

Abstract
Globoid cell leukodystrophy (GLD) (Krabbe disease) is an autosomal recessive, degenerative, lysosomal storage disease caused by a severe loss of galactocerebrosidase (GALC) enzymatic activity. Of the >70 disease-causing mutations in the GALC gene, most are located outside of the catalytic domain of the enzyme. To determine how GALC mutations impair enzymatic activity, we investigated the impact of multiple disease-causing mutations on GALC processing, localization, and enzymatic activity. Studies in mammalian cells revealed dramatic decreases in GALC activity and a lack of appropriate protein processing into an N-terminal GALC fragment for each of the mutants examined. Consistent with this, we observed significantly less GALC localized to the lysosome and impairment in either the secretion or reuptake of mutant GALC. Notably, the D528N mutation was found to induce hyperglycosylation and protein misfolding. Reversal of these conditions resulted in an increase in proper processing and GALC activity, suggesting that glycosylation may play a critical role in the disease process in patients with this mutation. Recent studies have shown that enzyme inhibitors can sometimes “chaperone” misfolded polypeptides to their appropriate target organelle, bypassing the normal cellular quality control machinery and resulting in enhanced activity. To determine whether this may also work for GLD, we examined the effect of α-lobeline, an inhibitor of GALC, on D528N mutant cells. After treatment, GALC activity was significantly increased. This study suggests that mutations in GALC can cause GLD by impairing protein processing and/or folding and that pharmacological chaperones may be potential therapeutic agents for patients carrying certain mutations.
Introduction
Globoid cell leukodystrophy (GLD), also known as Krabbe disease, is a devastating neurodegenerative disorder (Wenger et al., 2001). It is inherited as an autosomal recessive trait and is characterized by a deficiency in galactocerebrosidase (GALC) (EC 3.2.1.46) enzymatic activity (Suzuki and Suzuki, 1970). GALC enzymatic activity is predominantly localized in lysosomes, where it is essential for normal catabolism of galactolipids, including a major myelin component, galactocerebroside, and psychosine. GALC deficiency results in abnormal accumulation of psychosine, which is cytotoxic to oligodendrocytes and Schwann cells (Nagara et al., 1986; Tanaka et al., 1988). Loss of these myelin-forming cells causes demyelination in both the CNS and PNS during early developmental stages (Seitelberger, 1981; Kobayashi et al., 1988). Psychosine accumulation has also been proposed to cause axonal degeneration in the CNS and PNS in the twitcher mouse model of GLD, possibly by altering membrane microdomains (rafts) and some of the associated downstream signaling events (Galbiati et al., 2007; White et al., 2009).
To date, >70 disease-causing mutations in the GALC gene have been identified, many of which occur in compound heterozygote patterns in patients (De Gasperi et al., 1996; Furuya et al., 1997; Wenger et al., 1997, 2000; Fu et al., 1999; Selleri et al., 2000; Xu et al., 2006; Lissens et al., 2007). It has been difficult to establish genotype–phenotype relationships for GLD patients given dramatically varied clinical courses, even between individuals with similar or identical genotypes. To more effectively treat patients with diverse disease states, a more detailed understanding of individual GALC mutations must be established. The GALC gene was cloned in 1993 and the available sequence information provides a framework for studying GLD at the molecular level (Chen et al., 1993; Sakai et al., 1994). The precursor form of GALC contains 669 aa and is processed in lysosomes into two fragments, an N-terminal fragment (50 kDa) and a C-terminal fragment (30 kDa) (Nagano et al., 1998). GALC enzymatic activity in vitro has been correlated to the amount of the N-terminal (50–53 kDa) fragment present in a partially purified GALC fraction from human urine (Chen and Wenger, 1993). Little is known, however, about the molecular basis of the processing and the endocytosis of the GALC precursor into its lysosomal form. This information may help determine how disease-causing mutations impair the function of GALC at the molecular level, as a large number of disease-causing mutations are located outside of the catalytic domain of the enzyme but nonetheless cause substantial reductions (>95%) in residual enzymatic activity.
Herein, we focused on three mutations reported to cause GLD when inherited in the homozygous state: the D528N, I234T, and L629R. The D528N mutation has been reported as the primary mutation responsible for the high incidence of infantile GLD (1 in 100–150 live births) in two Moslem Arab villages near Jerusalem (Rafi et al., 1996). The I234T mutation was initially identified in a Greek GLD patient with disease onset at 28 months of age (De Gasperi et al., 1996). The L629R mutation was initially identified in a German GLD patient with symptom onset at 8 years of age (Jardim et al., 1999). These mutations are typically identified in a homozygous state, although the D528N mutation appears to always present with a common polymorphism, I546T, in GLD patients. Expression studies in COS-1 cells show that each of these mutations results in a substantial reduction in GALC activity compared with cells that express wild-type (WT) GALC (De Gasperi et al., 1996; Rafi et al., 1996; Jardim et al., 1999). In this study, we analyzed the effects of these mutations on GALC intracellular processing, secretion, uptake, and subcellular localization in mammalian cell lines. Furthermore, we specifically investigated the potential molecular mechanism by which the D528N mutation impairs GALC function. Finally, we describe the identification and use of the first reported GALC pharmacological chaperone (PC), α-lobeline, which partially rescues the impaired GALC function of the D528N mutant. We expect that these and similar studies may lead to the development of targeted therapeutics to restore GALC activity in GLD patients.
Materials and Methods
Cloning and construction of human GALC expression vectors.
We purchased full-length, human, wild-type GALC cDNA from Origene. The cDNA was amplified by Phusion high-fidelity DNA polymerase (New England Biolabs) according to the manufacturer's directions. Primers included an EcoRI site-containing forward primer (EcoRI-hGalc-START-F, 5′-CTCTCGGAATTCTGGCAACGCCGAGCGAAAGCTATGACTG-3′) and an XhoI site-containing reverse primer (XhoI-hGalc-noSTOP-R, 5′-CCTGTTAACTCGAGGCGTGTGGCTTCCACAAGAAAGTTGTC-3′). The amplified cDNA was inserted into the pcDNA4/V5-His A vector (Invitrogen) using standard procedures. DNA sequencing was performed by the Mayo Clinic Molecular Biology Core Facility to ensure that no random mutations were introduced during the cloning process. Site-directed mutagenesis was performed using the QuikChange II XL site-directed mutagenesis kit (Stratagene). The duplex primers (Integrated DNA Technologies) designed for generating the I234T, D528N, L629R, S530A, and D528N+S530A mutant constructs were as follows: T701C_forward, 5′-GTGGGTTGATGTTACAGGGGCTCATTATCC-3′; T701C_reverse, 5′-GGATAATGAGCCCCTGTAACATCAACCCAC-3′; G1582A_forward, 5′-CATTACGTGGGCTGCCAATGCATCCAACAC-3′; G1582A_reverse, 5′-GTGTTGGATGCATTGGCAGCCCACGTAATG-3′; T1886G_forward, 5′-CGCCTCTGGCATGCGGAATGACAAGTC-3′; T1886G_reverse, 5′-GACTTGTCATTCCGCATGCCAGAGGCG-3′; T1588G_forward, 5′-GTGGGCTGCCGATGCAGCCAACACAATCAG-3′; T1588G_reverse, 5′-CTGATTGTGTTGGCTGCATCGGCAGCCCAC-3′; G1582A+T1588G_forward, 5′-GTGGGCTGCCAATGCAGCCAACACAATCAG-3′; G1582A+T1588G_reverse, 5′-CTGATTGTGTTGGCTGCATTGGCAGCCCAC-3′. WT GALC cDNA, subcloned into the pcDNA4/V5-His vector, was used as the template for site-directed mutagenesis. All procedures were performed according to the manufacturer's directions. DNA sequencing was performed on all clones to ensure that no random mutations were introduced and that the desired mutation was present.
