

Abstract
In the olfactory epithelium (OE), injury induces ATP release, and subsequent activation of P2 purinergic receptors by ATP promotes neuroregeneration by increasing basal progenitor cell proliferation. The molecular mechanisms underlying ATP-induced increases in OE neuroregeneration have not been established. In the present study, the roles of neuroproliferative factors neuropeptide Y (NPY) and fibroblast growth factor2 (FGF2), and p44/42 extracellular signal-regulated kinase (ERK) on ATP-mediated increases of neuroregeneration in the OE were investigated. ATP increased basal progenitor cell proliferation in the OE via activation of P2 purinergic receptors in vitro and in vivo as monitored by incorporation of 5′-ethynyl-2′-deoxyuridine, a thymidine analog, into DNA, and proliferating cell nuclear antigen (PCNA) protein levels. ATP induced p44/42 ERK activation in globose basal cells (GBC) but not horizontal basal cells (HBC). ATP differentially regulated p44/42 ERK over time in the OE both in vitro and in vivo with transient inhibition (5–15 min) followed by activation (30 min – 1 hr) of p44/42 ERK. In addition, ATP indirectly activated p44/42 ERK in the OE via ATP-induced NPY release and subsequent activation of NPY Y1 receptors in the basal cells. There were no synergistic effects of ATP and NPY or FGF2 on OE neuroregeneration. These data clearly have implications for the pharmacological modulation of neuroregeneration in the olfactory epithelium.
Keywords: P2 purinergic receptors, NPY Y1 receptors, p44/42 ERK, globose basal cells, horizontal basal cells, synergistic effect
INTRODUCTION
The olfactory epithelium (OE) is a good model to study the mechanisms of injury-induced neuroregeneration as olfactory sensory neurons (OSNs) are easily damaged due to direct contact with airborne pollutants, toxicants and microbes and continuously regenerate throughout adulthood (Graziadei and Graziadei, 1979a; Graziadei and Graziadei, 1979b; Graziadei and Monti-Graziadei, 1978). After significant chemical, infectious or traumatic damage to the OE, the rate of neuroregeneration accelerates (Calof et al., 2002; Holcomb et al., 1995; Islam et al., 2006; Manglapus et al., 2004; Moon et al., 2009; Sultan-Styne et al., 2009). OSNs are regenerated to replace injured and dying OSNs by local restricted neuronal progenitor cells called basal cells. The two types of basal cells, globose basal cell (GBC) and horizontal basal cell (HBC), are located just above the basement membrane. In mature OE, basal cells proliferate into neuronal precursor cells and then differentiate into OSNs or non-neuronal cells (Carr and Farbman, 1992; Carter et al., 2004; Huard et al., 1998; Leung et al., 2007).
In the central nervous system (CNS), ATP is released from neurons and astrocytes upon injury and promotes neuroregeneration and cell proliferation via activation of P2 purinergic receptors (Franke and Illes, 2006; Neary and Zimmermann, 2009). In the OE, injury by toxic compounds such as nickel sulfate, satratoxin G or high concentrations of odorants induces ATP release and ATP promotes basal cell proliferation via activation of P2 purinergic receptors (Hegg and Lucero, 2006; Jia et al., 2010; Jia et al., 2011b). P2 purinergic receptors, including P2X and P2Y, are expressed in the OE (Hegg et al., 2003). ATP activation of these receptors evokes Ca2+ transients (Hassenklöver et al., 2009; Hegg et al., 2003; Hegg et al., 2009), releases trophic factors (Jia et al., 2011a; Kanekar et al., 2009), increases basal cell proliferation, differentiation and maturation of OSNs (Hassenklöver et al., 2009; Jia et al., 2009). Collectively, these data indicate that ATP is released and promotes OE neuroregeneration via activation of P2 purinergic receptors following injury. However, the molecular mechanisms underlying ATP-induced neuroregeneration in the OE are not known. In the CNS, P2 purinergic receptors activate p44/42 extracellular signal-regulated kinase (ERK) to induce cell proliferation (Franke and Illes, 2006; Neary and Zimmermann, 2009). The synergistic effects of ATP and polypeptide growth factors on cell proliferation are through parallel activation of p44/42 ERK signalling (Neary et al., 2008). In the OE, removal of the olfactory bulbs axotomizes the OSNs and induces a retrograde wave of OSN apoptosis within 3 days followed by a synchronized increase in basal cell proliferation in 2–3 weeks post-bulbectomy(Carter et al., 2004; Costanzo and Graziadei, 1983; Cowan et al., 2001; Schwob et al., 1992). While in the same time frame of 2–3 weeks post-bulbectomy, mitogen-activated protein kinase (MAPK) phosphatase-1, that inactivates MAPK, decreases greatly and phospho-p44/42 ERK robustly increases (Shinogami and Ishibashi, 2000), suggesting that activation of p44/42 ERK is involved in bulbectomy-induced increases in basal cell proliferation. The basal cells in the OE express P2Y purinergic receptors (Hegg et al., 2003). P2Y receptor activation of p44/42 ERK signaling promotes neuroregeneration in the CNS (Franke and Illes, 2006). Thus, we investigated the role of p44/42 ERK in the ATP-induced increase in basal cell proliferation in the OE in vitro and in vivo.
NPY, a 36-amino acid polypeptide widely expressed in the central and peripheral nervous systems, has been found to activate p44/42 ERK signaling and stimulate neuronal precursor cell proliferation in primary cultures isolated from the dentate gyrus, retina and subventricular zone (Agasse et al., 2008; Alvaro et al., 2008; Howell et al., 2005; Howell et al., 2003; Thiriet et al., 2011). In the OE, NPY is localized in non-neuronal sustentacular and microvillous cells which reside in the apical layer of OE and send long cell processes to the basement membrane (Hansel et al., 2001; Hegg et al., 2010; Jia and Hegg, 2010; Kanekar et al., 2009; Montani et al., 2006). A significant reduction in olfactory neuronal precursor proliferation occurs in NPY deficient mice (Hansel et al., 2001) and NPY Y1 receptor knockout mice (Doyle et al., 2008). Furthermore, NPY activation of Y1 receptor, expressed on basal cells, stimulates basal cell proliferation via activation of p44/42 ERK (Doyle et al., 2008; Hansel et al., 2001). We have previously demonstrated that ATP activation of P2 purinergic receptors up-regulates NPY expression, induces NPY release and promotes basal cell proliferation via activation of Y1 receptors in the OE (Jia and Hegg, 2010; Kanekar et al., 2009). Therefore, in the present study, we examined whether ATP-induced NPY release is involved in ATP-mediated regulation of p44/42 ERK in the OE. The synergistic effects of ATP and polypeptide growth factors, such as NPY and fibroblast growth factor 2 (FGF2), on OE neuroregeneration are also investigated.
MATERIALS AND METHODS
Animals
Adult (6–8 weeks) and young (postnatal day 21) male Swiss Webster mice were purchased from Charles River (Portage, MI) and housed in a temperature-, humidity-, and light-controlled room (12 hours light/dark cycle, lights on from 7:00 A.M. to 7:00 P.M.). Food and water were available ad libitum. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals as approved by Michigan State University Institutional Animal Care and Use Committee.
OE primary cell culture
The primary OE cells were obtained from olfactory epithelia of young male Swiss Webster mouse at postnatal day 21 due to high cell viability at this age vs. adult and cultured as described previously (Hegg et al., 2003). Our previous study shows that there is no age difference in ATP-induced cell proliferation in the OE (Jia et al., 2009). The cells were plated onto concanavalin A-coated 6-well plates or coverslips at 2 × 106 cells/ml and cultured in DMEM media supplemented with 5% FBS, 1% penicillin streptomycin, 1% insulin and 0.1% ascorbic acid at 37 °C. All treatments start at 5–7 days culture when the OE primary cell cultures contain 50% mature OSNs, 8% basal cells and 28% sustentacular and microvillous cells.
