

Abstract
Exposure of mitochondria to oxidative stress and elevated Ca2+ promotes opening of the mitochondrial permeability transition pore (PTP), resulting in membrane depolarization, uncoupling of oxidative phosphorylation, and potentially cell death. This study tested the hypothesis that treatment of rats with sulforaphane (SFP), an activator of the Nrf2 pathway of antioxidant gene expression, increases the resistance of liver mitochondria to redox-regulated PTP opening and elevates mitochondrial levels of antioxidants. Rats were injected with SFP or drug vehicle and liver mitochondria were isolated 40 hr later. Respiring mitochondria actively accumulated added Ca2+, which was then released through PTP opening induced by agents that either cause an oxidized shift in the mitochondrial redox state or that directly oxidize protein thiol groups. SFP treatment of rats inhibited the rate of pro-oxidant-induced mitochondrial Ca2+ release and increased expression of the glutathione peroxidase/reductase system, thioredoxin, and malic enzyme. These results are the first to demonstrate that SFP treatment of animals increases liver mitochondrial antioxidant defenses and inhibits redox-sensitive PTP opening. This novel form of preconditioning could protect against a variety of pathologies that include oxidative stress and mitochondrial dysfunction in their etiologies.
BACKGROUND
Oxidative stress contributes significantly to the pathophysiology of several disorders of the liver [1–5] and other vital organs [6–8]. Mitochondria are a primary target of oxidative stress in these and other pathologies, as reflected by oxidative damage to mitochondrial DNA [9], proteins [10] and lipids [11], oxidation and loss of mitochondrial glutathione [12], and an oxidized shift in mitochondrial redox state [13]. The combination of mitochondrial oxidative stress with exposure to abnormally high Ca2+ concentrations can lead to opening of the mitochondrial permeability transition pore (PTP) [14], which incapacitates mitochondrial ATP formation [15], often leading to metabolic failure and cell death [16].
PTP opening facilitates the flux of solutes of up to 1,500 Da across the inner membrane, resulting in membrane depolarization, uncoupling of oxidative phosphorylation, and mitochondrial osmotic swelling [17,18]. PTP-induced mitochondrial structural destabilization may also promote the release of mitochondrial pro-apoptotic proteins, e.g., cytochrome c and AIF [19,20]. The molecular identity of the PTP is unknown; however, cyclophilin D (CyD) is a target through which both Ca2+ and oxidative stress trigger PTP opening [21]. Moreover, mitochondria isolated from the livers of CyD knockout mice are resistant to Ca2+ induced PTP opening [22] and CyD −/− hepatocytes are resistant to H2O2 induced necrosis [23]. The potential clinical importance of PTP opening within liver cells is exemplified by the finding that CyD knockout mice are resistant to liver damage caused by toxic levels of acetaminophen [24].
Numerous antioxidant-based drugs have been used experimentally to reduce damage to the liver and other organs caused by oxidative stress [25]. While the use of specific antioxidants has been successful in various animal models of disease and injury [26], their effectiveness may be limited by their relatively small range of targets compared to the numerous reactive oxygen species (ROS) and molecular mechanisms of cellular toxicity induced by oxidative stress. An alternative approach is the use of chemicals or conditions that stimulate the expression of a wide variety of gene products representing the endogenous antioxidant defense system [27]. Compounds such as curcumin [28], carnosine, tert-butylhydroquinone [29] and sulforaphane (SFP) [30] exhibit the ability to induce such gene expression apparently through the transcriptional activating factor NF-E2-related factor 2 (Nrf2). Activation of Nrf2 involves its release from a binding protein, KEAP1, and Nrf2 serine phosphorylation. Nrf2 then translocates into the nucleus where it binds to antioxidant response elements (ARE) located upstream of the 5′ region of the promoter of many cytoprotective genes, e.g., as NAD(P)H:quinone oxidoreductase 1 (NQO1), heme-oxygenase 1 (HO1) and those that code for enzymes necessary for glutathione biosynthesis [31].
Considering the facts that PTP opening is promoted by ROS [32] and is inhibited by both exogenous antioxidants [33] and endogenous antioxidant enzymes, we hypothesized that mitochondria from animals treated with an Nrf2 activator would be resistant to redox-regulated PTP opening. We have observed a small inhibition of peroxide-induced PTP opening in mitochondria isolated from the brains of normal rats injected with sulforaphane [34]; however, no increase in likely Nrf2 targeted mitochondrial proteins was apparent (unpublished results). Since the blood brain barrier may limit access of sulforaphane to the brain and since only a fraction of brain mitochondria exhibit redox-sensitive PTP opening, we tested the much more redox-sensitive liver mitochondrial PTP for sensitivity to sulforaphane. Our results demonstrate that sulforaphane administration to rats dramatically inhibits redox-regulated PTP opening by liver mitochondria and also increases immunoreactive levels of mitochondrial antioxidant-related proteins. SFP treatment also increases mitochondrial peroxidase activity.
MATERIALS AND METHODS
Chemicals and Reagents
R,S-Sulforaphane was purchased from LKT Laboratories, Inc (St. Paul, MN). Mannitol, sucrose, EGTA, HEPES, Tris, potassium phosphate dibasic (K2HPO4), magnesium chloride (MgCl2), malate, glutamate, oligomycin, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), adenosine 5′-diphosphate (ADP), bovine serum albumin (BSA), succinate, rotenone, calcium, dimethylsulfoxide (DMSO), tert-butyl hydrogen peroxide, phenylarsine oxide, horseradish peroxidase and oxaoloacetate were obtained from Sigma–Aldrich (St. Louis, MO, USA). Potassium chloride suprapure was obtained from EM Sciences (Fort Washington, PA). Amplex red was obtained from Invitrogen (Carlsbad, CA).