Cell culture.
COS-1 cells (a monkey kidney, fibroblast-like cell line) and H4 cells (a human, CNS-derived cell line) were purchased from American Type Culture Collection. Both cell lines were cultured in Opti-MEM medium supplemented with 5% (v/v) fetal bovine serum (FBS), penicillin (100 IU/ml), and streptomycin (100 μg/ml). OLI-neu, an immortalized oligodendrocyte precursor cell line (Jung et al., 1995), was cultured in Opti-MEM medium supplemented with 5% (v/v) FBS, N2 supplement (1×), 500 pm tri-iodo-thyronine, and 5 μg/ml insulin. Culture media and all supplements were purchased from Invitrogen. The cells were maintained in a humidified incubator at 37°C and 5% CO2. For the GALC uptake study (see Figs. 2, 6), OLI-neu cells were seeded at 70% density onto six-well culture plates precoated with poly-l-lysine and were cultured in reduced serum medium (1% FBS) for 1 d before adding the GALC-containing media.
Figure 2.

Analysis of the secretion and endocytosis of WT and mutant GALC. Conditioned medium was collected from H4 cells stably transfected with WT GALC, mutant GALC (I234T, L629R, and D528N), or empty vector (Mock), and was then applied to OLI-neu oligodendrocyte precursor cells for 24 h to determine the efficiency of GALC uptake. A, Western blot (anti-V5 epitope tag) showing detectable levels of GALC protein in conditioned media from WT, I234T, and D528N GALC-transfected H4 cells. Secreted GALC was undetectable in conditioned media from empty vector (mock)-transfected and L629R GALC-transfected cells. B, Western blot (CL1021AP chicken anti-GALC) of lysates from recipient OLI-neu cells after incubation with conditioned medium from H4 cells expressing WT or mutant GALC. C, Summary of GALC activity determined by colorimetric assay from the H4 input media (A) and from the Oli-neu recipient lysates (B). Uptake index is defined as 100 times the ratio of the GALC activity measured in the cell lysate after uptake compared with the GALC activity in the corresponding input medium. The data in the table represent the mean values ± SE of triplicate samples in each group. GALC secretion and uptake were significantly decreased for each mutant compared with WT GALC (*p < 0.05).
Figure 6.
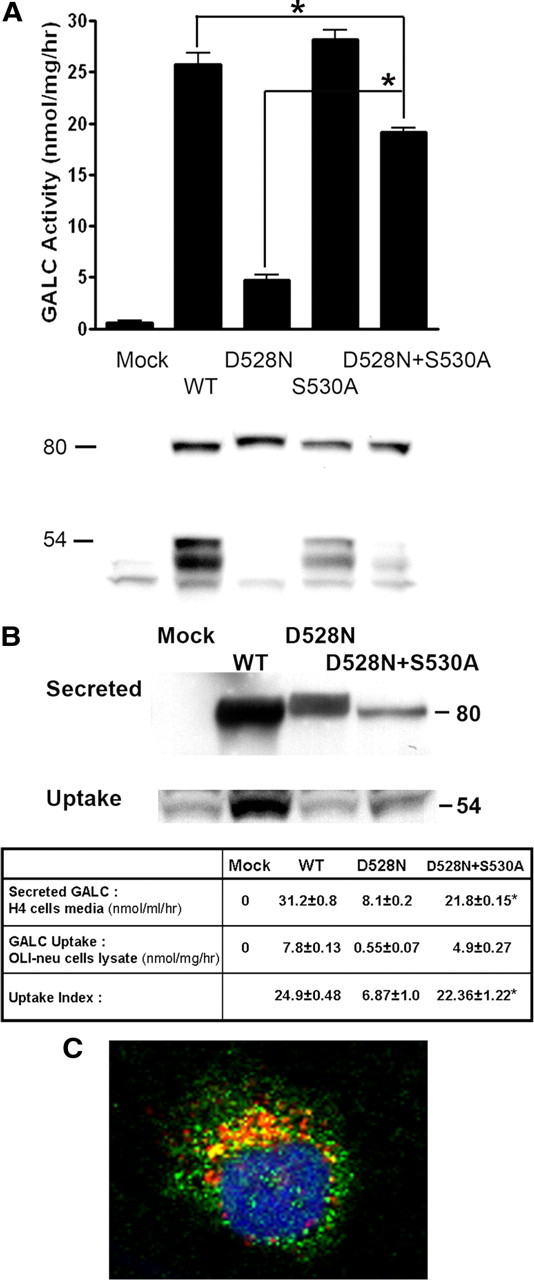
Expression study of D528N mutant GALC in the presence or absence of glycosylation at position 528. Partial rescue in D528N GALC function, processing, secretion, endocytosis, and lysosomal localization was observed in the absence of the glycosylation motif that is introduced with the D528N mutation. A, GALC activity in lysates from COS-1 cells transfected with empty vector (mock), WT or mutant GALC (D528N, S530A or D528N+S530A) (top, graph). Data plotted represent the mean ± SE of at least triplicate samples in each group. GALC activity was significantly increased in cells transfected with the double mutant D528N+S530A, compared with the D528N mutant alone (*p < 0.05). The same lysates were analyzed by Western blot using the CL1475 rabbit anti-GALC antibody (bottom, immunoblot). B, H4 cells stably transfected with WT and mutant GALC (D528N or D528N+S530S) were used to determine the influence of glycosylation on GALC secretion and uptake. Conditioned medium from the transfected H4 cells was applied to OLI-neu cells for 24 h. Media containing the secreted GALC protein (B, top immunoblot) and lysates from recipient OLI-neu cells (B, bottom immunoblot) were analyzed by Western blot using the anti-V5 antibody and the chicken anti-GALC antibody, CL1021AP, respectively. GALC activities from the input cell medium and from the recipient OLI-neu cell lysate were determined and summarized in table format (B, table). Uptake index is defined as 100 times the ratio of the GALC activity measured in the cell lysate after uptake compared with the GALC activity in the corresponding input medium. The data in the table represent the mean ± SE of triplicate samples in each group. *p < 0.05, comparison between the D528N GALC and the D528N+S530A GALC within the same row. C, The subcellular localization of the D528N+S530A mutant GALC expressed in H4 cells was examined by confocal microscopy. Localization of GALC in lysosomes was analyzed by double-immunofluorescent staining using CL1475 rabbit antibody against GALC and anti-lamp 2 to label lysosomes, respectively. GALC is in green. Lamp 2 is in red. Nucleus (DAPI) is in blue. Magnification, 40×. The images are representative of a field (n = 3) of one of three independent experiments.