Measurement of ATP-induced increases in neuroregeneration
OE primary cells were incubated with saline or ATP (10, 50, 100, 200 and 500 μM) for 2 hours. Each treatment had 3–5 replications. The 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay was performed using the Click-iT EdU® Alexa Fluor 488 imaging kit following manufacturer’s instructions (Invitrogen, Carlsbad, CA). The advantage of the Click-iT EdU® assay is that EdU detection does not require DNA denaturation, which not only disrupts dsDNA integrity but also affects nuclear counterstaining and destroys cell morphology. We chose EdU in these experiments to allow co-localization of EdU-labeling with nuclei counterstained with DAPI (20 nM; 5–10 min). Co-localization of EdU with DAPI using Metamorph 7.5 (MDS Analytical technologies, Sunnyvale, CA) was determined by measuring the area of overlap between two fluorescent probes at 488 and 405nm wavelengths from 3–5 non-overlapping fields of view per replication taken using a 10X objective (0.3 n. a.) on a Nikon TE2000-U inverted fluorescence microscope (Nikon, Melville, NY). The data was expressed as the percentage of area of overlap to area of DAPI. To measure protein levels of proliferating cell nuclear antigen (PCNA), OE primary cells were incubated with saline vehicle or ATP (100 μM) for 2 hours. Some cells were pre-treated with a mixture of P2 purinergic receptor antagonists (PPADS, 25 μM and suramin, 100 μM) for 30 min shown previously to inhibit P2 receptors in the OE (Hegg et al., 2003). Following treatment, cells were washed with ice-cold phosphate-buffered saline (PBS), collected, and centrifuged at 10,000g for 3 min at 4 °C. The pellets were stored at −80 °C for further western blot analysis. Each treatment had 3 replications.
Anesthetized adult mice (4% isoflurane, n = 4 mice/group) were intranasally instilled with saline vehicle or P2 purinergic receptor antagonists (PPADS, 50 nmoles/kg and suramin, 200 nmoles/kg), followed by saline or ATP (400 nmoles/kg) 30 min later. Intranasal instillation was performed via a pipette tip placed immediately above the nares while the mouse was in a supine position. Test compounds (50 μl) were intranasally delivered dropwise to the nares, and were subsequently aspirated. The OE tissue was collected 48 hours post-instillation of ATP for immunohistochemistry as described previously (Jia et al., 2010). Briefly, mice were cardiac-perfused with ice-cold 0.1M PBS followed by 4% paraformaldehyde (PFA). The tissue was postfixed overnight in 4% PFA and decalcified with EDTA (0.5 M, pH = 8) for 4–5 days. Frozen coronal sections of OE (10 or 20 μm) were collected from level 3 of the mouse nasal cavity taken at the level of the second palatal ridge (Young, 1981).
Time course of ATP-induced modulation of p44/42 ERK
OE primary cells were incubated with saline or ATP (100 μM) for 5, 15, 30 min, 1, 2 or 3 hours. Then cells were washed with ice-cold PBS, collected by cell scraper and centrifuged at 10,000g for 3 min at 4 °C. The pellets were stored at −80 °C. Each treatment included 4 replications. Anesthetized adult mice (4% isoflurane, n = 4 mice/group) were intranasally instilled with saline or ATP (400 nmoles/kg) and OE tissue was dissected and collected by decapitation at 5, 15, 30 min, 1, 6 and 24 hours post-instillation and stored at −80 °C for further western blot analysis.
Co-localization of ATP-induced activation of p44/42 ERK with basal cell markers
OE primary cells were incubated with saline or ATP (100 μM) for 30 min and fixed with 4% PFA for 10 min. In vivo, anesthetized adult mice (4% isoflurane) were intranasally instilled with saline or ATP (400 nmoles/kg). The OE tissue was collected 1 hour post-instillation for immunohistochemistry as described above.
The p44/42 ERK activation mediates ATP-induced increase in neuroregeneration
Anesthetized adult mice (4% isoflurane, n = 3 mice/group) were intranasally instilled with saline vehicle or MEK inhibitor U0126 (400 nmoles/kg) followed by saline or ATP (400 nmoles/kg) instillation 1 hour later. The OE tissue was collected 48 hours post-instillation of ATP for immunohistochemistry as described above.
NPY release mediates ATP-induced p44/42 ERK activation
Anesthetized adult mice (4% isoflurane, n = 4 mice/group) were intranasally instilled with saline vehicle or ATP (400 nmoles/kg) followed by saline vehicle or NPY Y1 receptor antagonist BIBP3226 (20 nmoles/kg) 5 min later. The OE tissue was dissected and collected 1 hour post-instillation of ATP by decapitation and stored at −80 °C for further western blot analysis.
The synergistic effect of ATP and NPY or FGF2 on neuroregeneration
Anesthetized adult mice (4% isoflurane, n = 3–4 mice/group) were intranasally instilled with saline, ATP (400 nmoles/kg), NPY (4 nmoles/kg), FGF2 (12 pmoles/kg), a cocktail of ATP and NPY or FGF2 and followed by BrdU injection (i.p., total 216 mg/kg) at 42, 44, 46 hours later. The OE tissue was collected 2 hours after last BrdU injection for immunohistochemistry as described above. For measuring the levels of phospho-p44/42 ERK and p44/42 ERK in vitro, OE primary cells were incubated with saline, NPY (1 μM) or FGF2 (3 μM) for 5, 30 min and 1 hour. Then cells were washed with ice-cold PBS, collected and centrifuged at 10,000g for 3 min at 4 °C. The pellets were stored at −80 °C for western blot analysis. Each treatment had 4 replications.
Western blot
The OE primary cells were lysed and OE tissue was homogenized by sonication in Tris buffer with a protease inhibitor cocktail (1:1000, Sigma-Aldrich, St. Louis, MO). The concentration of protein in the homogenate and cell lysates was measured using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). Homogenates or cell lysates (20 μg/lane or 7.5 μg/lane) were resolved by electrophoresis and transferred to a nitrocellulose membrane. After incubation with blocking buffer containing 0.1% Tween-20 and 5% BSA, the membranes were probed with mouse anti-PCNA(1:1000, Millipore, Billerica, MA) or mouse anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibody (1:2000, Cell signaling, Danvers, MA) overnight at 4°C. After washing with wash buffer containing 0.1% Tween-20, the membranes were incubated with HRP-labeled secondary antibody (Jackson Laboratory, West Grove, PA, USA). Immunoreactive proteins were detected with a chemiluminescence reagent (ECL, Amersham Biosciences, Piscataway, NJ) and then exposed to Kodak X-ray film. The membranes were then stripped with restore™ plus western blot stripping buffer (Thermo Scientific, Rockford, IL), re-blocked with 0.05% Tween-20 and 0.2% I-block (Millipore, Bedford, MA), re-probed with rabbit anti-p44/42 MAPK antibody (1:1000, Cell signaling, Danvers, MA) and mouse anti-actin antibody (1:5000, Sigma, St Louis, MS) overnight at 4°C followed by HRP-labeled secondary antibody. Immunoreactive proteins were detected with ECL and then exposed to Kodak X-ray film. Films were analyzed by Image J (NIH). Integrated optical densities (IOD)/μg protein in treatment groups were normalized to saline vehicle animals. The value of phospho-p44/42 ERK for each animal was then normalized to the value of p44/42 ERK. Since the levels of p44/42 ERK in some groups of cell lysate were reduced, we used actin as loading control in vitro and hence the value of phospho-p44/42 ERK was then normalized to the value of actin. The value of PCNA for each animal was normalized to the value of actin. Each sample was measured on three independent gels.
Immunohistochemistry
For detection of PCNA, tissue sections were rehydrated with PBS and antigen retrieval was performed by placing sections in a citrate buffer (pH = 6) and heated in a microwave oven (700 Watt) at low power for 6 min, 2 times and cooled down for 20 min. Then the sections were permeabilized with 0.01% triton X-100, blocked with 5% BSA and incubated with rabbit anti-PCNA IgG (1:50, Abcam, Cambridge, MA) overnight at 4 °C. For detection of BrdU, tissue sections were permeabilized with 0.3% triton X-100, blocked with 10% normal donkey serum, incubated in 2 M HCl for 30 min at 65°C to denature DNA, and then incubated with rat anti-BrdU IgG (1:100, Abcam, Cambridge, MA) overnight at 4°C. For detection of phospho-p44/42 ERK, the tissue sections were permeabilized with 0.3% Triton X-100 for 20 min and followed by 1 hour blocking with 10% normal donkey serum containing 0.3% Triton X-100. Tissue sections were incubated with mouse anti-phospho-p44/42 MAPK antibody (1:400, Cell signaling, Danvers, MA) for 120 hours at 4 °C. For double-labeling detection, tissue sections were incubated with the mixture of rabbit anti-phospho-p44/42 MAPK (1:400, Cell signaling, Danvers, MA) and mouse anti-MASH1 (1:30, BD Pharmingen, San Diego, CA) antibody or mouse anti-phospho-p44/42 MAPK antibody (1:400) and rabbit anti-cytokeratin 5 (1:100, Abcam, Cambridge, MA) for 120 hours at 4 °C. Immunoreactivity was detected using TRITC or FITC-conjugated donkey anti-rabbit or mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). The nuclei were counterstained with Vectashield mounting medium for fluorescence with DAPI (Vector Laboratory, Burlingame, CA). Immunoreactivity was visualized on an Olympus FV1000 confocal laser scanning microscope. Antibody specificity was examined by omitting the primary antibody or secondary antibody. No immunoreactivity was observed in any of the controls. The number of PCNA+ or BrdU+ cells per linear millimeter of ecto-turbinate 2 and endo-turbinate II on three consecutive coronal sections of OE between level 3 and 4 in each animal were counted by an experimenter blind to the treatments and then normalized to the length of OE on which the immunopositive cells were scored.