Administration of sulforaphane to rats
All experimental procedures were approved by the University of Maryland, Baltimore Institutional Animal Care and Use Committee and conducted in accordance with the recommendations provided in the Guide for the Care and Use of Laboratory Animals. Ten male 300 g Fischer 344 rats (Charles River) were injected intraperitoneally with sulforaphane at 10 mg/kg in 200 μL solution containing 40% DMSO and 60% isotonic saline. Ten animals received DMSO plus saline as the vehicle control. The mitochondria obtained from any one animal were not sufficient to perform all the different measurements; therefore, the number of animals used for each paired comparison varied between 4/group to 10/group.
Isolation of liver mitochondria
Rats were fasted overnight and euthanized by decapitation at 40 hr after injection of rats with sulforaphane or drug vehicle. Liver mitochondria were isolated as previously described [35]. Rats were fasted overnight and euthanized by decapitation and all four lobes of the liver rapidly removed, chopped and homogenized in ice-cold isolation buffer (210 mM mannitol, 70 mM sucrose, 5 mM Hepes, 0.5 mg/ml fatty acid free BSA, 1 mM EGTA, pH 7.2, at 4°C). The homogenate was centrifuged in a Beckman JA20 rotor at 2800 rpm for 12 min. A thin layer of lipid was aspirated from the top of the tube and the supernatant collected and centrifuged at 9300 rpm for 12 min. The pellets were re-suspended in media and centrifuged at 12,000 rpm for 12 min. The re-suspended pellets were subjected to a final centrifugation at 12,000 rpm for 12 min. The final combined pellet was re-suspended in 1 mL isolation media not containing EGTA. Mitochondrial protein concentrations were measured using a Lowry DC kit (Bio-Rad, Hercules, CA, USA) with bovine serum albumin used as concentration standards. The respiratory activities of isolated liver mitochondria were measured with a Clark-type oxygen electrode as described previously [34], using 5 mM glutamate plus 0.1 mM malate as respiratory substrates present in a KCl-based medium containing 2.0 mM KH2PO4 and 1.0 mM MgCl2 at pH 7.0 and 30 °C [34]. State 3 respiration was initiated by the addition of 0.5 mM ADP and State 4 respiration was obtained by the subsequent addition of the ATP synthetase inhibitor oligomycin at 2.5 μg/ml.
Concurrent measurements of mitochondrial Ca2+-uptake and release and pyridine nucleotide redox state
Experiments were performed as previously described for isolated brain mitochondria [34]. Briefly, rat liver mitochondria were suspended at a protein concentration of 0.5 mg/ml in 2 ml of assay buffer (125 mM supra-pure KCl, 2 mM K2HPO4, 1 mM MgCl2, 20 mM Tris-HCl, 5 mM succinate, and 4 μM rotenone, pH 7.4 at 30°C). The use of the electron transport chain complex II substrate succinate in the presence of the complex I inhibitor rotenone allows for the mitochondrial pyridine nucleotide redox state (NAD(P)H/NAD(P)+) to be varied between totally reduced (no added pro-oxidant) to oxidized (after addition of pro-oxidants) independent of mitochondrial respiration and membrane potential. Mitochondria were added to the medium containing succinate and allowed to equilibrate, bioenergetically, based on stabilization of the medium free [Ca2+] and NADP(H) autofluorescence. CaCl2 was then added to the medium at a level that the mitochondria were capable of completely accumulating. The rate of spontaneous Ca2+ release, if any, was then quantified in the absence of further additions. In other experiments, the rate of release was measured after the addition of a chemical pro-oxidant. The pro-oxidants used in these experiments were tert-butyl hydroperoxide, which induces oxidation of NAD(P)H by its metabolism via mitochondrial glutathione peroxidase and reductase, oxaloacetate, which drives the oxidation of NADH through the malate dehydrogenase reaction, and phenylarsine oxide, which selectively and directly oxidizes sulfhydryl groups. In a few experiments, cyclosporin A, a permeability transition inhibitor, was present to confirm that mitochondrial Ca2+ release was a consequence of permeability transition pore opening. Respiration-dependent mitochondrial uptake of Ca2+ and subsequent release, either spontaneous or induced by addition of pro-oxidants, were measured by monitoring the free Ca2+ concentration present in the medium in which the mitochondria were suspended. The medium free Ca2+ was measured fluorimetrically in the presence of the Ca2+-sensitive fluorescent dye Calcium Green-5N (0.1 μM), using excitation and emission wavelengths of 506 nm and 532nm, respectively.
Mitochondrial pyridine nucleotide redox state
Autofluorescence of reduced pyridine nucleotides (NADH plus NADPH) in the mitochondrial suspensions was measured concurrently with the Calcium Green 5N measurements of medium [Ca2+] using excitation and emission wavelengths of 350 nm and 460 nm, respectively, cycling every 2 seconds.
Immunoblot analysis
Freshly Isolated liver mitochondria and liver homogenates were lysed in RIPA buffer containing a cocktail of protease and phosphatase inhibitors (Calbiochem). Equal amounts of mitochondrial protein from each sample were separated by SDS-PAGE (4–12% Bis-Tris gels) (Invitrogen) and transferred to PVDF membranes (Invitrogen), and then incubated with primary antibodies (overnight at 4°C) with NQO1 1:500 (Santa Cruz), Cyclophilin D 1:30000 (Mitosciences), Glutathione Peroxidase 1:500 (Abcam), Thioredoxin 2 1:150000 (Santa Cruz), Malic Enzyme 3 1:5000 (Sigma), Isocitrate Dehydrogenase 2 1:2000 (Santa Cruz), Manganese Superoxide Dismutase 1:250 (Santa Cruz), GAPDH 1:500000 (Abcam), VDAC 1:50,000–1:300,000 (Mitosciences). The membranes were then washed with PBST and incubated for 1 h at RT in HRP conjugated antibodies at 1:2000 dilution for 1 hr at RT. The washed blots were then treated with an enhanced chemiluminiscence detection reagent (Amersham Bioscience, UK). Densitometric analysis of the protein bands was performed using the Image J software.