Transfection and cell harvesting.
Subconfluent (∼8 × 105) densities of COS-1 or H4 cells were seeded onto six-well culture plates 1 d before transfection. Cell transfection using the Lipofectamine 2000 reagent (Invitrogen) was performed according to the manufacturer's directions. Plasmids including empty vector (pcDNA4/V5-His A), wild-type GALC cDNA, and GALC containing the disease-causing mutations were purified to transfection grade using an EndoFree plasmid maxi kit (QIAGEN). For transiently transfected COS-1 cells, cells were harvested at 48 h after transfection. To establish stable, transfected H4 cells, 10% of the transfected cells were passed to new dishes, which contained zeocin (400 μg/ml; Invitrogen)-supplemented growth medium. The transfected H4 cells were selected for ∼3 weeks until zeocin-resistant stable colonies were established. Polyclonal stable H4 cell lines expressing wild-type GALC or GALC mutants were either frozen down for storage or propagated for experiments. Harvested cells were prewashed three times with PBS, pH 7.4, and lysed in the M-Per reagent (Pierce) supplemented with a protease inhibitor mixture (Sigma-Aldrich) for 15 min at 4°C. Cell medium was collected and centrifuged at 500 × g for 5 min before assays. The protein concentration in the lysate was determined by BCA protein assay (Pierce) to normalize protein amounts for Western blot and GALC enzymatic activity assays.
Western blot analysis.
One hundred micrograms of total protein from cell lysates or 20 μl of cell medium was loaded per lane on a 10% Novex Tris-glycine precast gel (Invitrogen). Protein standards were purchased from New England Biolabs. We performed SDS-PAGE, protein transblotting, and antibody staining using standard methodology (Yager et al., 2002). We used the following antibodies: affinity-purified anti-GALC rabbit polyclonal antibody (CL1475) at 1:1000 (Lee et al., 2005); affinity-purified anti-GALC chicken polyclonal antibody (CL1021AP) at 1:1000; anti-V5 epitope tag antibody at 1:1000 (Abcam). Secondary antibodies included an anti-rabbit IgG horseradish peroxidase-linked antibody (GE Healthcare) and an anti-chicken IgY horseradish peroxidase-linked antibody (Sigma-Aldrich). Both were used at 1:5000 dilution.
GALC enzymatic activity assay.
GALC activity assays were performed by modifying the method described previously by Gal et al. (1977). Duplicates from each sample, containing 50 μg of total protein or 17.5 μl of cell medium, were mixed with a reaction mixture [sodium citrate solution at pH 4, sodium taurocholate-oleic acid solution and substrate, 2-hexadecanoylamino-4-nitrophenyl-β-d-galactopyranoside (HNG)]. The reaction components were mixed on a 96-well, round-bottom plate and incubated at 37°C at a time period from 30 min to 16 h depending on the GALC activity of the samples. After incubation, stop solution and absolute ethanol were added and mixed followed by centrifugation (3220 × g, 10 min) to remove precipitates. The absorbance of the supernatant was measured at 410 nm using a SpectraMax M5e microplate reader (Molecular Devices). GALC-specific activity was calculated based on 0.0712 absorbance units measured at 410 nm being equal to 1 nmol of hydrolyzed HNG product in 175 μl.
Identification of α-lobeline as an enzyme inhibitor to GALC.
The described GALC enzymatic activity assay was applied as the screening method to identify GALC inhibitors. A mixture solution containing the reaction buffer, HNG substrate, and recombinant human GALC protein (0.1 μg per reaction), was prepared immediately before it was added to tested drugs (GenPlus drug library; Microsource) or vehicle controls [DMSO, 4% (v/v)]. The final concentration of the drug in each reaction was 0.4 mm and each drug was tested in triplicate wells. The procedures to produce GALC protein were the same as described previously (Lee et al., 2005). Instead of incubating for 4 h, the reaction was incubated for 30 min. Tested drugs that showed a >25% inhibition of GALC activity relative to the control were selected as hits from the primary screen. A dose–response assay was performed to reconfirm the effect of the hits. α-Lobeline (Sigma-Aldrich) was prepared as a 20 mg/ml (60 mm) stock solution. It was filtered sterile and stored under −20°C before use.
Confocal immunocytofluorescence analysis.
Subconfluent densities (∼20%) of H4 or H4-GALC cells were seeded on glass coverslips in a 35 mm culture dish 2 d before staining. Under room temperature conditions, the cells were washed three times with prewarmed PBS and were fixed in 4% paraformaldehyde for 15 min. Blocking solution (PBS at pH 7.4, 0.05% Tween 20, 5% goat serum, 0.04% saponin) was added and incubated for 1 h. Primary antibodies were then added as follows: anti-V5 tag antibody (Abcam) at a 1:100 dilution, anti-GALC antibody, CL1475, at a 1:500 dilution, anti-lamp 2 antibody (BD Biosciences Pharmingen) at a 1:500 dilution, anti-calnexin antibody (Abcam) at a 1:500 dilution, and anti-58k Golgi protein antibody (Abcam) at a 1:100 dilution. These were allowed to incubate for 2 h and then were washed three times with PBS. Secondary antibodies (Invitrogen), Alexa Fluor 488 anti-rabbit IgG (for CL1475 and anti-V5 tag antibodies), and Alexa Fluor 488 anti-mouse IgG1 antibody (for anti-lamp2, anti-calnexin, and anti-58k Golgi protein antibodies), were used at a 1:1000 dilution and allowed to incubate for 1 h. After secondary antibody binding, the cells on coverslips were washed three times with PBS, dried on paper towels, and immediately sealed onto glass slides with a drop of Vectashield mounting media containing 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories). The slides were allowed to dry overnight at room temperature before examination. All procedures were performed in a light-protected environment after secondary antibodies were added. The slides were examined and captured using a Zeiss LSM 510 META confocal microscope. Each image is representative of a field (n = 3) of one of three independent experiments from each cell line.