Immunocytochemistry
The cells were washed with PBS and permeabilized with 0.3% Triton X-100 for 20 min and followed by 1 hr blocking with 10% normal donkey serum containing 0.3% Triton X-100. Cells were then incubated with mouse anti-phospho-p44/42 MAPK antibody (1:800, Cell signaling, Danvers, MA) overnight at 4 °C. For double-labeling immunofluorescence, the cells were incubated with the mixture of rabbit anti-phospho-p44/42 MAPK (1:800, Cell signaling, Danvers, MA) and mouse anti-MASH1 (1:50, BD Pharmingen, San Diego, CA) antibody or mouse anti-phospho-p44/42 MAPK antibody (1:800) and rabbit anti-cytokeratin 5 (1:200, Abcam, Cambridge, MA) overnight at 4 °C. Immunoreactivity was detected using TRITC- or FITC-conjugated donkey anti-rabbit or mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). The nuclei were counterstained using Vectashield mounting medium for fluorescence with DAPI (Vector Laboratory, Burlingame, CA). Antibody specificity was examined by omitting the primary antibody or secondary antibody. No immunoreactivity was observed in any of the controls. Co-localization of phospho-pERK with DAPI in vehicle- and ATP-treated cells and phospho-pERK with MASH1 or Cy5 in ATP-treated cells was analyzed using Metamorph 7.5 (MDS Analytical technologies, Sunnyvale, CA) from 3–5 non-overlapping fields of view per replication taken using a 20X objective (0.5 n. a.) on a Nikon TE2000-U inverted fluorescence microscope (Nikon, Melville, NY). Each treatment had 3 replications.
Statistical analysis
Student’s t test and one or two-way analysis of variance (ANOVA) were performed and followed by Newman-Keuls post-hoc multiple comparison test using GB-Stat v9.0 (Dynamic Microsystems, Silver Spring, MD).
RESULTS
ATP increases neuroregeneration in the OE via activation of P2 purinergic receptors in vitro and in vivo
In the mature OE, injured and dying OSNs are continuously regenerated by basal cells that proliferate into neuronal precursor cells and then differentiate into OSNs. We examined the effect of ATP on neuroregeneration in OE primary cell culture using the Click-iT EdU® assay that detects the incorporation of a modified nucleoside, 5-ethynyl-2′-deoxyuridine (EdU), during DNA synthesis. Incubation of ATP with OE primary cell cultures for 2 hours increased EdU-incorporation in a dose-dependent manner (Figure 1A–G). Compared to saline vehicle (18.5 ± 2.1), the levels of EdU+ cells were significantly (p < 0.05) elevated following 100, 200 and 500 μM ATP incubation (34.6 ± 1.9, 35.5 ± 3.7 and 41.3 ± 2.9 respectively). In order to test whether P2 purinergic receptors are involved in the ATP-induced increase in cell proliferation in vitro, we incubated OE primary cell cultures with P2 purinergic receptor antagonists, PPADS (25 μM) and suramin (100 μM), for 30 min prior to ATP treatment and then measured the protein levels of PCNA by western blot. Compared to vehicle (107.5 ± 6.6), incubation of OE primary cell cultures with 100 μM ATP significantly (p < 0.01) increased the protein level of PCNA (Figure 1H–I, 184.6 ± 14.9). Pre-treatment of the cells with P2 purinergic receptor antagonists significantly (p < 0.05) blocked ATP-induced elevation of PCNA (134.6 ± 16.4). Taken together, these data indicate that ATP increases cell proliferation in OE primary cell culture via activation of P2 purinergic receptors.
Figure 1. ATP increases cell proliferation in OE primary cell culture via activation of P2 purinergic receptors.

(A–G) ATP dose-dependently increases cell proliferation in OE primary cell culture. OE primary cell cultures were incubated with saline vehicle or ATP (10, 50, 100, 200 or 500 μM) for 2 hours and then subjected to the EdU incorporation assay. (A–F) Representative images of EdU incorporation (green) and DAPI nuclear staining (blue) for each treatment are shown. Scale bar = 25 μm. (G) Quantification of EdU and DAPI co-localization from 3–5 non-overlapping fields of view per replication. Each treatment had 3–5 replications. * indicates significant differences from vehicle at p < 0.05 or 0.01. (H–I) OE primary cell cultures were pre-treated with saline vehicle or P2 purinergic receptor antagonists, PPADS (25 μM) and suramin (100 μM) 30 minutes prior to incubation with saline vehicle or ATP (100 μM) for 2 hours. (H) Representative immunoblots of PCNA and actin. (I) Quantification of PCNA levels from three replications. The levels of PCNA are normalized to actin and data are expressed as a ratio to saline vehicle. * indicates a significant difference from vehicle at p<0.01. # indicates a significant difference from ATP at p < 0.05 (one-way ANOVA followed by Newman-Keuls post hoc test).
To validate the use of OE cell cultures in these studies, we next used basal cell proliferation as a marker to measure ATP-induced increases in OE neuroregeneration in vivo. We measured the number of PCNA+ cells in the basal layer of OE at 2 days post-instillation of ATP. Compared to saline vehicle treatment (62.3 ± 4.6), intranasal instillation of ATP (400 nmoles/kg) significantly (p < 0.01) increased the number of PCNA+ cells in the basal layer (BL) of mouse OE (Figure 2A, B and E, 107 ± 4.6). Pre-treatment with P2 purinergic receptor antagonists, PPADS (50 nmoles/kg) and suramin (200 nmoles/kg), significantly (p < 0.01) blocked ATP-induced increases in the number of PCNA+ cells in the basal layer of OE (Figure 2D–E, 67.9 ± 2.6), while P2 antagonists alone did not alter the levels of PCNA+ cells (Figure 2C and E, 76.7 ± 1.6). These data indicate that ATP increases neuroregeneration in the OE via activation of P2 purinergic receptors. In addition, intranasal instillation of ATP significantly (p < 0.05) increased the number of PCNA+ cells in the apical cell layer (AL) where the non-neuronal sustentacular and microvillous cells reside (5.5 ± 0.8 vs. 8.4 ± 0.2). Pretreatment with P2 purinergic receptor antagonists significantly (p < 0.01) blocked ATP-induced increases in apical PCNA+ cell (Figure 2F, 8.4 ± 0.2 vs. 5.0 ± 0.5).
Figure 2. ATP activation of P2 purinergic receptors increases basal and apical cell proliferation in mouse OE in vivo.

Mice were intranasally instilled with saline vehicle (A–B) or P2 purinergic receptor antagonist, PPADS (50 nmoles/kg) and suramin (200 nmoles/kg), (C–D) 30 min prior to saline vehicle (A and C) or ATP (400 nmoles/kg, B and D) and tissue was collected 48 hours post-instillation of ATP. (A–D) Representative images of PCNA immunostaining. Dotted white line depicts the apical (AL), neuron (NL) and basal layer (BL). Arrows indicate PCNA+ cells in the apical layer. Scale bar = 5 μm. (E–F) Quantification of PCNA+ cells in the basal (E) and apical (F) layer of OE (n = 12 sections from 4 mice/group). * indicates significant differences from vehicle at p < 0.05. # indicates significant differences from ATP at p < 0.01 (two-way ANOVA followed by Newman-Keuls post hoc test).