Peroxidase Activity
Isolated mitochondria were suspended at 0.5 mg/ml in assay buffer and incubated with 1 mM tBOOH for 1 min at 30°. A 1 μl aliquot was then withdrawn and added to 2 mL of assay buffer at 30° containing 10 μm amplex red and 25 U/mL horseradish peroxidase. Fluorescence of resorufin, the oxidized form of amplex red generated by peroxidation of tBOOH, was measured using excitation and emission wavelengths of 563 nm and 587 nm, respectively [36].
Measurements of total reduced glutathione
Glutathione measurements in are measured using ThioGloTM-1 (exicitation-388nm emission-500nm) as described elsewhere [37]. Glutathione was measured by the resulting fluorescence of the sample combined with cumene hydroperoxide and glutathione peroxidase subtracted from the fluorescence of the sample containing neither.
Data Analysis
Data are expressed as means ± standard error (SE) obtained from “n” different liver mitochondria or homogenates obtained from either drug- or drug vehicle-treated animals. Differences between groups were assessed by a two-tailed t-test. For non-parametic data, e.g. ratios, the data were transformed by taking the square root. All statistical analyses used Prism, version 5.0 software. Comparisons where p<0.05 was considered to be statistically significant.
RESULTS
Evidence for Nrf2 activation in livers from rats treated with sulforaphane
NAD(P)H quinone oxidoreductase 1 is an antioxidant enzyme whose gene and protein expression is generally up-regulated by compounds that activate the Nrf2/ARE pathway, including sulforaphane [38]. In order to verify that this Nrf2 pathway biomarker protein was elevated in rats 40 hr after intraperitoneal injection of sulforaphane at 10 mg/kg, liver homogenates were used for immunoblot analysis. This dose of sulforaphane and the time between injection of drug and liver acquisition were based on what were used previously for the analysis of brain mitochondria. Fig. 1A provides a sample immunoblot for NQO1 and the house-keeping protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for liver homogenates from a rat treated with drug vehicle and a rat treated with sulforaphane. As expected, the densitometric ratio of NQO1/GAPDH was significantly greater (more than 100%) in the homogenates from sulforaphane-treated rats (Fig. 1B).
Fig. 1.
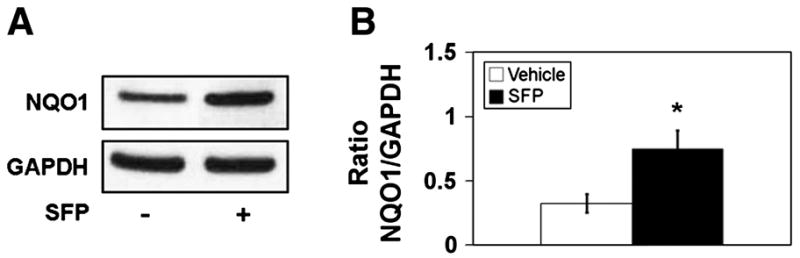
NAD(P)H quinone oxidoreductase 1 (NQO1) immunoreactivity in liver homogenates from vehicle- and sulforaphane (SFP) treated rats. A. Representative immunoblots for NQO1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). B. Densitometric ratios for NQO1/VDAC for n = 4–6 animals per group. * p<0.05, t=2.81
Inhibition of redox-regulated mitochondrial permeability transition pore opening by treatment of rats with sulforaphane
After each mitochondrial preparation, O2 electrode measurements were made of State 3 (ADP-stimulated) and State 4 (ATP synthase-inhibited) respiration as described in Materials and Methods. There were no significant differences in State 3 rates for mitochondria isolated from the livers of vehicle- and sulforaphane-treated rats, using a representative n=4 animals/group (50.5 ± 5.3 nmol O2/min/mg vs. 47.0 ± 5.3, t=0.47). No differences were also observed for State 4 rates (9.3 ± 1.1 vs. 7.0 ± 0.6, t=1.8). The respiratory control ratios, defined as State 3 divided by State 4 respiration were also not different (5.5 ± 0,4 vs. 7.0 ± 1.2, t=1.1). These data therefore indicate that the bioenergetic quality of the mitochondria isolated from both animal groups was good and not significantly different.
Figure 2A describes the respiration-dependent uptake and peroxide-induced release of Ca2+ by mitochondria isolated from the livers of rats inoculated 40 hr earlier with either sulforaphane or drug vehicle. Fig 2B describes the concurrent measurements of NAD(P)H autofluorescence as an indicator of mitochondrial redox state. The addition of 25 μM CaCl2 to the suspension of mitochondria caused an immediate increase in the fluorescence of the Ca2+ indicator Calcium Green 5N, followed by a reduction back to near baseline. Electrophoretic uptake of Ca2+ into mitochondria through the Ca2+ uniporter is driven by succinate-driven, respiratory generation of the electrochemical gradient of protons, with a positive-outside membrane potential. In the absence of further additions, this mitochondrial load of 50 nmol Ca2+/mg protein was retained within the mitochondria for at least 8 min with only a very slow rate of release into the medium (Fig. 2A, tracing 1). There was no change in NAD(P)H autofluorescence during this time (Fig. 2B, tracing 1), as expected since the presence of the electron transport chain Complex I inhibitor rotenone blocks the oxidation of NADH that would otherwise be stimulated by respiration-dependent activities like Ca2+ accumulation. Both the Calcium Green 5N and NAD(P)H fluorescent measurements were very similar for mitochondria from rats treated with vehicle and those treated with sulforaphane (Fig. 2A & B, tracings 1 and 4).
Fig. 2.