Affinity purification by Ni-nitrilotriacetic acid agarose and in vitro deglycosylation of GALC proteins.
Approximately 8 × 106 GALC-transfected, COS-1 cells were lysed in 1 ml of buffer A (50 mm sodium phosphate, 300 mm sodium chloride) containing 1% Triton X-100 and 1X protease inhibitor mixture. The supernatant was collected after centrifugation at 20,000 × g for 10 min at 4°C. Next, 100 μl of Ni-NTA (nitrilotriacetic acid) agarose slurry was added to each 200 μl of supernatant. Binding proceeded at 4°C for 1 h on an end-to-end shaker. Agarose beads were washed three times with buffer A supplemented with 20 mm imidazole followed by successive centrifugations at 200 × g for 3 min. GALC was released by incubating the beads in 50 μl of buffer A supplemented with 250 mm imidazole. GALC was deglycosylated with endoglycosidase H (Endo H) (1000 U) or peptide:N-glycosidase F (PNG F) (1000 U) (New England Biolabs) at 37°C for 16 h. Deglycosylation was stopped by adding an equal volume of 2× Laemmli sample buffer, and the reaction was heated to 85°C for 5 min before SDS-PAGE and Western blotting.
Statistical analysis.
Two-tailed, unpaired t test was applied to analyze the difference between each mutant GALC group and WT GALC group (as in Figs. 1A,D, 2C) and between the D528N GALC and the D528N+S530A GALC groups (as in Fig. 6A,B). The same statistical analysis was used in the temperature sensitivity study to analyze the effect of subphysiological temperature on GALC activity in the H4-GALC cell lines (see Fig. 7A). In Figure 8, one-way ANOVA, followed by the Tukey's post hoc test, was applied to test for differences in GALC activity between α-lobeline-treated groups and control group within the same cell line. For the “uptake index” shown in Figures 2C and 6B, the data in all groups had passed the Kolmogorov–Smirnov normality test before being analyzed using an unpaired t test. All data are presented as mean values ± SE of at least triplicate samples from representative experiments. Each experiment was independently repeated at least three times.
Figure 1.

Expression of WT and mutant GALC in mammalian cells. A–F, Empty pcDNA4 vector (Mock), human wild-type GALC cDNA (WT), or GLD-causing GALC mutations (I234T, L629R, and D528N) were transiently transfected into COS-1 cells (A–C) or stably transfected into H4 cells (D–F). GALC enzymatic activity, measured by in vitro colorimetric assay, was significantly reduced in lysates from cells transfected with each mutant GALC, compared with WT GALC (*p < 0.05) (A, D). The precursor and processed forms of GALC were detected by Western blot using the CL1475 rabbit antibody against the N terminus of GALC (B, E), and the anti-V5 epitope tag antibody against the C terminus of GALC (C, F). Error bars indicate SEM.
Figure 7.

Activity and processing of GALC proteins in H4 cells under subphysiological temperature. WT and mutant GALC (D528N, I234T, L629R, and D528N+S530A) transfected H4 cells were cultured at 37 or 30°C for 72 h. A, GALC enzymatic activity in lysates was determined by in vitro colorimetric assay. Data shown represent the mean ± SE of at least triplicate samples in each group (*p < 0.05). B, Processed N-terminal GALC fragments (50–54 kDa) from cell lysate were analyzed by Western blot using the chicken anti-GALC antibody, CL1021AP.
Figure 8.

Effect of α-lobeline on human GALC in vitro and in H4 cells expressing WT and mutant GALC (I234T, L629R, and D528N). A, The inhibition of recombinant GALC by α-lobeline was analyzed using an in vitro colorimetric enzymatic activity assay. B, C, H4 cells expressing WT GALC (B) or D528N GALC (C) were treated with different concentrations of α-lobeline (60–240 μm) for 72 h. GALC enzymatic activity in the lysates (black bars) and in the culture media (white bars) was analyzed. Data shown represent the mean ± SE of at least triplicate samples in each group. Statistical analysis was performed by one-way ANOVA test, followed by the Tukey's post hoc test (*p < 0.05).
Results
GLD mutations result in GALC misprocessing and a reduction in residual enzymatic activity
To determine how the GLD mutations influence GALC activity, we generated WT and mutant GALC constructs containing D528N, I234T, and L629R mutations fused to a V5 epitope tag followed by a six-histidine (6-His) tag at the C terminus for analysis. Expression of mutant constructs in COS-1 cells resulted in low, yet detectable, levels of GALC activity in the lysates (Fig. 1A). The WT GALC transfectants exhibited a ∼47-fold increase in GALC-specific activity compared with the mock transfectants. The D528N, I234T, and L629R mutants had ∼18, 7, and 6% of the GALC-specific activity observed in the WT control, respectively. Identical expression studies were repeated in the human CNS derived cell line, H4 (Fig. 1D). Higher GALC activity was observed in WT H4-GALC cells compared with WT COS1-GALC cells. The D528N, I234T, and L629R H4-GALC mutants expressed ∼6, 2.6, and 1.6% of the GALC activity found in the H4-GALC WT control, respectively.
We next sought to determine whether or not these changes in specific activity in the mutant cells were attributable to changes in processing of the GALC precursor. Western blot analyses were performed on cell lysates using an anti-GALC antibody, CL1475 (Fig. 1B,E), and an anti-V5 epitope antibody (Fig. 1C, F), that detect the N-terminal and C-terminal ends of GALC, respectively. As expected, WT GALC was normally processed from its 80 kDa precursor form into an N-terminal (50–54 kDa) fragment, as detected by the CL1475 antibody (Fig. 1B,E). Whereas the 80 kDa precursor form of GALC was detected in both H4 and COS-1 cells transfected with mutant forms of the enzyme (Fig. 1C,F), the 50–54 kDa N-terminal GALC fragment was not detected in any of the three mutants examined in either cell type (Fig. 1B,E). These data strongly suggest that these mutations alter protein processing. Interestingly, the D528N mutant enzyme also showed an increase in the apparent molecular weight of the 80 kDa precursor form of GALC by SDS-PAGE compared with either of the other two mutants or the wild-type enzyme. We used the V5 antibody in this series of experiments (Fig. 1C,F) and did not detect the 30 kDa C-terminal fragment of GALC reported by previous studies (Chen and Wenger, 1993).