ATP differentially modulates p44/42 ERK over time both in vitro and in vivo
We investigated the molecular mechanisms underlying the ATP-induced increase in OE neuroregeneration, focusing on the role of p44/42 ERK. Compared to saline vehicle, incubation of OE primary cells with ATP (100 μM) for 5 and 15 min produced significant reductions in the levels of phospho-p44/42 ERK (phospho-p42, p < 0.01 and 0.05 respectively; phospho-p44, p < 0.01) followed by a significant (p < 0.01) increase at 30 min (Figure 3A–C, phospho-p42, 104.6 ± 7.4, 23.3 ± 5.9, 48.9 ± 8.7 and 191.3 ± 29.9, and phospho-p44, 106.4 ± 7.5, 24.1 ± 6.8, 39.1 ± 5.2 and 285.1 ± 29.9 for vehicle, ATP at 5, 15 and 30 min respectively). The levels of phospho-p44/42 ERK went back to the control level at 1 hour (Figure 3A–C, phospho-p42, 102.4 ± 29.7 and phospho-p44, 94.4 ± 25.4). Similarly, in vivo, intranasal instillation of ATP (400 nmoles/kg) significantly reduced the levels of phospho-p44/42 ERK at 5 and 15 min compared to saline vehicle (phospho-p42, p < 0.01; phospho-p44, p < 0.05) followed by a significant (p < 0.01) increase of phospho-p44/42 ERK at 1 hour (Figure 3D–F, phospho-p42, 102.5 ± 3.8, 62.3 ± 5.6, 56.6 ± 7.2 and 159.1 ± 15.1, and phospho-p44, 101.2 ± 3.4, 61.4 ± 6.8, 56.9 ± 6.2 and 197.9 ± 27.9 for vehicle, ATP at 5, 15 and 1 hour respectively). The levels of phospho-p44/42 ERK went back to the control level at 6 hours (phospho-p42, 89.9 ± 5.9 and phospho-p44, 102.9 ± 29.6). These data indicate that ATP differentially modulates p44/42 ERK signaling at different times in the OE both in vitro and in vivo with initial transient inhibition followed by later activation of p44/42 ERK.
Figure 3. ATP differentially modulates p44/42 ERK over time in mouse OE both in vitro and in vivo.

(A–C) OE primary cell cultures were incubated with saline vehicle (0 min) or ATP (100 μM) for 5, 15, 30 min, 1, 2 and 3 hours. (A) Representative immunoblots for phospho-p44/42 ERK, p44/42 ERK and actin. (B–C) Quantification of phospho-p42 ERK and phospho-p44 ERK from 4 replications. The levels of phospho-p44/42 ERK were normalized to actin and data are expressed as a ratio to 0 min. * indicates significant differences from 0 min at p<0.05. (D–F) Mice were intranasally instilled with saline vehicle (0 min) or ATP (400 nmoles/kg) and OE tissue was collected at 5, 15, 30 min, 1, 6 and 24 hours post-instillation. (D) Representative immunoblots for phospho-p44/42 ERK and p44/42 ERK. (E–F) Quantification of phospho-p42 ERK and phospho-p44 ERK (n = 4 mice/group). The levels of phospho-p44/42 ERK were normalized to p44/42 ERK and data are expressed as ratios over 0 min. * indicates significant differences from 0 min at p<0.05 (one-way ANOVA followed by Newman-Keuls post hoc test).
ATP activates p44/42 ERK in GBCs but not HBCs in the OE in vitro and in vivo
The cell types in which ATP induces p44/42 ERK activation in the OE was investigated by immunocytochemistry and immunohistochemistry. Incubation of OE primary cell cultures with ATP significantly increased the number of cells with positive immunoreactivity to phospho-p44/42 ERK (green, Figure 4A, B and I; 9.3 ± 1.2 vs. 29.2 ± 3.4; p < 0.01). The phospho-p44/42 ERK immunoreactivity co-localized with GBC marker MASH1 in 84% of the ATP-treated cells (red, Figure 4C and J) and co-localized with HBC marker cytokeratin 5 in only 4% of the ATP-treated cells (red, Figure 4D and J). These data indicate that ATP activates p44/42 ERK in GBCs but not HBCs in OE primary cell culture. In vivo, positive immunoreactivity to phospho-p44/42 ERK was sparsely observed in the basal layer (BL), neuron layer (NL), and lamina propria (LP) of vehicle-treated animals (Figure 4E). Intranasal instillation of ATP increased the number of phospho-p44/42 ERK positive cells (red) in the basal layer of OE (Figure 4F), indicating that ATP activates p44/42 ERK in the basal cells of OE in vivo. The immunoreactivity to phospho-p44/42 ERK (red) in the basal layer of OE in ATP-treated animals was co-localized with GBC marker MASH1 (green, Figure 4G) but not HBC marker cytokeratin 5 (green, Figure 4H), indicating that ATP activates p44/42 ERK in GBC but not HBC in the basal layer of OE. Collectively, these data demonstrate that ATP activates p44/42 ERK in GBC but not HBC in the OE in vitro and in vivo.
Figure 4. ATP induces activation of p44/42 ERK in GBCs but not HBCs in vitro and in vivo.

(A–D) OE primary cell cultures were incubated with (A) saline vehicle or (B–D) ATP (100 μM) for 30 min. Representative images of phospho-p44/42 ERK immunoreactivity (green) alone (A–B) or co-localized with globose basal cell marker, MASH1 (red, C) or horizontal basal cell marker, cytokeratin 5 (red, D) are shown. Nuclei were counterstained with DAPI (blue). Arrows in A, B and D indicate cells with phospho-p44/42 ERK immunoreactivity and arrows in C indicate co-localization of phospho-p44/42 ERK with MASH1. Arrowheads in D indicate cytokeratin 5 immunoreactive cells. Scale bar = 25 μM. (E–H) Mice were intranasally instilled with (E) saline vehicle or (F–H) ATP (400 nmoles/kg) and tissues were collected 1 hour post-instillation of ATP. Representative images of phospho-p44/42 ERK immunoreactivity (red) alone (E–F) or co-localized with globose basal cell marker, MASH1 (green, G) or horizontal basal cell marker, cytokeratin 5 (green, H) are shown. Nuclei were counterstained with DAPI (blue). Dotted white line depicts the apical (AL), neuron (NL), basal layer (BL) and lamina propria (LP). The inserted images in upper right corner show the co-localization of phospho-p44/42 (green) with MASH1 (red, G) but not cytokeratin 5 (red, H) without DAPI staining. Arrows in E, F and H indicate immunoreactivity of p44/42 ERK in the basal layer of OE. Arrowhead in E indicates the positive staining of p44/42 ERK located in OSN layer. Thick arrow in E indicates the positive staining of p44/42 in lamina propria. Arrows in G indicate the co-localization of phospho-p44/42 ERK with MASH1. Arrowheads in H indicate cytokeratin 5 immunoreactive cells. Scale bar = 10 μM. (I) Quantification of co-localization of phospho-pERK with DAPI in vehicle- and ATP-treated cells. (J) Quantification of co-localization of phospho-pERK with MASH1 or Cy5 in ATP-treated cells. The co-localization was analyzed from 3–5 non-overlapping fields of view per replication. Each treatment had 3 replications. * indicates significant differences at p < 0.01 (Student’s t test).
The p44/42 ERK activation mediates ATP-induced increases in OE neuroregeneration
In order to investigate whether activation of p44/42 ERK mediates the ATP-induced increase in neuroregeneration in the OE, U0126, an inhibitor of MEK, the upstream activator of ERK, was used. We intranasally instilled U0126 (100 nmoles/kg) 1 hour prior to ATP treatment. In vehicle-pretreated animals, ATP significantly (p < 0.01) increased the number of PCNA+ cells in the basal layer of OE (Figure 5, 59.6 ± 4.3 vs. 105.9 ± 7.1). U0126 alone did not significantly alter the levels of PCNA+ cell in the OE (69.3 ± 5.6). However, U0126 significantly (p < 0.05) blocked ATP-induced increases in the levels of PCNA+ cells in the basal layer of OE (64.4 ± 3.4), indicating that blockade of p44/42 ERK cascade significantly abolishes ATP-induced increase in basal cell proliferation. These data demonstrate that the ATP-induced increase of neuroregeneration in the OE is via activation of p44/42 ERK signaling in GBCs.