Peroxide-induced permeability pore transition (PTP) opening and pyridine nucleotide oxidation in rat liver mitochondria. A. Representative tracings of respiration-dependent mitochondrial Ca2+ uptake and subsequent PTP-dependent release. Extramitochondrial free Ca2+ was monitored with the Calcium Green 5N fluorescent dye in medium containing succinate as a respiratory substrate and the respiratory chain complex I inhibitor, rotenone. At time 0 mitochondria (0.5 mg/ml) from vehicle-treated or sulforaphane (SFP)-treated rats were loaded with 25 μM calcium, which is a level that was retained almost completely during the duration of the measurements (Line 1 & 4). At 250 sec, tert-butyl hydroperoxide (tBOOH) was added at either 5 μM (Line 2) or 250 μM (Line 3), resulting in an almost immediate increase in calcium green 5N fluorescence, representing mitochondrial Ca2+ release. Ca2+ efflux from control mitochondria was completely inhibited by the presence of 1 μM cyclosporin A (CsA) (Line 7) and was substantially greater than that observed with mitochondria from sulforaphane-treated rats (Lines 5 & 6). At 450 sec, ionomycin, a non-specific calcium ionophore, was added to completely release the small, residual mitochondrial Ca2+ (Line 3). B. Representative tracings of NAD(P)H autofluorescence in the absence and presence of tBOOH. Pyridine nucleotide redox state for mitochondria from either vehicle- or sulforaphane-treated rats remained highly reduced and stable after the addition of Ca2+ alone (Lines 1 & 4). The addition of 5 μM (Line 2) or 250 μM (Line 3) tBOOH caused immediate and complete oxidation of NAD(P)H within vehicle control mitochondria. Treatment of rats with SFP inhibited NAD(P)H oxidation at 5 μM tBOOH (Line 5), but did not inhibit oxidation at 250 μM tBOOH (Line 6).
Mitochondrial PTP opening is promoted by the presence of peroxides through their metabolism via glutathione peroxidase and subsequent oxidation of reduced glutathione, pyridine nucleotides, and protein cysteine sulfhydryl groups [39]. Glutathione peroxidase requires the reduced form of glutathione (GSH) formed from oxidized glutathione (GSSG) via NADPH-dependent glutathione reductase. At rates of peroxidation that exceed maximal rates of NADP+ and GSSG reduction, an oxidized shift in redox state occurs, as reflected by a decrease in NAD(P)H autofluorescence. When a 250 μM final concentration of tert-butyl hydroperoxide (tBOOH) was added to the suspension of mitochondria from a vehicle-treated rat at 250 sec after the addition of Ca2+, a rapid and continuous release of Ca2+ calcium ensued (Fig. 2A, tracing 3). This release of Ca2+ was nearly complete, as indicated by an only small further increase in Calcium Green 5N fluorescence after the subsequent addition of the Ca2+ ionophore, ionomycin (Fig. 2A, tracing 3). The tBOOH-induced Ca2+ release is due to PTP opening, as evidenced by the fact that the presence of 1 μM cyclosporin A completely blocked mitochondrial Ca2+ efflux (Fig. 2A, tracing 7). The rate at which Ca2+ was released upon addition of tBOOH was slightly slower than the rate at which the NAD(P)H fluorescence declined (Fig. 2B, tracing 3). In comparison to the mitochondria from vehicle-treated rats, the tBOOH-induced Ca2+ release by mitochondria from sulforaphane-treated rats was substantially slower (Fig. 2A, tracing 6) even though the rate and extent of NAD(P)H oxidation was very similar (Fig. 2B, tracing 3). Even at a much lower, 5 μM addition of tBOOH, the release of Ca2+ and the NAD(P)H oxidation for mitochondria from a vehicle-treated rat were very similar to those observed with 250 μM tBOOH (Fig. 2A & B, tracings 2). However, the addition of 5 μM tBOOH to mitochondria from sulforaphane-treated rats resulted in only a transient, minor release of accumulated Ca2+ and transient NAD(P)H oxidation, followed by a re-reduction to a level of maximal fluorescence (Fig. 2A & B, tracings 5).
Initial rates of the increase in Calcium Green 5N fluorescence starting 250 sec after the addition of Ca2+ alone or immediately after the subsequent addition of 5, 50, and 250 μM tBOOH are shown in Fig. 3A for n = 6 different mitochondrial preparations from either sulforaphane or vehicle-treated rats. Even though the Ca2+ release rate was very slow in the absence of tBOOH, it was significantly slower for sulforaphane-treated rats. The release rate was also significantly slower at each concentration of tBOOH with the largest reduction (75%) observed at 5 μM tBOOH. The final reductions in NAD(P)H autofluorescence observed at the end of these experiments is shown in Fig. 3B. No difference was observed after the addition of Ca2+ alone for mitochondria from vehicle and sulforaphane-treated rats. There were also no significant differences between animal groups for the extent of NAD(P)H oxidation elicited at either 50 or 250 μM tBOOH. At 5 μM, however, there was a significant, approximately 50% decrease in NAD(P)H oxidation observed with mitochondria from sulforaphane-treated rats.
Fig. 3.

Comparison of peroxide induced Ca2+ release rates and pyridine nucleotide oxidation for mitochondria from sulforaphane treated and vehicle treated rats (n = 7/group). A. Initial rates of rise in Calcium Green 5N fluorescence after addition of 0, 5, 50, and 250 μM tBOOH were all significantly slower for mitochondria from sulforaphane-treated rats. * p<0.05, t=2.16–4.91. Extent to which NAD(P)H autofluorescence decreased after additions of different concentrations of tBOOH was significantly less at 5 μM tBOOH. * p<0.05, t=0.82–2.20.