GLD mutations lead to impaired GALC protein secretion and/or reduced cellular reuptake
The precursor form of GALC is normally secreted and then taken up by cells via the mannose-6-phosphate receptor for delivery to lysosomes and additional processing (Nagano et al., 1998). To determine the influence of GLD mutations on the secretion of GALC, we next performed Western blots on the culture media of WT and mutant GALC-transfected H4 cells. The GALC precursor (80 kDa) was readily detectable in medium secreted from WT GALC-transfected H4 cells (Fig. 2A) and was enzymatically active (Fig. 2C). The I234T and D528N GALC mutants were also secreted by H4 cells at detectable levels, but GALC was not detected in medium from cells transfected with L629R GALC, indicating that this mutation causes a severe lack-of-secretion phenotype.
An oligodendrocyte precursor cell line, OLI-neu, was used to examine uptake of WT and mutant GALC secreted by the H4-GALC cells. When the conditioned medium from GALC-transfected cells was applied to OLI-neu cells for 24 h, the cells took up and processed the WT GALC precursor and generated the expected 50–54 kDa N-terminal fragment. The protein band resulting from the uptake and processing of the exogenously added GALC comigrated with endogenous GALC in OLI-neu cells, resulting in an apparent increase in band intensity compared with the mock control (Fig. 2B). We were unable to detect the 80 kDa precursor form in the cell lysates at the 24 h time point, suggesting that the precursor is efficiently processed after it enters the cell. The uptake and processing of exogenous WT GALC by OLI-neu cells was associated with a net increase in GALC activity compared with control (Fig. 2C). The “uptake index” is defined as the ratio of GALC activity detected in the cell lysate after uptake divided by the GALC activity in the corresponding input medium. This ratio is then multiplied by 100 and is presented as the “uptake index.” For WT GALC, the uptake index was 25. As the L629R mutant GALC is not secreted from H4 cells at detectable levels, we were unable to analyze this mutation in a similar fashion. The I234T GALC mutant was secreted by H4 cells at levels comparable with WT GALC (Fig. 2A). However, it appears to be mostly enzymatically inactive (Fig. 2C) and was not taken up by OLI-neu cells (Fig. 2B). The secreted form of D528N mutant was detected in the conditioned medium (Fig. 2A) and was enzymatically active (Fig. 2C), but it also was not efficiently taken up by OLI-neu cells (Fig. 2B,C). These results demonstrate that, although protein misprocessing appears to be common among the mutations examined, the precise mechanisms differ. These data indicate that, unlike WT GALC, cells expressing I234T, D528N, and L629R mutations are unlikely to supply nearby cells with significant GALC enzymatic activity.
GLD mutations I234T, L629R, and D528N impair localization of GALC
To determine whether the GLD-causing GALC mutations influence the lysosomal localization of the enzyme in cells, we performed double-immunofluorescent labeling experiments and confocal image analysis using the CL1475 rabbit anti-GALC antibody to detect both the precursor form and the mature form of GALC, or anti-V5 tag antibody to detect predominantly the precursor form of GALC. For subcellular organelle markers, we used antibodies to detect the lysosomal marker, lysosomal-associated membrane protein 2 (lamp 2), or the endoplasmic reticulum (ER) marker, calnexin, or the Golgi apparatus marker, 58k Golgi protein. As shown in Figure 3, we detected colocalization of GALC (CL1475 antibody) with lamp 2 in the WT H4-GALC cells, but not in cells expressing any of the three GLD mutations examined. Double labeling with anti-calnexin (ER) and the anti-V5 tag antibody to GALC showed colocalization only in the L629R mutant, indicating that the L629R mutant potentially accumulates in the ER, which is entirely consistent with its lack-of-secretion phenotype (Fig. 2A). WT GALC and I234T and D528N mutants were found to colocalize with the Golgi marker but not with the ER marker, suggesting that the precursor form of GALC, with or without these mutations, traffics through the ER and reaches the Golgi apparatus. However, it appears that only WT GALC is transported to the lysosome to an extent that colocalization studies can readily detect. Collectively, these data indicate that the mutations all influence the level of GALC in lysosomes, where it is needed to maintain normal function.
Figure 3.

Confocal immunocytofluorescent analysis of GALC in H4-GALC cells. H4 cells (mock), WT GALC, and mutant (I234T, L629R and D528N) GALC-expressing H4 cells were seeded at subconfluent density on glass coverslips 2 d before staining. The subcellular localization of GALC was analyzed by costaining of GALC (CL1475 rabbit anti-GALC antibody) and lamp 2 for lysosomes, costaining of GALC (anti-V5 antibody) and calnexin for ER, and costaining of GALC (anti-V5 antibody) and 58k Golgi protein for Golgi apparatus. GALC and the organelle markers were illustrated in green and red colors, respectively. Nuclei were labeled with DAPI and illustrated in blue color. The yellow color indicates colocalization of GALC with the organelle markers. All images were captured in 40× magnification using a Zeiss LSM 510 META confocal microscope.
The D528N mutant is hyperglycosylated in transfected COS-1 cells
The asparagine residue at position 528 of the D528N GALC mutant is flanked in the forward direction by alanine and serine, which combine to fulfill the triplet sequence requirement for a potential N-glycosylation site (Fig. 4). To examine whether this mutation-induced, potential N-glycosylation site resulted in aberrant glycosylation of the D528N mutant, we first analyzed the apparent molecular weights of various species of COS-1 expressed GALC by Western blot as detected by the anti-V5 tag antibody. Consistent with an increase in glycosylation, we noticed an increase in the apparent molecular weight of the D528N 80 kDa GALC precursor (Figs. 1, 2, 5B). We also observed an increase in the molecular weight of a 25 kDa C-terminal fragment (Fig. 5C) and a larger band migrating at ∼170 kDa (Fig. 5A). The C-terminal fragment of GALC was apparent only when the lysates were affinity-purified and concentrated. A similar amount of the C-terminal fragment was found in the D528N cells compared with the WT GALC cells. This suggests that the mutation has no effect on the generation of this fragment. The identity of the larger band may represent a dimer or larger aggregate of the GALC precursor. Deglycosylation with either Endo H or PNG F resulted in a decrease in the apparent molecular weights of all three mutant bands so that they migrated similarly to the deglycosylated wild-type enzyme (Fig. 5). These data provide strong support that the additional, putative N-glycosylation site created by the D528N mutation is indeed glycosylated.
Figure 4.

Amino acid sequences near the D528N mutation in GALC. The aspartic acid residue (D) at position 528 was substituted by an asparagine residue (N), which creates an extra N-glycosylation site in the D528N GALC mutant. A double mutant, D528N+S530A, was generated to disrupt the extra N-glycosylation site created by the D528N mutation by replacing the serine residue at position 530 with an alanine residue. A construct containing the S530A mutation only was also generated for an assay control.
