Chemical synthesis and biological activity of a novel fluorescent etoposide derivative. Biochem. Pharmacol. 1997;53:715–722. doi: 10.1016/s0006-2952(96)00906-9. [DOI] [PubMed] [Google Scholar]
- [10].Zhu XK, Guan J, Tachibana Y, Bastow KF, Cho SJ, Cheng HH, Cheng YC, Gurwith M, Lee KH. Antitumor agents. 194. Synthesis and biological evaluations of 4-beta-mono-, -di-, and -trisubstituted aniline-4 '-O-demethyl-podophyllotoxin and related compounds with improved pharmacological profiles. J. Med. Chem. 1999;42:2441–2446. doi: 10.1021/jm990055f. [DOI] [PubMed] [Google Scholar]
- [11].Xiao ZY, Xiao YD, Feng J, Golbraikh A, Tropsha A, Lee KH. Antitumor agents. 213. Modeling of epipodophyllotoxin derivatives using variable selection k nearest neighbor QSAR method. J. Med. Chem. 2002;45:2294–2309. doi: 10.1021/jm0105427. [DOI] [PubMed] [Google Scholar]
- [12].You YJ. Podophyllotoxin derivatives: Current synthetic approaches for new anticancer agents. Curr. Pharm. Des. 2005;11:1695–1717. doi: 10.2174/1381612053764724. [DOI] [PubMed] [Google Scholar]
- [13].Gordaliza M, Castro MA, del Corral JMM, San Feliciano A. Anti-tumor properties of podophyllotoxin and related compounds. Curr. Pharm. Des. 2000;6:1811–1839. doi: 10.2174/1381612003398582. [DOI] [PubMed] [Google Scholar]
- [14].Hitotsuyanagi Y, Fukuyo M, Tsuda K, Kobayashi M, Ozeki A, Itokawa H, Takeya K. 4-aza-2,3-dehydro-4-deoxypodophyllotoxins: Simple aza-podophyllotoxin analogues possessing potent cytotoxicity. Bioorg. Med. Chem. Lett. 2000;10:315–317. doi: 10.1016/s0960-894x(99)00693-9. [DOI] [PubMed] [Google Scholar]
- [15].Gensler WJ, Murthy CD, Trammell MH. Non-enolizable podophyllotoxin derivatives. J. Med. Chem. 1977;20:635–644. doi: 10.1021/jm00215a004. [DOI] [PubMed] [Google Scholar]
- [16].Pearce HL, Bach NJ, Cramer TL. Synthesis of 2-azapodophyllotoxin. Tetrahedron Lett. 1989;30:907–910. [Google Scholar]
- [17].Tomioka K, Kubota Y, Koga K. Synthesis and antitumor-activity of podophyllotoxin aza-analogues. Tetrahedron Lett. 1989;30:2953–2954. [Google Scholar]
- [18].Tomioka K, Kubota Y, Koga K. Design, synthesis, and antitumor activity-absolute configuration relationships of podophyllotoxin aza-analogs. Tetrahedron. 1993;49:1891–1900. [Google Scholar]
- [19].Tomioka K, Kubota Y, Koga K. Efficient stereoselective synthesis of 2-aza-4'-demethylepipodophyllotoxin. J. Chem. Soc., Chem. Commun. 1989:1622–1624. [Google Scholar]
- [20].Van der Eeycken J, Bosmans JP, Vanhaver D, Vandewalle M, Hulkenberg A, Veerman W, Nieuwenhuizen R. The synthesis of 4-desoxy-2-azapodophyllotoxins. Tetrahedron Lett. 1989;30:3873–3876. [Google Scholar]
- [21].Bosmans JP, Vandereycken J, Vandewalle M, Hulkenberg A, Vanhes R, Veerman W. The synthesis of 2-azapodophyllotoxins. Tetrahedron Lett. 1989;30:3877–3880. [Google Scholar]
- [22].Hitotsuyanagi Y, Ichihara Y, Takeya K, Itokawa H. Synthesis of 4-oxa-2-azapodophyllotoxin, a novel analog of the antitumor lignan podophyllotoxin. Tetrahedron Lett. 1994;35:9401–9402. [Google Scholar]
- [23].Hitotsuyanagi Y, Naka Y, Yamagami K, Fujii A, Tahara T. Synthesis of 2,4-di-aza-4-deoxypodophyllotoxin, a new analog of podophyllotoxin possessing antitumor-activity. J. Chem. Soc., Chem. Commun. 1995:49–50. [Google Scholar]
- [24].Hitotsuyanagi Y, Yamagami K, Fujii A, Naka Y, Ito Y, Tahara T. Syntheses and biological properties of novel aza-podophyllotoxin analogs possessing pronounced antitumor-activity. Bioorg. Med. Chem. Lett. 1995;5:1039–1042. [Google Scholar]
- [25].Hitotsuyanagi Y, Kobayashi M, Morita H, Itokawa H, Takeya K. Synthesis of (−)-4-aza-4-deoxypodophyllotoxin from (−)-podophyllotoxin. Tetrahedron Lett. 1999;40:9107–9110. [Google Scholar]
- [26].Hitotsuyanagi Y, Kobayashi M, Fukuyo M, Takeya K, Itokawa H. A facile synthesis of the 4-aza-analogs of 1-arylnaphthalene lignans - Chinensin, justicidin B, and Taiwanin C. Tetrahedron Lett. 1997;38:8295–8296. [Google Scholar]