To determine if treatment of rats protects against mitochondrial PTP opening promoted by oxidative stress other than that generated by peroxide metabolism, we measured Ca2+ release rates in response to either phenylarsine oxide (PhAsO) or oxaloacetate (OAA). PhAsO induces PTP opening by directly oxidizing protein thiol groups associated with PTP components, e.g., CyD. Oxaloacetate indirectly causes PTP opening through its reduction to malate via the malate dehydrogenase reaction [40], which oxidizes NADH directly and NADPH indirectly, thus also causing an oxidized shift in the GSH and protein thiol redox states [41]. As shown in Fig. 4, sulforaphane treatment of rats reduced the rate of OAA-induced Ca2+ release by approximately 60%, which is similar to what was observed with the saturating concentrations of tBOOH. Sulforaphane treatment also significantly inhibited the rate of Ca2+ release induced by the addition of 30 μM PhAsO, albeit by only 25%. As with tBOOH, Ca2+ release elicited by the addition of either OAA or PhAsO was inhibited by the presence of the PTP inhibitor cyclosporin A (not shown).
Fig. 4.
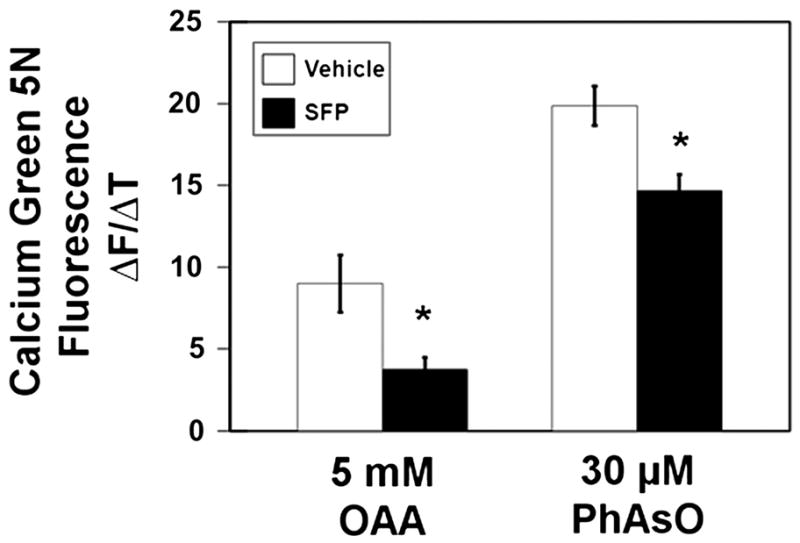
Inhibition of oxaloacetate- or phenylarsine oxide-induced Ca2+ release by rat liver mitochondria (n=4/group). A. Initial rates of rise in Calcium Green 5N fluorescence after the addition of 5 mM oxaloacetate (OAA) or B. 30 μM phenylarsine oxide (PhAsO) were significantly slower for sulforaphane-treated animals. * p<0.05, t=2.77–2.83.
Treatment of rats with sulforaphane does not alter cyclophilin D protein expression
One possible explanation for inhibition of PTP opening by sulforaphane treatment is down-regulation of CyD expression, since mitochondria from CyD knockout mice exhibit little if any PTP activity [22]. A sample immunoblot and densitometric quantification of CyD levels, expressed as the ratio of CyD to the mitochondrial voltage dependent anion channel (VDAC), are shown in Fig. 5A &B. These results indicate that sulforaphane treatment has no effect on levels of CyD in rat liver mitochondria.
Fig. 5.

Effects of sulforaphane treatment on immunoreactive levels of mitochondrial proteins related to regulation of permeability transition pore opening (n=6–7/group). Representative immunoblots and densitometry of A, B. cyclophilin D (CyD)/voltage dependent anion channel (VDAC), t=1.14, C, D. glutathione peroxidase 1 (GPX1)/VDAC, t=2.47, E, F. malic enzyme 3 (ME3)/VDAC, t=2.25, G, H. isocitrate dehydrogenase 2 (IDH2)/VDAC, t=1.19, I, J. thioredoxin 2 (Trx2)/VDAC, t=4.05, K, L. superoxide dismutase 2 (SOD2)/VDAC, 0.52. * p<0.05
Sulforaphane treatment selectively increases expression of proteins involved in mitochondrial defenses against oxidative stress
Sulforaphane could potentially affect the level of several mitochondrial proteins that could contribute to the inhibition of redox-sensitive PTP opening. For instance, an elevation of glutathione peroxidase could increase the rate at which added tBOOH is fully metabolized, resulting in a transient rather than a prolonged oxidized shift in mitochondrial redox state, as demonstrated in Fig. 2B. We therefore performed immunoblots for glutathione peroxidase 1 (GPX1). which is located in both liver cytosol and mitochondria [42]. As shown in Fig. 5C & D, sulforaphane treatment caused a significant, approximately 50% increase in GPX1 immunoreactivity present in isolated liver mitochondria. Evidence that this increase in protein is associated with an increase in activity came from measurements of peroxidase activity performed with freshly isolated mitochondria, as described in Materials and Methods. The resorufin fluorescence value shown in Fig. 6A represents the fraction of tBOOH present in a mitochondrial suspension after one minute incubation since the fluorescence value in the absence of mitochondria was 1000 units. There was significantly less fluorescence present after incubation with mitochondria from sulforaphane treated compared to vehicle treated rats. Based on the differences between maximal and post-incubation fluorescence of approximately 300 and 600 units for control and sulforaphane mitochondria, respectively, treatment of rats with sulforaphane resulted in an approximate 100% increase in total mitochondrial peroxidase activity.
Fig. 6.

Effects of sulforaphane treatment on mitochondrial peroxidase activity and glutathione content (n=7–10/group). A. Liver mitochondria were incubated with 1 mM tBOOH for one minute. An aliquot was added to a cuvette containing horseradish peroxidase (HRP) and amplex red. Any residual tBOOH not reduced to H2O reacts with HRP to oxidize amplex red into its fluorescent product, rezorufin. * p<0.05, t=2.39. B. Comparision of the content of reduced glutathione in liver mitochondria from either vehicle or sulforaphane treated rats. * p<0.05, t=2.93.