Figure 5.

Western blot analysis of the hyperglycosylated D528N GALC mutant overexpressed in COS-1 cells. A–C, “Polymeric” GALC (A), monomeric GALC precursors (B), and C-terminal GALC fragments (C) were purified and analyzed without treatment (control) or after deglycosylation by Endo H or by PNG F.
Enzyme kinetics is not altered in the D528N mutant
To test whether the extra N-glycosylation at the D528N residue alters the enzyme kinetics of the mutant GALC protein, Km and Vmax values of WT and D528N mutant GALC were determined with various concentration of HNG substrate (from 14 to 900 μm) in 0.1 m sodium citrate buffer, pH 4, at 37°C using Lineweaver–Burk plots (data not shown). The Km and Vmax values of WT GALC from culture medium of H4-GALC cells were 1.35 mm and 0.6 nmol/h, respectively. The D528N mutant was determined to have a similar Km and Vmax values of 1.71 mm and 0.64 nmol/h, respectively, indicating that GALC enzyme kinetics is not apparently altered by the mutation.
Disruption of the extra D528N N-glycosylation site results in restored protein processing, increased residual GALC activity, increased GALC secretion and uptake, and lysosomal localization
Next, we investigated the impact of the additional N-glycosylation site on GALC enzyme activity, reduced levels of secretion, lack of extracellular uptake, and lack of lysosomal localization observed in cells transfected with the D528N mutant. To accomplish this, we designed a GALC-expression construct in which the extra N-glycosylation site created by the D528N mutation was disrupted by a second mutation, S530A (Fig. 4). This double mutant, D528N+S530A, was processed similarly to WT GALC with significant increases in the levels of 50–54 kDa N-terminal fragment detected in cells, as opposed to the D528N mutation alone (Fig. 6A, immnoblot). This was accompanied by a significant increase in enzymatic activity in the double mutant (Fig. 6A, graph). Additionally, there was a significant increase in secreted GALC enzymatic activity of GALC in the H4 cells even though the amount of GALC detected by Western blot was not apparently increased (Fig. 6B, table). The uptake efficiency of the double mutant was also increased to a level comparable with that in WT GALC by OLI-neu cells as indicated by the “uptake index” (Fig. 6B, table). Finally, the D528N+S530A GALC was observed to colocalize with lamp2 (as shown in yellow color), indicating that lysosomal localization was partially restored (Fig. 6C). These results suggest that the molecular defects in the D528N GALC mutant are likely related, at least in part, to N-glycosylation at the mutation site and that reversing this glycosylation can result in substantial improvements, even though the original mutation is still present in this construct.
The D528N mutant GALC is stabilized and properly processed under reduced temperature
To examine whether or not protein misfolding is involved in the GALC protein misprocessing and impairment of GALC activity in the I234T, L629R, D528N, and D528N+S530A mutant cell lines, cells were cultured under physiological (37°C) or reduced (30°C) temperature for 72 h, a condition known to facilitate protein folding (Denning et al., 1992; Berson et al., 2000; Vollrath and Liu, 2006). Processing of WT GALC into the 54 kDa N-terminal fragment and associated enzyme activity was significantly decreased by ∼31% at reduced temperature (Fig. 7A,B). This is consistent with reduced temperature not being ideal for normal GALC processing. In contrast, processing of GALC precursor into the N-terminal fragment was apparently increased in the D528N mutant when the cells were cultured at reduced temperature (Fig. 7B). This increase in processing was accompanied by a significant 50% increase in GALC activity (Fig. 7A). No significant change in activity was found in I234T, L629R, and D528N+S530A mutants (Fig. 7A). Collectively, these results suggest that protein misfolding may also be involved in the molecular mechanism by which the D528N mutation impairs the function of GALC.
Identification of α-lobeline to rescue the function of D528N GALC mutant
A novel, therapeutic approach to treat lysosomal storage diseases (LSDs) includes the use of enzyme inhibitors as ligands to protect misfolded, newly synthesized polypeptides from degradation. These enzyme inhibitors, also known as pharmacological chaperones, have been reported to rescue partial function of mutant lysosomal enzymes that are responsible for the onset of various LSDs [reviewed in part in the study by Fan (2008)]. To identify potential PCs to treat GLD, we screened for GALC inhibitors with a modified, 96-well microplate in vitro enzymatic activity assay (Gal et al., 1977). α-Lobeline, a Food and Drug Administration category 3 drug with a known ability to cross the blood–brain barrier, was identified as a relatively weak inhibitor of GALC (Fig. 8A). To determine whether this weak inhibitor might act as a chemical chaperone, we treated the D528N mutant H4-GALC cells with α-lobeline at 60, 120, and 240 μm for 72 h and analyzed GALC enzymatic activity in lysates and culture media. As a control, the WT GALC cells were also treated and analyzed under the same conditions. The intracellular (lysates) and the extracellular (media) GALC activity in D528N mutant were increased significantly by 52 and 64%, respectively, at the highest α-lobeline dose (Fig. 8C). In contrast, the intracellular GALC activity in WT cells was not changed and the extracellular GALC activity was increased by a more modest 26%, under the same conditions (Fig. 8B). We did not see similar effects with the I234T and L629R mutants, suggesting that the effect of α-lobeline on the D528N mutant was mutation specific (data not shown) (Sawkar et al., 2005). Our results indicate that α-lobeline preferentially increases the intracellular GALC activity of the D528N mutant, which suggests that α-lobeline has the ability to function as a pharmacological chaperone to rescue the function of this mutant.
Discussion
In this report, we have shown that three different mutations in GALC result in protein misprocessing (Fig. 1), altered protein secretion and/or reuptake of the enzyme (Fig. 2), and/or lack of trafficking to lysosomes of the mutant enzyme (Fig. 3). Of the three disease-causing mutations we examined, all were produced at the protein level and could be detected as an 80 kDa precursor, but none of the mutants was detected as the processed 50–54 kDa N-terminal fragment. These data clearly indicate that GLD can be caused by protein misprocessing of GALC.
Although it may be reasonable to expect a direct correlation of residual GALC enzyme activity with disease phenotype (i.e., a higher residual activity should present a less severe phenotype or later symptom onset), it appears that it is not always the case with GLD. For example, residual GALC activity in both leukocyte and skin fibroblast samples from the patient with the L629R mutation, who was diagnosed with late-onset GLD, is indistinguishable from residual GALC activity in infantile patients who have the most severe form of GLD (Jardim et al., 1999). The I234T mutation, which was also found in a late-onset patient, was confirmed to have an undetectable level of residual activity (De Gasperi et al., 1996). These observations suggest that, in addition to the level of residual GALC activity, genetic background and potentially environmental factors may also contribute to the severity of GLD.