Figure 5. The MEK inhibitor U0126 significantly blocks ATP-induced increases in basal cell proliferation.
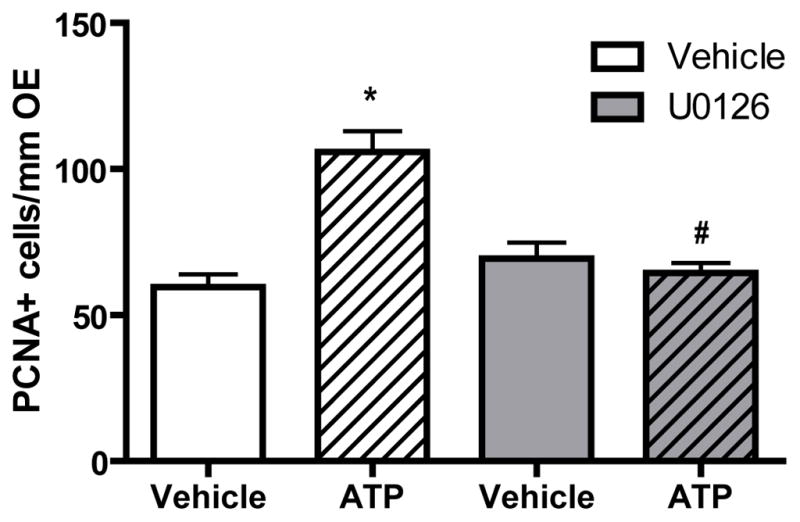
The MEK inhibitor U0126 (400 nmoles/kg) was intranasally instilled 1 hour prior to ATP treatment (400 nmoles/kg) and tissue was collected 48 hours post-instillation of ATP. Quantification of PCNA+ cells in the basal layer of the OE were from 9 sections of 3 mice/group. * indicates a significant difference from vehicle-vehicle at p < 0.01. # indicates a significant difference from vehicle-ATP at p < 0.05 (two-way ANOVA followed by Newman-Keuls post hoc test).
ATP-induced p44/42 ERK activation is due to an indirect effect of NPY release
Our previous study indicated that ATP induces NPY release from neonatal OE slices at 1 hour, the time at which ATP activates p44/42 ERK in the OE (Kanekar et al., 2009). We hypothesized that the activation of p44/42 ERK by ATP in GBCs was via an indirect effect: ATP induced NPY release that subsequently activated p44/42 ERK via NPY Y1 receptors expressed in basal cells. We tested this hypothesis using Y1 receptor antagonist BIBP3226 previously shown to inhibit both Y1 receptors and ATP-induced increases in basal cell proliferation in the OE (Jia and Hegg, 2010). We intranasally instilled BIBP3226 (10 μM) 5 min post-instillation of ATP and then quantified the levels of phospho-p44/42 ERK at 1 hour post-instillation of ATP (Figure 6A–C). Compared to saline vehicle-treated animals, ATP instillation significantly (p < 0.05) increased the levels of phospho-p44/42 ERK (phospho-p42, 101.8 ± 2.2 vs. 149.7 ± 19.3 and phospho-p44, 99.6 ± 6.5 vs. 153.0 ± 8.4). BIBP3226 post-treatment alone did not alter the levels of phospho-p44/42 ERK (phospho-p42, 110.5 ± 6.7 and phospho-p44, 87.6 ± 8.8). However, BIBP3226 significantly (p < 0.05) blocked ATP-induced increases in the levels of phospho-p44/42 ERK back to control levels (phospho-p42, 100.5 ± 9.3 and phospho-p44, 94.9 ± 14.6)., indicating that NPY Y1 receptors mediate ATP-induced activation of p44/42 ERK. These data demonstrate that ATP-induced p44/42 ERK activation in GBCs is due to an indirect effect of NPY release and subsequent activation of NPY Y1 receptors in basal cells.
Figure 6. NPY Y1 receptor antagonist BIBP3226 significantly blocks ATP-induced activation of p44/42 ERK in mouse OE.

Mice were intranasally instilled with ATP (400 nmoles/kg) followed by BIBP3226 instillation (20 nmoles/kg) 5 min later. Tissue was collected 1 hour post-instillation of ATP. (A) Representative immunoblots for phospho-p44/42 ERK and p44/42 ERK. (B–C) Quantification of phospho-p42 ERK and phospho-p44 ERK (n = 4 mice/group). The levels of phospho-p44/42 ERK were normalized to p44/42 ERK and data are expressed as ratios to vehicle-vehicle animals. * indicates significant differences from vehicle-vehicle at p < 0.05. # indicates significant differences from ATP-vehicle at p < 0.05 (two-way ANOVA followed by Newman-Keuls post hoc test).
The synergistic effect of ATP and NPY or FGF2 on neuroregeneration in the OE
In the CNS, ATP and polypeptide growth factors synergistically increase cell proliferation through parallel activation of p44/42 ERK signalling (Neary et al., 2008). Even though our data strongly support that ATP-induced p44/42 ERK activation in GBCs is via an indirect effect of NPY release, we can not conclude that ATP does not activate p44/42 ERK in GBCs via direct activation of P2 purinergic receptors since P2 purinergic receptors are expressed on basal cells in the OE (Hegg et al., 2003). If this is the case, activation of P2 purinergic receptors and NPY Y1 receptors expressed in basal cells will activate p44/42 ERK in parallel and synergistically increase basal cell proliferation in the OE. Therefore, instillation of ATP and NPY together should synergistically increase basal cell proliferation in the OE via parallel activation of p44/42 ERK in GBCs through P2 purinergic receptors and NPY Y1 receptors. To test this hypothesis, we intranasally instilled saline vehicle, ATP (400 nmoles/kg), NPY (4 nmoles/kg) or ATP + NPY and then measured basal cell proliferation via the BrdU incorporation assay 48 hours post-instillation (Figure 7A–D and G). Compared to saline vehicle instillation (27.2 ± 1.4), ATP and NPY alone significantly increased the number of BrdU+ cells in the basal layer of OE, indicating that ATP or NPY alone increases OE neuroregeneration via promoting basal cell proliferation (ATP, 61.6 ± 9.9, p < 0.05; NPY, 77.3 ± 3.9, p < 0.01). However, the number of BrdU+ cells was comparable between ATP and ATP+NPY treated groups (61.6 ± 9.9 vs. 47.8 ± 7.8), indicating that simultaneous instillation of ATP with NPY does not synergistically enhance basal cell proliferation. Surprisely, the number of BrdU+ cells in the ATP+NPY-instilled group was significantly (p < 0.05) reduced when compared to NPY-instilled group, suggesting that instillation of ATP with NPY may compromise NPY-induced increases in basal cell proliferation. To further examine the potential synergism of ATP and polypeptide growth factors in OE neuroregeneration, we used FGF2 that has been shown to promote GBC proliferation in the OE in vivo (Nishikawa et al., 2009) and in vitro (Newman et al., 2000). Our recent data indicate that FGF2 is released following ATP application and is partially responsible for the ATP-induced increase in OE neuroregeneration (Jia et al., 2011a). Compared to saline vehicle, intranasal instillation of FGF2 alone (12 pmoles/kg) significantly (p < 0.05) increased the number of BrdU+ cells in the basal layer of OE (Figure 7E–G, 57.3 ± 5.1). However, compared to both ATP and FGF2 alone, co-administration of FGF2 and ATP did not synergistically enhance basal cell proliferation (44.8 ± 5.9). Taken together, these data indicate that ATP does not potentiate the polypeptide growth factor-induced neuroregeneration in the OE.
Figure 7. ATP does not interact synergistically with NPY or FGF2 to induce basal cell proliferation in the OE.

Mice were intranasally instilled with saline vehicle or ATP (400 nmoles/kg), NPY (4 nmoles/kg), FGF2 (12 pmoles/kg), or a cocktail of ATP and NPY or FGF2. BrdU was injected (i.p., 216 mg/kg total) at 42, 44 and 46 hours and the tissue was collected 48 hours post-instillation. (A–F) Representative images of BrdU immunoreactivity for each group. Dotted white line depicts the apical (AL), neuron (NL) and basal layer (BL). Scale bar = 10 μm. (G) Quantification of BrdU+ cells in the basal layer of OE (n= 9–12 sections from 3–4 mice/group). * indicates significant differences from vehicle at p < 0.05. # indicates a significant difference from NPY at p < 0.05 (two-way ANOVA followed by Newman-Keuls post hoc test).