Glutathione peroxidase activity in situ requires the enzyme, reduced glutathione, and the glutathione reductase and NADPH necessary to re-reduce glutathione as it is oxidized. Although antibodies to mitochondrial glutathione reductase were not available, we did measure total mitochondrial glutathione and immunoreactivity for two of the enzymes responsible for reduction of NADP+ to form NADPH. Fig. 6B demonstrates that there was a small, albeit significant increase in total reduced glutathione present in mitochondria from sulforaphane- compared to vehicle-treated rats. Sample immunoblots and densitometry results for NADP+-dependent mitochondrial malic enzyme 3 (ME3) and NADP+-dependent isocitrate dehydrogenase 2 (IDH2) indicate that there was a significant, >100% increased ME3 immunoreactivity in mitochondria from sulforaphane treated rats (Fig. 5E & F) but no difference in IDH2 levels for the two animal groups (Fig. 5G & H).
Mitochondria contain other antioxidant defense enzmes, including thioredoxin 2 (Trx2), thioredoxin reductase 2, and thioredoxin peroxidase, also known as peroxiredoxin 3 and 5. Thioredoxins in the mitochondria serve a dual function of being able to both scavenge free radicals as well as keep protein thiols reduced. Trx2 protein levels were significantly higher in mitochondria from sulforaphane-treated rats (Fig. 5I & J).
We also tested for the effects of sulforaphane treatment on the immunoreactivity of the mitochondria-specific superoxide dismutase (SOD2), based on the findings that the tBOOH-induced PTP is more active in both liver and heart mitochondria from SOD2 +/− mice [43], and that sulforaphane increases total mitochondrial SOD activity in rat aortic muscle cells [44]. As shown in Fig. 5K & L, sulforaphane had no effect on rat liver mitochondrial SOD2 immunoreactivity.
DISCUSSION
This is the first in-depth study of the regulation of mitochondrial PTP opening and antioxidant-related mitochondrial proteins by a pharmacologic activator of the Nrf2 pathway of cytoprotective gene expression. The primary conclusion reached from this study is that treatment of rats with sulforaphane results in a robust inhibition of PTP opening in rat liver mitochondria triggered by several forms of oxidative stress. These forms include a shift in redox state induced through either glutathione-dependent peroxide metabolism or direct oxidation of pyridine nucleotides by NADH-dependent reduction of oxaloacetate. In addition, sulforaphane treatment also inhibits PTP opening by direct oxidation of sulfhydryl groups mediated by phenylarsine oxide. An additional novel finding from this study is that sulforaphane treatment selectively increases several important mitochondrial proteins and molecules that serve as direct antioxidants, e.g. glutathione peroxidase, thioredoxin, and glutathione, or that generate the reducing power in the form of NADPH that is necessary for driving these antioxidant activities, i.e., malic enzyme.
A number of findings provide insight into molecular mechanisms responsible for inhibition of PTP opening by treatment of rats with sulforaphane. The > 100% increase in NQO1 immunoreactivity in liver homogenates strongly suggests that sulforaphane activates the Nrf2 pathway of gene expression [45]. While the pattern of mitochondrial protein expression that is elevated by sulforaphane treatment supports this conclusion, additional comparisons between mitochondria from Nrf2 +/+ and Nrf2 −/− mice will be necessary to prove that Nrf2 activation is necessary for sulforaphane inhibition of PTP opening [46]. One mechanism of action that can be ruled out is a general influence of sulforaphane treatment on basic mitochondrial bioenergetics, based on no differences observed for rates of state 3 or state 4 respiration. These findings do not, however, address the possibility that sulforaphane and other Nrf2 activators can stimulate mitochondrial biogenesis, as described by Piantadosi et al. [47].
The possibility that sulforaphane treatment inhibits PTP opening by reducing the expression of PTP components remains an open question, due mainly to the fact that the identity of these components is unresolved. We focused our measurements on CyD since the relationship between its expression and PTP activity is best characterized and because it has no apparent direct influence over mitochondrial bioenergetics like other putative PTP components, e.g., the adenine nucleotide transporter and the phosphate transporter. We found that sulforaphane treatment has no effect on CyD immunoreactivity in rat liver mitochondria, which is consistent with the lack of an effect reported earlier for rat brain mitochondria [34].
Insight into mechanisms that are applicable to the inhibition of PTP opening by sulforaphane comes from comparisons between release of accumulated Ca2+ and oxidization of pyridine nucleotides at different concentrations of tBOOH. Similar rates of tBOOH-induced mitochondrial Ca2+ release and complete NAD(P)H oxidation were observed at 5, 50, and 250 μM tBOOH for mitochondria from vehicle-treated rats (Fig. 3). While Ca2+ release rates and pyridine nucleotide oxidation were also similar at both 50 and 250 μM tBOOH with mitochondria from sulforaphane-treated rats, both were much reduced at 5 μM tBOOH and also much lower than the values obtained with mitochondria from vehicle-treated animals. This comparison is similar to what we reported when comparing redox- sensitive PTP activity for mitochondria for normal rat liver and AS-30D hepatoma mitochondria. Compared to normal liver mitochondria, hepatoma mitochondria are resistant to both tBOOH-induced NAD(P)H oxidation and Ca2+ release [48]. We determined that the cause for this resistance is increased mitochondrial malic enzyme activity, which produces NADPH and increases the intramitochondrial redox buffering power. The fact that ME3 immunoreactivity is more than twice as high in the liver mitochondria from rats treated with sulforaphane (Fig. 5) is consistent with the redox buffering power being responsible for resistance to PTP opening, at least at the low tBOOH concentration of 5 μM. Another possible explanation is that tBOOH is metabolized so quickly that the redox state is only temporarily disturbed, resulting in only transient PTP opening. The transient rise in medium [Ca2+] concentration and reduction in NAD(P)H autofluorescence described in Fig. 2 for mitochondria from sulforaphane-treated rats is consistent with this mechanism. This hypothesis is also supported by the finding that the total peroxidase activity of these mitochondria is significantly greater than that of mitochondria from vehicle-treated rats (Fig. 6).