Interestingly, one of the mutations examined, the D528N mutation, appears to create a novel glycosylation site. Disrupting this site by mutating the adjacent amino acid can partially restore enzyme function even though the protein still contains the original mutation (Fig. 6). According to the Human Gene Mutation Database, ∼142 of 10,047 potential mutations (∼1.4%) in 77 of 577 genes (∼13.3%) create new, potential N-glycosylation sites (Vogt et al., 2005). Some of these mutations are linked to pathogenic effects in various inherited diseases such as congenital dysfibrinogenemia (Yamazumi et al., 1989), neonatal Marfan syndrome (Lönnqvist et al., 1996), and type I antithrombin deficiency (Fitches et al., 2001). Similarly, the D528N mutation appears to influence glycosylation and alter protein processing, secretion, reuptake, and trafficking to lysosomes in a way that reduces GALC function and causes disease.
Previous studies have shown that protein misfolding is a common molecular mechanism that accounts for many monogenetic-inherited disorders, including many LSDs (Butters, 2007; Fan and Ishii, 2007). Temperature-sensitive folding (TSF) mutations, originally found in the tailspike protein of Phage P22 (Yu and King, 1988), have been defined as amino acid substitutions that destabilize intracellular protein folding by kinetically trapping it in an intermediate, nascent polypeptide state at physiological temperature. Under subphysiological temperature, proteins bearing TSF mutations can resume folding into their mature form without significant loss of function. To determine whether any of the GLD mutations in this study are temperature sensitive, we examined the processing and activity of the GALC mutants at reduced temperature (Fig. 7). Consistent with influence of temperature on other TSF mutations, an increase in protein processing to the 54 kDa N-terminal fragment, accompanied by an increase in GALC activity, was found in cells expressing the D528N mutant when cultured under reduced temperature.
PCs, often in the form of enzyme inhibitors that bind and stabilize the native conformation of misfolded proteins, are emerging therapeutic agents for protein misfolding diseases. To attempt to determine whether PCs might be useful therapeutics for GLD, we tested whether or not α-lobeline, a relatively weak GALC inhibitor, could influence protein processing and enzymatic activity in cells expressing the D528N mutation. α-Lobeline treatment preferentially increased the intracellular GALC activity of the D528N mutant (Fig. 8C) but not WT GALC cells (Fig. 8B) and caused a dose-dependent increase in GALC secretion in the D528N mutant (Fig. 8B). These in vitro data suggest that α-lobeline is a potential PC that can partially enhance GALC function in cells carrying the D528N GLD mutation. To validate this same effect in vivo, new animal models containing this mutation are needed. Collectively, these data indicate that GLD can be caused by protein misfolding/misprocessing and that, at least with the D528N mutation, this can be partially corrected through the use of pharmacological chaperones.
Footnotes
This work was supported by funding from the Mayo Clinic Foundation for Medical Education and Research and National Institute of Neurological Disorders and Stroke Grant NS44230 (C.B.E.). We also acknowledge Holly Hammond for proofreading this manuscript.
A patent has been filed regarding the use of α-lobeline as a potential pharmacological agent to treat Krabbe disease.
References
- Berson JF, Frank DW, Calvo PA, Bieler BM, Marks MS. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J Biol Chem. 2000;275:12281–12289. doi: 10.1074/jbc.275.16.12281. [DOI] [PubMed] [Google Scholar]
- Butters TD. Gaucher disease. Curr Opin Chem Biol. 2007;11:412–418. doi: 10.1016/j.cbpa.2007.05.035. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Wenger DA. Galactocerebrosidase from human urine: purification and partial characterization. Biochim Biophys Acta. 1993;1170:53–61. doi: 10.1016/0005-2760(93)90175-9. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Rafi MA, de Gala G, Wenger DA. Cloning and expression of cDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum Mol Genet. 1993;2:1841–1845. doi: 10.1093/hmg/2.11.1841. [DOI] [PubMed] [Google Scholar]
- De Gasperi R, Gama Sosa MA, Sartorato EL, Battistini S, MacFarlane H, Gusella JF, Krivit W, Kolodny EH. Molecular heterogeneity of late-onset forms of globoid-cell leukodystrophy. Am J Hum Genet. 1996;59:1233–1242. [PMC free article] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Fan JQ. A counterintuitive approach to treat enzyme deficiencies: use of enzyme inhibitors for restoring mutant enzyme activity. Biol Chem. 2008;389:1–11. doi: 10.1515/BC.2008.009. [DOI] [PubMed] [Google Scholar]
- Fan JQ, Ishii S. Active-site-specific chaperone therapy for Fabry disease. Yin and Yang of enzyme inhibitors. FEBS J. 2007;274:4962–4971. doi: 10.1111/j.1742-4658.2007.06041.x. [DOI] [PubMed] [Google Scholar]
- Fitches AC, Lewandowski K, Olds RJ. Creation of an additional glycosylation site as a mechanism for type I antithrombin deficiency. Thromb Haemost. 2001;86:1023–1027. [PubMed] [Google Scholar]
- Fu L, Inui K, Nishigaki T, Tatsumi N, Tsukamoto H, Kokubu C, Muramatsu T, Okada S. Molecular heterogeneity of Krabbe disease. J Inherit Metab Dis. 1999;22:155–162. doi: 10.1023/a:1005449919660. [DOI] [PubMed] [Google Scholar]
- Furuya H, Kukita Y, Nagano S, Sakai Y, Yamashita Y, Fukuyama H, Inatomi Y, Saito Y, Koike R, Tsuji S, Fukumaki Y, Hayashi K, Kobayashi T. Adult onset globoid cell leukodystrophy (Krabbe disease): analysis of galactosylceramidase cDNA from four Japanese patients. Hum Genet. 1997;100:450–456. doi: 10.1007/s004390050532. [DOI] [PubMed] [Google Scholar]