Polypeptide growth factors activate p44/42 ERK in the OE
NPY induces basal cell proliferation via activation of p44/42 ERK in vitro (Hansel et al., 2001). Here, we investigated the time frame of p44/42 activation. Incubation of OE primary cells with NPY (1μM) transiently activates p44/42 ERK at 5 min (Figure 8A–C, phospho-p44, 100.4 ± 3.1 vs. 187.5 ± 36.9 and phospho-p42, 98.3 ± 3.9 vs. 195.4 ± 21.6), in the same time frame as ATP inhibition of p44/42 ERK in the OE (Figure 3). Incubation of OE primary cell cultures with FGF2 (3 μM) significantly (p < 0.01, 0.05 and 0.05 respectively) activated p44/42 ERK at 5, 30 min and 1 hour (Figure 8D–E, phospho-p44, 97.9 ± 9.9 vs. 268.2 ± 38.5, 244.8 ± 32.7 and 220.5 ± 45.9 respectively; phospho-p42, 109.8 ± 10.0 vs. 365.2 ± 57.9, 340.2 ± 55.8 and 295.1 ± 37.1 respectively). Taken together, the lack of a synergistic effect of ATP and polypeptide growth factors on OE neuroregeneration (e.g., Figure 7G) could be due to opposing regulation of p44/42 ERK in basal cells by P2 purinergic receptors and growth factor receptors. Activation of P2 purinergic receptors in basal cells by ATP transiently inhibits p44/42 ERK, while growth factor receptors in basal cells activate p44/42 ERK.
Figure 8. NPY transiently activates p44/42 ERK at 5 min and FGF2 produces prolonged activation of p44/42 ERK in the OE in vitro.

OE primary cell cultures were incubated with saline vehicle (0 min), NPY (1 μM, A–C) or FGF2 (3 μM, D–F) for 5, 30 min and 1 hour. (A and D) Representative immunoblots for phospho-p44/42 ERK and actin. (B–C and E–F) Quantification of phospho-p44/42 ERK was made from 4 replications. The levels of phospho-p44/42 ERK were normalized to actin. Data are expressed as ratios over 0 min. * indicates significant differences from 0 min at p<0.05.
DISCUSSION
The present study investigated the molecular mechanism underlying injury-induced neuroregeneration in the OE. We specifically focused on ATP, which has been shown to be released upon injury and promote neuroregeneration in the OE, (Hegg et al., 2003; Jia et al., 2009; Jia et al., 2010) and the role of p44/42 ERK on the ATP-induced increase in OE neuroregeneration in vitro and in vivo.
ATP activation of P2 purinergic receptors induces neuroregeneration in the OE
ATP activation of P2 purinergic receptors increased (1) cell proliferation in the OE in vitro, measured by EdU incorporation and protein levels of PCNA and (2) basal cell proliferation in the OE in vivo measured by the number of PCNA+ cells in the basal layer of OE. These data indicate that ATP promotes neuroregeneration via activation of P2 purinergic receptors in the OE. We previously showed that ATP activation of P2 purinergic receptors (1) increases BrdU-incorporation in basal cells in neonatal and adult mouse OE at post-instillation day 2, (2) elevates BrdU+ cells expressing markers for immature neurons at post-instillation day 9 and (3) increases BrdU+ cells expressing markers for mature OSNs at post-instillation day 16 in adult mouse OE (Jia et al., 2009). In the present study, we used basal cell proliferation as a marker to measure neuroregeneration in adult mouse OE in vivo. We used the same doses of ATP and P2 purinergic receptor antagonists as previously published (Jia et al., 2009). These doses were chosen as they are physiologically relevant and within the EC50 range of purinergic receptors (Jia et al., 2009). BrdU and EdU are markers of cell proliferation as they are thymidine analogs that are incorporated into newly synthesized DNA of replicating cells. PCNA is a protein that acts as a factor for DNA polymerase delta and expressed in the nuclei of cells during DNA synthesis phase of the cell cycle. Collectively, the data from the present study confirmed our previous in vivo observations using the BrdU-incorporation assay and demonstrate that ATP promotes neuroregeneration in the OE via activation of P2 purinergic receptors. Furthermore, we also observed that ATP significantly increased PCNA+ cells in the apical layer that includes non-neuronal sustentacular and microvillous cells at 48 hours post-instillation. Sustentacular cells are generated either from basal cell proliferation, differentiation and migration to the apical layer, a ~7 day process, or from self-proliferation, a < 2 day process (Jia et al., 2009). Thus, these results indicate that ATP also promotes self-proliferation of sustentacular and microvillous cells in the OE. Collectively, these data demonstrate that ATP serves as a signal to be released upon injury and triggers recovery by promoting neuroregeneration and non-neuronal cell proliferation in the OE.
The functional significance of ATP-mediated regulation of p44/42 ERK in the OE
In the OE, ATP differentially regulates p44/42 ERK signaling over time both in vitro and in vivo in a similar manner with transient inhibition (5–15 min) followed by activation of p44/42 ERK (30 min – 1 hr). In mammals, an ecto-5′-nucleotidase enzyme degrades ATP to ADP, AMP and adenosine. In the OE, ecto-5′-nucleotidase is found in horizontal basal cells, microvillous cells and the inner lining of Bowman’s gland ducts (Braun and Zimmermann, 1998). The half-life of ATP in the OE ex vivo measured by luciferin-luciferase assay (163 seconds; unpublished data) is significantly shorter than the timeframe of ATP-mediated p44/42 activation (30 min –1 hour). The timeframe of ATP gaining access to purinergic receptors may be similar in vitro and in vivo as the times of ATP-induced p44/42 ERK inhibition are similar in vitro and in vivo. In addition, ATP-mediated p44/42 ERK activation is mediated by NPY release, thus, the 30 min difference between ATP-induced p44/42 ERK activation in vitro and in vivo may reflect the difference in time between NPY release and NPY diffusion to GBCs in vitro and in vivo.
Both in vitro and in vivo data in the present study indicate that ATP transiently inhibits p44/42 ERK activation at 5–15 min. Our previous reports demonstrate that ATP is neuroprotective in the OE. Over a period of hours to days, ATP initiates a heat shock protein-mediated stress signaling cascade that facilitates cell survival (Hegg and Lucero, 2006), and blockade of P2 purinergic receptors increases DNA fragmentation in the OE (Jia et al., 2011b), indicating that activation of P2 purinergic receptors is involved in cell protection. Inhibition of p44/42 ERK with the MEK inhibitor PD98059, leads to a reduction of cells that are double-labeled with TUNEL and mature OSN marker olfactory marker protein (OMP) in primary OSN culture, indicating ERK inhibition decreases OSN apoptosis (Simpson et al., 2003) and is neuroprotective. Taken together with the results of the present study, these data suggest that ATP-induced p44/42 ERK inhibition in the OE may be involved in P2 purinergic receptor-mediated neuroprotection. The ATP-induced reductions in the levels of phospho-p44/42 ERK could be due to the increased activity of MAPK phosphatase-1 that is expressed abundantly in the OE and inactivates p44/42 ERK by dephosphorylation (Shinogami and Ishibashi, 2000). Future studies will investigate whether p44/42 ERK inhibition is involved in purinergic receptor-mediated cell protection by using phosphatase inhibitors. Identification of the cell types that exhibit p44/42 ERK inhibition is problematic. However, our data, as detailed below, suggest that activation of P2 purinergic receptors inhibits p44/42 ERK in basal cells of OE (Figure 9A).
Figure 9. ATP-induced NPY release and subsequent NPY Y1 receptor-mediated p44/42 ERK activation in GBCs underlies injury-induced neuroregeneration in the OE.

Schematic diagram showing: (A) Extracellular or injury-released ATP transiently inhibits p44/42 ERK via activation of purinergic receptors in the OE. We don’t know the cell types in which ATP transiently inhibits p44/42 ERK. Our data suggest that activation of purinergic receptors in GBCs by ATP produces transient inhibition of p44/42 ERK. (B) Extracellular or injury-released ATP induces NPY release from sustentacular (SC) and microvillous cells (MC) via activation of purinergic receptors. Released NPY activates p44/42 ERK in GBCs via NPY Y1 receptors and subsequently promotes basal cell proliferation. (C) Simultaneous application of ATP with polypeptide growth factors, such as NPY or FGF2 does not synergistically increase basal cell proliferation since growth factor-mediated activation of p44/42 ERK in GBCs is attenuated by purinergic receptor-mediated inhibition of p44/42 ERK.