- [27].Tratrat C, Giorgi-Renault S, Husson HP. A multicomponent reaction for the one-pot synthesis of 4-aza-2,3-didehydropodophyllotoxin and derivatives. Org. Lett. 2002;4:3187–3189. doi: 10.1021/ol0200908. [DOI] [PubMed] [Google Scholar]
- [28].Magedov IV, Manpadi M, van Slambrouck S, Steelant WFA, Rozhkova E, Przheval'skii NM, Rogelj S, Kornienko A. Discovery and investigation of antiproliferative and apoptosis-inducing properties of new heterocyclic podophyllotoxin analogues accessible by a one-step multicomponent synthesis. J. Med. Chem. 2007;50:5183–5192. doi: 10.1021/jm070528f. [DOI] [PubMed] [Google Scholar]
- [29].Shi CL, Shi DQ, Kim SH, Huang ZB, Ji SJ, Ji M. A novel and efficient one-pot synthesis of furo[3 ',4 ': 5,6]pyrido[2,3-c]pyrazole derivatives using organocatalysts. Tetrahedron. 2008;64:2425–2432. [Google Scholar]
- [30].Madec D, Mingoia F, Prestat G, Poli G. N-substituted tetronamides as ambident nucleophilic building blocks for the synthesis of new 4-aza-2,3-didehydropodophyllotoxins. Synlett. 2008:1475–1478. [Google Scholar]
- [31].Labruere R, Helissey P, Desbene-Finck S, Giorgi-Renault S. Design and effective synthesis of the first 4-aza-2,3-didehydropodophyllotoxin rigid aminologue: A N-methyl-4-[(3,4,5-trimethoxyphenyl)amino)]-1,2-dihydroquinoline-lactone. J. Org. Chem. 2008;73:3642–3645. doi: 10.1021/jo800166b. [DOI] [PubMed] [Google Scholar]
- [32].Shi CL, Wang JX, Chen H, Shi DQ. Regioselective synthesis and in vitro anticancer activity of 4-aza-podophyllotoxin derivatives catalyzed by L-proline. J. Comb. Chem. 2010;12:430–434. doi: 10.1021/cc100003c. [DOI] [PubMed] [Google Scholar]
- [33].Labruere R, Gautier B, Testud M, Seguin J, Lenoir C, Desbene-Finck S, Helissey P, Garbay C, Chabot GG, Vidal M, Giorgi-Renault S. Design, synthesis, and biological evaluation of the first podophyllotoxin analogues as potential vascular-disrupting agents. Chemmedchem. 2010;5:2016–2025. doi: 10.1002/cmdc.201000305. [DOI] [PubMed] [Google Scholar]
- [34].Kumar A, Alegria AE. Synthesis of novel functionalized 4-aza-2,3-didehydropodophyllotoxin derivatives with potential antitumor activity. J. Heterocycl. Chem. 2010;47:1275–1282. doi: 10.1002/jhet.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tu SJ, Zhang Y, Jiang B, Jia RH, Zhang JY, Zhang JP, Ji SJ. One-pot synthesis of N-substituted azapodophyllotoxin derivatives under microwave irradiation. Synthesis. 2006:3874–3882. [Google Scholar]
- [36].Tu SJ, Zhang Y, Zhang JY, Jiang B, Jia RH, Zhang JP, Ji SJ. A simple procedure for the synthesis of 4-aza-podophyllotoxin derivatives in water under microwave irradiation conditions. Synlett. 2006:2785–2790. [Google Scholar]
- [37].Shi F, Zhang G, Zhang Y, Ma N, Jiang B, Tu SJ. A facile and efficient synthesis of N-substituted furo[3,4-b]indeno[2,1-e]pyridine analogues of azapodophyllotoxin via microwave-assisted multicomponent reactions. J. Heterocyc. Chem. 2009;46:965–970. [Google Scholar]
- [38].Tu SJ, Zhang XH, Han ZG, Cao XD, Wu SS, Yan S, Hao WJ, Zhang G, Ma N. Synthesis of isoxazolo[5,4-b]pyridines by microwave-assisted multi-component reactions in water. J. Comb. Chem. 2009;11:428–432. doi: 10.1021/cc800212v. [DOI] [PubMed] [Google Scholar]
- [39].Shi F, Ma N, Zhang Y, Zhang G, Jiang B, Tu SJ. Unexpected and green synthesis of azapodophyllotoxin derivatives via microwave-assisted multicomponent reactions in ammonia water. Synth. Commun. 2010;40:235–241. [Google Scholar]
- [40].Tu SJ, Cao LJ, Zhang Y, Shao QQ, Zhou DX, Li CM. An efficient synthesis of pyrido[2,3-d]pyrimidine derivatives and related compounds under ultrasound irradiation without catalyst. Ultrason. Sonochem. 2008;15:217–221. doi: 10.1016/j.ultsonch.2007.03.002. [DOI] [PubMed] [Google Scholar]
- [41].Schwartz EL. Antivascular actions of microtubule-binding drugs. Clinical Cancer Res. 2009;15:2594–2601. doi: 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gaya AM, Rustin GJS. Vascular disrupting agents: A new class of drug in cancer therapy. Clin. Oncol. 2005;17:277–290. doi: 10.1016/j.clon.2004.11.011. [DOI] [PubMed] [Google Scholar]