- Gal AE, Brady RO, Pentchev PG, Furbish FS, Suzuki K, Tanaka H, Schneider EL. A practical chromogenic procedure for the diagnosis of Krabbe's disease. Clin Chim Acta. 1977;77:53–59. doi: 10.1016/0009-8981(77)90401-6. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Basso V, Cantuti L, Givogri MI, Lopez-Rosas A, Perez N, Vasu C, Cao H, van Breemen R, Mondino A, Bongarzone ER. Autonomic denervation of lymphoid organs leads to epigenetic immune atrophy in a mouse model of Krabbe disease. J Neurosci. 2007;27:13730–13738. doi: 10.1523/JNEUROSCI.3379-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardim LB, Giugliani R, Pires RF, Haussen S, Burin MG, Rafi MA, Wenger DA. Protracted course of Krabbe disease in an adult patient bearing a novel mutation. Arch Neurol. 1999;56:1014–1017. doi: 10.1001/archneur.56.8.1014. [DOI] [PubMed] [Google Scholar]
- Jung M, Krämer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, Khazaie K, Chlichlia K, von Blankenfeld G, Kettenmann H, Trotter J. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. Eur J Neurosci. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Katayama M, Satoh J, Suzuki K, Suzuki K. The twitcher mouse. An alteration of the unmyelinated fibers in the PNS. Am J Pathol. 1988;131:308–319. [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Courtenay A, Troendle FJ, Stallings-Mann ML, Dickey CA, DeLucia MW, Dickson DW, Eckman CB. Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. FASEB J. 2005;19:1549–1551. doi: 10.1096/fj.05-3826fje. [DOI] [PubMed] [Google Scholar]
- Lissens W, Arena A, Seneca S, Rafi M, Sorge G, Liebaers I, Wenger D, Fiumara A. A single mutation in the GALC gene is responsible for the majority of late onset Krabbe disease patients in the Catania (Sicily, Italy) region. Hum Mutat. 2007;28:742. doi: 10.1002/humu.9500. [DOI] [PubMed] [Google Scholar]
- Lönnqvist L, Karttunen L, Rantamäki T, Kielty C, Raghunath M, Peltonen L. A point mutation creating an extra N-glycosylation site in fibrillin-1 results in neonatal Marfan syndrome. Genomics. 1996;36:468–475. doi: 10.1006/geno.1996.0492. [DOI] [PubMed] [Google Scholar]
- Nagano S, Yamada T, Shinnoh N, Furuya H, Taniwaki T, Kira J. Expression and processing of recombinant human galactosylceramidase. Clin Chim Acta. 1998;276:53–61. doi: 10.1016/s0009-8981(98)00095-3. [DOI] [PubMed] [Google Scholar]
- Nagara H, Ogawa H, Sato Y, Kobayashi T, Suzuki K. The twitcher mouse: degeneration of oligodendrocytes in vitro. Brain Res. 1986;391:79–84. doi: 10.1016/0165-3806(86)90009-x. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Zlotogora J, Wenger DA. Two different mutations are responsible for Krabbe disease in the Druze and Moslem Arab populations in Israel. Hum Genet. 1996;97:304–308. doi: 10.1007/BF02185759. [DOI] [PubMed] [Google Scholar]
- Sakai N, Inui K, Fujii N, Fukushima H, Nishimoto J, Yanagihara I, Isegawa Y, Iwamatsu A, Okada S. Krabbe disease: isolation and characterization of a full-length cDNA for human galactocerebrosidase. Biochem Biophys Res Commun. 1994;198:485–491. doi: 10.1006/bbrc.1994.1071. [DOI] [PubMed] [Google Scholar]
- Sawkar AR, Adamski-Werner SL, Cheng WC, Wong CH, Beutler E, Zimmer KP, Kelly JW. Gaucher disease-associated glucocerebrosidases show mutation-dependent chemical chaperoning profiles. Chem Biol. 2005;12:1235–1244. doi: 10.1016/j.chembiol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Seitelberger F. Demyelination and leukodystrophy at an early age (in Spanish) Bol Estud Med Biol. 1981;31:373–382. [PubMed] [Google Scholar]
- Selleri S, Torchiana E, Pareyson D, Lulli L, Bertagnolio B, Savoiardo M, Farina L, Carrara F, Filocamo M, Gatti R, Sghirlanzoni A, Uziel G, Finocchiaro G. Deletion of exons 11–17 and novel mutations of the galactocerebrosidase gene in adult- and early-onset patients with Krabbe disease. J Neurol. 2000;247:875–877. doi: 10.1007/s004150070076. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Suzuki Y. Globoid cell leucodystrophy (Krabbe's disease): deficiency of galactocerebroside β-galactosidase. Proc Natl Acad Sci U S A. 1970;66:302–309. doi: 10.1073/pnas.66.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Nagara H, Kobayashi T, Goto I. The twitcher mouse: accumulation of galactosylsphingosine and pathology of the sciatic nerve. Brain Res. 1988;454:340–346. doi: 10.1016/0006-8993(88)90835-9. [DOI] [PubMed] [Google Scholar]
- Vogt G, Chapgier A, Yang K, Chuzhanova N, Feinberg J, Fieschi C, Boisson-Dupuis S, Alcais A, Filipe-Santos O, Bustamante J, de Beaucoudrey L, Al-Mohsen I, Al-Hajjar S, Al-Ghonaium A, Adimi P, Mirsaeidi M, Khalilzadeh S, Rosenzweig S, de la Calle Martin O, Bauer TR, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- Vollrath D, Liu Y. Temperature sensitive secretion of mutant myocilins. Exp Eye Res. 2006;82:1030–1036. doi: 10.1016/j.exer.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat. 1997;10:268–279. doi: 10.1002/(SICI)1098-1004(1997)10:4<268::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab. 2000;70:1–9. doi: 10.1006/mgme.2000.2990. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Suzuki Ku, Suzuki Y, Suzuki Ki. Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease) In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3669–3694. [Google Scholar]
- White AB, Givogri MI, Lopez-Rosas A, Cao H, van Breemen R, Thinakaran G, Bongarzone ER. Psychosine accumulates in membrane microdomains in the brain of Krabbe patients, disrupting the raft architecture. J Neurosci. 2009;29:6068–6077. doi: 10.1523/JNEUROSCI.5597-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Sakai N, Taniike M, Inui K, Ozono K. Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation. J Hum Genet. 2006;51:548–554. doi: 10.1007/s10038-006-0396-3. [DOI] [PubMed] [Google Scholar]
- Yager D, Watson M, Healy B, Eckman EA, Eckman CB. Natural product extracts that reduce accumulation of the Alzheimer's amyloid β peptide: selective reduction in Aβ42. J Mol Neurosci. 2002;19:129–133. doi: 10.1007/s12031-002-0023-5. [DOI] [PubMed] [Google Scholar]
- Yamazumi K, Shimura K, Terukina S, Takahashi N, Matsuda M. A gamma methionine-310 to threonine substitution and consequent N-glycosylation at gamma asparagine-308 identified in a congenital dysfibrinogenemia associated with posttraumatic bleeding, fibrinogen Asahi. J Clin Invest. 1989;83:1590–1597. doi: 10.1172/JCI114056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu MH, King J. Surface amino acids as sites of temperature-sensitive folding mutations in the P22 tailspike protein. J Biol Chem. 1988;263:1424–1431. [PubMed] [Google Scholar]