The increase in either malic enzyme or mitochondrial peroxidases could explain sulforaphane-induced resistance of PTP opening at low tBOOH levels but is unlikely to be responsible at concentrations of 50 μM and above. At these higher tBOOH levels, complete and sustained pyridine nucleotide oxidation occurs with mitochondria from both vehicle- and sulforaphane treated rats, thus overcoming any elevated rates of NADPH production or peroxide metabolism. The increase in mitochondrial total reduced glutathione observed after sulforaphane administration could contribute to increased sulfhydryl buffering power; however the 25% increase is modest (Fig. 6). Another possible mechanism may lie distal to redox buffering and be limited kinetically rather than thermodynamically by reduction of protein thiol groups. The increased thioredoxin immunoreactivity observed in mitochondria from sulforaphane-treated rats is consistent with this possibility (Fig. 5). There are several other proteins that could also contribute to maintenance of reduced sulfhydryl redox state for PTP associated proteins like CyD. These proteins include but are not limited to thioredoxin reductase, glutaredoxin and glutaredoxin reductase [49].
In summary, measurements of peroxide induced release of accumulated Ca2+ and pyridine nucleotide oxidation demonstrate that redox-sensitive PTP opening by isolated liver mitochondria is substantially inhibited by treatment of rats with i.p. sulforaphane over one and a half days earlier, particularly at lower levels of added tBOOH that are likely most relevant to levels of oxidative stress that occur in situ within cells. Several mechanisms of action are probably responsible for this inhibition, including increased redox buffering power, more rapid disposition of added peroxide, and more effective use of mitochondrial redox buffering to maintain PTP associated protein sulfhydryls in the reduced redox state. Experiments are in progress to further characterize the relative contribution of each of these mechanisms in this system. Considering the over 100 genes whose expression are induced by Nrf2 activators like sulforaphane, it is not surprising that the mitochondrion, like the entire cell, responds in multiple ways to Nrf2 activation. This multifactorial response probably confers mitochondria with protection against different forms of oxidative stress and many sequelae, including PTP opening and associated metabolic dysfunction.
Acknowledgments
This study was supported by NIH RO1 NS34152
LIST OF ABBREVIATIONS
- ADP
adenosine 5′-diphosphate
- ARE
antioxidant response element
- CSA
cyclosporin A
- CyD
cyclophilin D
- DMSO
dimethylsulfoxide
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- GPX1
glutathione peroxidase 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSH
reduced glutathione
- GSR
glutathione reductase
- GSSG
oxidized glutathione
- HO-1
heme oxygenase 1
- IDH2
isocitrate dehydrogenase 2
- ME3
malic enzyme 3
- NQO1, NAD(P)H
quinone oxidoreductase 1
- Nrf2
NF-E2-related factor 2
- OAA
oxaloacetate
- PhAsO
phenylarsine oxide
- PTP
mitochondrial permeability transition pore
- SOD2
superoxide dismutase 2
- SFP
sulforaphane
- tBOOH
tert-butyl hydroperoxide
- Trx2
thioredoxin 2
- VDAC
voltage dependent anion channel
Reference List
- 1.Bianchi G, Marchesini G, Fabbri A, Ronchi M, Chianese R, Grossi G. Lipoperoxide plasma levels in patients with liver cirrhosis. Hepatogastroenterology. 1997;44:784–788. [PubMed] [Google Scholar]
- 2.Yamamoto Y, Yamashita S, Fujisawa A, Kokura S, Yoshikawa T. Oxidative stress in patients with hepatitis, cirrhosis, and hepatoma evaluated by plasma antioxidants. Biochem Biophys Res Commun. 1998;247:166–170. doi: 10.1006/bbrc.1998.8752. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Flecha B, Cutrin JC, Boveris A. Time course and mechanism of oxidative stress and tissue damage in rat liver subjected to in vivo ischemia-reperfusion. J Clin Invest. 1993;91:456–464. doi: 10.1172/JCI116223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Packer MA, Murphy MP. Peroxynitrite causes calcium efflux from mitochondria which is prevented by Cyclosporin A. FEBS Lett. 1994;345:237–240. doi: 10.1016/0014-5793(94)00461-7. [DOI] [PubMed] [Google Scholar]
- 5.Moore M, Thor H, Moore G, Nelson S, Moldeus P, Orrenius S. The toxicity of acetaminophen and N-acetyl-p-benzoquinone imine in isolated hepatocytes is associated with thiol depletion and increased cytosolic Ca2+ J Biol Chem. 1985;260:13035–13040. [PubMed] [Google Scholar]
- 6.Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–810. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazzeo AT, Beat A, Singh A, Bullock MR. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp Neurol. 2009;218:363–370. doi: 10.1016/j.expneurol.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 8.Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38:841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 9.Kukielka E, Dicker E, Cederbaum AI. Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch Biochem Biophys. 1994;309:377–386. doi: 10.1006/abbi.1994.1127. [DOI] [PubMed] [Google Scholar]
- 10.Devi BG, Henderson GI, Frosto TA, Schenker S. Effect of acute ethanol exposure on cultured fetal rat hepatocytes: relation to mitochondrial function. Alcohol Clin Exp Res. 1994;18:1436–1442. doi: 10.1111/j.1530-0277.1994.tb01447.x. [DOI] [PubMed] [Google Scholar]
- 11.Ponsoda X, Bort R, Jover R, Gomez-Lechon MJ, Castell JV. Molecular mechanism of diclofenac hepatotoxicity: Association of cell injury with oxidative metabolism and decrease in ATP levels. Toxicol In Vitro. 1995;9:439–444. doi: 10.1016/0887-2333(95)00035-7. [DOI] [PubMed] [Google Scholar]
- 12.Shigesawa T, Sato C, Marumo F. Significance of plasma glutathione determination in patients with alcoholic and non-alcoholic liver disease. J Gastroenterol Hepatol. 1992;7:7–11. doi: 10.1111/j.1440-1746.1992.tb00926.x. [DOI] [PubMed] [Google Scholar]