Conversely, activation of p44/42 ERK is robustly increased 2–3 weeks after bulbectomy, a procedure that temporally increases neuronal proliferation from basal cells, suggesting that p44/42 ERK activation is involved in bulbectomy-induced neuroregeneration (Shinogami and Ishibashi, 2000). We also observed ATP-induced p44/42 ERK activation in the OE at 30 min in vitro and at 1 hour in vivo. By using MEK inhibitor U0126 we demonstrate that p44/42 ERK activation mediates the ATP-induced increase in OE neuroregeneration. However, inhibition of p44/42 ERK by MEK inhibitor PD98059 significantly blocks neutrotrophin-3-induced reduction of Ki67 positive cells that are co-localized with neuron-specific tubulin in primary OSN culture, indicating that p44/42 ERK activation reduces neuronal precursor proliferation (Simpson et al., 2003). Additionally, activation of p44/42 ERK mediates odorant-enhanced survival of OSNs in vitro and in vivo (Miwa and Storm, 2005; Watt et al., 2004; Watt and Storm, 2001), indicating that p44/2 ERK activation in OSNs is neuroprotective. Whether or not ATP activates or inhibits p44/42 ERK may depend on the cell type and the functional outcomes of p44/42 ERK activation in different cell types in the OE may be different.
Role of NPY in ATP-induced p44/42 ERK regulation
Using the NPY Y1 receptor antagonist BIBP3226, we demonstrated that ATP-induced p44/42 ERK activation is due to an indirect effect of NPY release and subsequent activation of Y1 receptors in basal cells (Figure 9B). In the OE, NPY is expressed in low levels in sustentacular cells and to a greater extent in a subpopulation of microvillous cells, both of which have processes extending to the basement membrane (Hansel et al., 2001; Hegg et al., 2010; Jia and Hegg, 2010; Kanekar et al., 2009; Montani et al., 2006). ATP increases intracellular Ca2+ and up-regulates NPY expression in these cells (Hegg et al., 2003; Jia and Hegg, 2010; Kanekar et al., 2009). ATP also promotes NPY release from OE (Kanekar et al., 2009). NPY Y1 receptors are expressed in the basal cell layer where the basal progenitor cells are located (Hansel et al., 2001). Collectively, these data indicate that NPY is ideally situated to have stimulus-induced release and promote basal cell proliferation and ATP can serve as one of the stimuli. We have shown that ATP induced NPY release from OE slices of neonatal mouse after 1 hour exposure (Kanekar et al., 2009), which is the same time point with ATP-induced p44/42 ERK activation in the OE in vivo. Furthermore, NPY Y1 receptor antagonist, BIBP3226, significantly blocked ATP-induced increase in BrdU-incorporated basal cell proliferation in adult mouse OE (Jia and Hegg, 2010), indicating that ATP-induced increase in OE neuroregeneration is via NPY release and subsequent activation of NPY Y1 receptors. In vitro, NPY binds to NPY Y1 receptors in basal cells and promotes cell proliferation via activation of p44/42 ERK signaling in OE primary cell culture (Doyle et al., 2008; Hansel et al., 2001). These data support ATP-induced p44/42 ERK activation in the OE via an indirect effect of NPY release. In conclusion, the ATP-induced increase in OE neuroregeneration is through NPY release, NPY Y1 receptor activation of p44/42 ERK in basal cells, and increased basal cell proliferation. However, the effect could theoretically be due to different neurochemicals released by ATP application. In addition to NPY, we found that ATP also induced FGF2 release (Jia et al., 2011a), although FGF2 has little probability of activating a NPY receptor. Another member of the NPY family is peptide YY (PYY) that shares extensive sequence homology with NPY. Recently, PYY expression has been found in the OE (Doyle et al., 2008). Therefore, PYY could mediate the ATP effect. But further investigation is required to determine whether ATP triggers PYY release in the OE.
Activation of p44/42 ERK in globose basal cells
ATP-induced p44/42 ERK activation was co-localized with GBC marker, MASH1, but not HBC marker, cytokeratin 5, indicating that ATP-induced p44/42 ERK activation is in GBCs but not HBCs. Since ATP-induced activation of p44/42 ERK is due to an indirect effect of NPY release, the differential activation of p44/42 ERK in GBCs and HBCs could be due to the differential distribution of NPY Y1 receptors in these cells. NPY Y1 receptors have been found in basal cells of OE and promote basal cell proliferation via activation of p44/42 ERK (Hansel et al., 2001). However, there is no direct evidence showing differential distribution of NPY Y1 receptors in GBC and HBC. Doyle et al. (2008) report that knockout of NPY Y1 receptors reduces the number of MASH1+ GBCs and OSNs but has no effect on HBC, suggesting that NPY Y1 receptors are expressed in GBCs and mediate neuronal proliferation in the OE.
The GBCs are the major proliferating population in the OE during normal neuronal turnover and after acute, selective loss of mature OSNs. The GBCs are functionally heterogeneous and multipotent progenitors (Chen et al., 2004; Goldstein and Schwob, 1996; Guo et al., 2010; Huard et al., 1998; Manglapus et al., 2004). The composition of the whole populations of GBCs is not fully understood. FACS-purified GBCs derived from normal OE can give rise to GBCs, neurons, sustentacular cells, gland cells and respiratory epithelium cells following transplantation into the methyl bromide-lesioned OE (Chen et al., 2004; Goldstein and Schwob, 1996; Huard et al., 1998; Manglapus et al., 2004). MASH1 is a well established marker for GBCs. The MASH1+ GBC is neuron-committed and gives rise to transit amplifying cells that differentiate into OSNs (Caggiano et al., 1994; Chen et al., 2004; Huard et al., 1998). However, the percentage of MASH1+ GBCs in the whole population of GBCs is not known. HBCs serve as reservoirs of long-lived progenitors that remain largely quiescent. After extensive injuries that deplete GBCs, HBCs transiently proliferate and their progeny fully reconstitute the OE (Leung et al., 2007). Our recent data showed that ATP was constitutively released in the OE under normal physiological condition (unpublished data). Taken together, these data suggest that ATP-mediated indirect activation of p44/42 ERK by NPY is underlying mechanism for normal neuronal turnover and neuroregeneration following injury in the OE.
The synergistic effect of ATP and polypeptide growth factors on OE neuroregeneration
In adult mouse OE, basal cells only express G protein-coupled P2Y purinergic receptors that have been reported to transiently activate p44/42 ERK in the CNS (Franke and Illes, 2006). It is possible that ATP transiently activates p44/42 ERK in the OE via direct activation of P2Y purinergic receptors in basal cells. In amphibian OE, activation of P2Y receptors in the basal cells induces calcium signaling that leads to increased basal cell proliferation (Hassenklöver et al., 2009). The ATP-induced increase in basal cell proliferation could be due to dual effects: direct activation of P2Y purinergic receptors in basal cells and indirect effect of NPY release from non-neuronal cells. If so, we should detect two phases of p44/42 ERK activation in the OE following ATP: the early activation by direct activation of P2 purinergic receptors and the late activation by indirect NPY release. However, in this study we detected inhibition of p44/42 ERK via activation of P2Y purinergic receptors in the OE between 5–15 min. This result may be due to transient inhibitory effects of ATP on p44/42 ERK in other cell types, such as OSNs, sustentacular and microvillous cells.