- 13.Thorniley MS, Simpkin S, Fuller B, Jenabzadeh MZ, Green CJ. Monitoring of surface mitochondrial NADH levels as an indication of ischemia during liver isograft transplantation. Hepatology. 1995;21:1602–1609. [PubMed] [Google Scholar]
- 14.Takeyama N, Matsuo N, Tanaka T. Oxidative damage to mitochondria is mediated by the Ca(2+)-dependent inner-membrane permeability transition. Biochem J. 1993;294:719–725. doi: 10.1042/bj2940719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nieminen AL, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem J. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Herman B. The mitochondrial permeability transition in toxic, hypoxic and reperfusion injury. Mol Cell Biochem. 1997;174:159–165. [PubMed] [Google Scholar]
- 17.Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84:153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- 18.Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- 19.Norberg E, Gogvadze V, Ott M, Horn M, Uhlen P, Orrenius S, Zhivotovsky B. An increase in intracellular Ca2+ is required for the activation of mitochondrial calpain to release AIF during cell death. Cell Death Differ. 2008;15:1857–1864. doi: 10.1038/cdd.2008.123. [DOI] [PubMed] [Google Scholar]
- 20.Gogvadze V, Norberg E, Orrenius S, Zhivotovsky B. Involvement of Ca2+ and ROS in alpha-tocopheryl succinate-induced mitochondrial permeabilization. Int J Cancer. 2010;127:1823–1832. doi: 10.1002/ijc.25204. [DOI] [PubMed] [Google Scholar]
- 21.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 24.Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 2011;45:156–164. doi: 10.3109/10715762.2010.520319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol. 2009;6:542–553. doi: 10.1038/nrgastro.2009.127. [DOI] [PubMed] [Google Scholar]
- 26.Liu SL, Degli ES, Yao T, Diehl AM, Zern MA. Vitamin E therapy of acute CCl4-induced hepatic injury in mice is associated with inhibition of nuclear factor kappa B binding. Hepatology. 1995;22:1474–1481. [PubMed] [Google Scholar]
- 27.Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol. 2007;35:459–473. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- 28.Balogun E, Hoque M, Gong P, Killeen E, Green CJ, Foresti R, Alam J, Motterlini R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J. 2003;371:887–895. doi: 10.1042/BJ20021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JM, Moehlenkamp JD, Hanson JM, Johnson JA. Nrf2-dependent activation of the antioxidant responsive element by tert-butylhydroquinone is independent of oxidative stress in IMR-32 human neuroblastoma cells. Biochem Biophys Res Commun. 2001;280:286–292. doi: 10.1006/bbrc.2000.4106. [DOI] [PubMed] [Google Scholar]
- 30.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 31.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 32.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 33.Halestrap AP, Kerr PM, Javadov S, Woodfield KY. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim Biophys Acta. 1998;1366:79–94. doi: 10.1016/s0005-2728(98)00122-4. [DOI] [PubMed] [Google Scholar]
- 34.Greco T, Fiskum G. Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. J Bioenerg Biomembr. 2010;42:491–497. doi: 10.1007/s10863-010-9312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreyev A, Fiskum G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999;6:825–832. doi: 10.1038/sj.cdd.4400565. [DOI] [PubMed] [Google Scholar]
- 36.Kowaltowski AJ, Fenton RG, Fiskum G. Bcl-2 family proteins regulate mitochondrial reactive oxygen production and protect against oxidative stress. Free Radic Biol Med. 2004;37:1845–1853. doi: 10.1016/j.freeradbiomed.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RS, Kochanek PM, Kagan VE. Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol. 2007;62:154–169. doi: 10.1002/ana.21168. [DOI] [PubMed] [Google Scholar]
- 38.Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52 (Suppl 1):S128–38. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- 39.Toman J, Fiskum G. Influence of aging on membrane permeability transition in brain mitochondria. J Bioenerg Biomembr. 2011;43:3–10. doi: 10.1007/s10863-011-9337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehninger AL, Vercesi A, Bababunmi EA. Regulation of Ca2+ release from mitochondria by the oxidation-reduction state of pyridine nucleotides. Proc Natl Acad Sci U S A. 1978;75:1690–1694. doi: 10.1073/pnas.75.4.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vercesi AE. Possible participation of membrane thiol groups on the mechanism of NAD(P)+-stimulated Ca2+ efflux from mitochondria. Biochem Biophys Res Commun. 1984;119:305–310. doi: 10.1016/0006-291x(84)91652-8. [DOI] [PubMed] [Google Scholar]
- 42.Esworthy RS, Ho YS, Chu FF. The Gpx1 gene encodes mitochondrial glutathione peroxidase in the mouse liver. Arch Biochem Biophys. 1997;340:59–63. doi: 10.1006/abbi.1997.9901. [DOI] [PubMed] [Google Scholar]
- 43.Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;281:H1422–H1432. doi: 10.1152/ajpheart.2001.281.3.H1422. [DOI] [PubMed] [Google Scholar]
- 44.Zhu H, Jia Z, Strobl JS, Ehrich M, Misra HP, Li Y. Potent induction of total cellular and mitochondrial antioxidants and phase 2 enzymes by cruciferous sulforaphane in rat aortic smooth muscle cells: cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol. 2008;8:115–125. doi: 10.1007/s12012-008-9020-4. [DOI] [PubMed] [Google Scholar]
- 45.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;501:116–123. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 47.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiskum G, Pease A. Hydroperoxide-stimulated release of calcium from rat liver and AS-30D hepatoma mitochondria. Cancer Res. 1986;46:3459–3463. [PubMed] [Google Scholar]
- 49.Koehler CM, Beverly KN, Leverich EP. Redox pathways of the mitochondrion. Antioxid Redox Signal. 2006;8:813–822. doi: 10.1089/ars.2006.8.813. [DOI] [PubMed] [Google Scholar]