The synergistic effect of P2 purinergic and polypeptide growth factor receptors on neuronal proliferation via parallel activation of p44/42 ERK has been well established in the CNS. ATP synergistically enhances the mitogenic activity of FGF2 through parallel activation of p44/42 ERK signalling (Lin et al., 2007; Mishra et al., 2006; Neary et al., 2005; Neary et al., 1996). In cultured subventricular zone-derived neuronal progenitor cells, both purinergic and growth factor receptors activate p44/42 ERK and synergistically induce neuronal proliferation (Grimm et al., 2009). If ATP transiently activates p44/42 ERK via activation of P2Y receptors in basal cells, we would expect that ATP and growth factors may synergistically increase basal cell proliferation via parallel activation of p44/42 ERK. We chose NPY and FGF2 to investigate this hypothesis as both NPY Y1 and FGF2 receptors are expressed in basal cells (Hansel et al., 2001; Hsu et al., 2001). Further, NPY activation of Y1 receptors increases phospho-p44/42 ERK and basal cell proliferation (Hansel et al., 2001) and FGF2 stimulates the proliferation of GBCs in vitro (Barraud et al., 2007; Goldstein et al., 1997; Newman et al., 2000) and in vivo (Nishikawa et al., 2009). In the CNS, FGF2 promotes cell proliferation through activation of p44/42 ERK signalling (Neary et al., 2008) and ATP potentiates the mitogenic effect of FGF2 by parallel activation of p44/42 ERK via P2 purinergic receptors (Grimm et al., 2009; Neary et al., 2005; Neary et al., 1996). However, we did not observe the synergistic effects on BrdU-labeled basal cell proliferation with ATP and NPY or FGF2, suggesting that, as with single administration, co-administration activates P2 purinergic receptors in basal cells and transiently inhibits p44/42 ERK as measured by western blot. This transient inhibition of p44/42 ERK attenuates further polypeptide growth factor-mediated activation of p44/42 ERK in basal cells, which leads to the reduction of growth factor-induced basal cell proliferation in the OE (Figure 9C). Further studies are needed to confirm absence of p44/42 ERK activation in basal cells at 5 and 15 min post-instillation of ATP.
Conclusions
Our study shows that ATP-induced increases in neuroregeneration in the OE is via p44/42 ERK activation in GBCs that result from an indirect effect of NPY release and subsequent activation of NPY Y1 receptors in basal cells. ATP is constitutively released in the OE. Noxious stimulation or injury of the olfactory system leads to increased release of ATP. ATP activation of purinergic receptors stimulates a linear and/or network of intracellular signalling cascades to protect and regenerate OSNs. Identification of signalling molecules that protect neurons and promote neuronal proliferation in the physiological conditions will lead to investigation of the mechanisms responsible for initiating enhanced neuroregeneration under injurious and pathological conditions. New tools for improving the functional sense of smell and recovery after injury by enhancing neuronal proliferation and preventing apoptosis will lead to therapeutic approaches in injury-related anosmia. This work is also relevant to postnatal neuronal regeneration in the CNS. The mechanisms that control neuronal death and regeneration in the olfactory system will be of great value in understanding repopulation of neurons in the CNS.
Acknowledgments
This work was supported by NIH DC006897 (CCH) and by Michigan State University Institutional Funds (CCH). We thank Dr. Stephanie W. Watts, Brian Jespersen, and the Pharmacology and Toxicology Departmental Core Facilities for technical support.
Abbreviation
- OE
Olfactory epithelium
- NPY
Neuropeptide Y
- ERK
Extracellular signal-regulated kinase
- OSN
Olfactory sensory neuron
- GBC
Globose basal cell
- HBC
Horizontal basal cell
- CNS
Central nervous system
- MAPKs
Mitogen-activated protein kinases
- PPADS
Pyridoxalphosphate-6-azophenyl-20, 40-disulfonic acid
- PCNA
Proliferating cell nuclear antigen
- PBS
Phosphate-buffered saline
- FGF2
Fibroblast growth factor 2
- PFA
Paraformaldehyde
- BrdU
Bromodeoxyuridine
- EdU
5-ethynyl-2′-deoxyuridine
- MASH1
Mammalian Achaete-Scute Homolog 1
- OMP
Olfactory marker protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agasse F, Bernardino L, Kristiansen H, Christiansen SH, Ferreira R, Silva B, Grade S, Woldbye DP, Malva JO. Neuropeptide Y promotes neurogenesis in murine subventricular zone. Stem Cells. 2008;26:1636–1645. doi: 10.1634/stemcells.2008-0056. [DOI] [PubMed] [Google Scholar]
- Alvaro AR, Martins J, Araujo IM, Rosmaninho-Salgado J, Ambrosio AF, Cavadas C. Neuropeptide Y stimulates retinal neural cell proliferation - involvement of nitric oxide. J Neurochem. 2008;105(6):2501–10. doi: 10.1111/j.1471-4159.2008.05334.x. [DOI] [PubMed] [Google Scholar]
- Barraud P, He X, Zhao C, Ibanez C, Raha-Chowdhury R, Caldwell MA, Franklin RJ. Contrasting effects of basic fibroblast growth factor and epidermal growth factor on mouse neonatal olfactory mucosa cells. Eur J Neurosci. 2007;26:3345–3357. doi: 10.1111/j.1460-9568.2007.05950.x. [DOI] [PubMed] [Google Scholar]
- Braun N, Zimmermann H. Association of ecto-5′-nucleotidase with specific cell types in the adult and developing rat olfactory organ. J Comp Neurol. 1998;393:528–537. doi: 10.1002/(sici)1096-9861(19980420)393:4<528::aid-cne10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Caggiano M, Kauer JS, Hunter DD. Globose basal cells are neuronal progenitors in the olfactory epithelium: a lineage analysis using a replication-incompetent retrovirus. Neuron. 1994;13:339–352. doi: 10.1016/0896-6273(94)90351-4. [DOI] [PubMed] [Google Scholar]
- Calof AL, Bonnin A, Crocker C, Kawauchi S, Murray RC, Shou J, Wu HH. Progenitor cells of the olfactory receptor neuron lineage. Microsc Res Tech. 2002;58:176–188. doi: 10.1002/jemt.10147. [DOI] [PubMed] [Google Scholar]
- Carr VM, Farbman AI. Ablation of the olfactory bulb up-regulates the rate of neurogenesis and induces precocious cell death in olfactory epithelium. Exp Neurol. 1992;115:55–59. doi: 10.1016/0014-4886(92)90221-b. [DOI] [PubMed] [Google Scholar]
- Carter LA, MacDonald JL, Roskams AJ. Olfactory horizontal basal cells demonstrate a conserved multipotent progenitor phenotype. Journal of Neuroscience. 2004;24:5670–5683. doi: 10.1523/JNEUROSCI.0330-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Fang H, Schwob JE. Multipotency of purified, transplanted globose basal cells in olfactory epithelium. J Comp Neurol. 2004;469:457–474. doi: 10.1002/cne.11031. [DOI] [PubMed] [Google Scholar]
- Costanzo RM, Graziadei PP. A quantitative analysis of changes in the olfactory epithelium following bulbectomy in hamster. J Comp Neurol. 1983;215:370–381. doi: 10.1002/cne.902150403. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Thai J, Krajewski S, Reed JC, Nicholson DW, Kaufmann SH, Roskams AJ. Caspases 3 and 9 send a pro-apoptotic signal from synapse to cell body in olfactory receptor neurons. J Neurosci. 2001;21:7099–7109. doi: 10.1523/JNEUROSCI.21-18-07099.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle KL, Karl T, Hort Y, Duffy L, Shine J, Herzog H. Y1 receptors are critical for the proliferation of adult mouse precursor cells in the olfactory neuroepithelium. J Neurochem. 2008;105:641–652. doi: 10.1111/j.1471-4159.2007.05188.x. [DOI] [PubMed] [Google Scholar]
- Franke H, Illes P. Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol Ther. 2006;109:297–324. doi: 10.1016/j.pharmthera.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Goldstein BJ, Schwob JE. Analysis of the globose basal cell compartment in rat olfactory epithelium using GBC-1, a new monoclonal antibody against globose basal cells. J Neurosci. 1996;16:4005–4016. doi: 10.1523/JNEUROSCI.16-12-04005.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BJ, Wolozin BL, Schwob JE. FGF2 suppresses neuronogenesis of a cell line derived from rat olfactory epithelium. J Neurobiol. 1997;33:411–428. [PubMed] [Google Scholar]
- Graziadei GA, Graziadei PP. Neurogenesis and neuron regeneration in the olfactory system of mammals. II Degeneration and reconstitution of the olfactory sensory neurons after axotomy. J Neurocytol. 1979a;8:197–213. doi: 10.1007/BF01175561. [DOI] [PubMed] [Google Scholar]
- Graziadei PP, Graziadei GA. Neurogenesis and neuron regeneration in the olfactory system of mammals. I Morphological aspects of differentiation and structural organization of the olfactory sensory neurons. J Neurocytol. 1979b;8:1–18. doi: 10.1007/BF01206454. [DOI] [PubMed] [Google Scholar]
- Graziadei PPC, Monti-Graziadei GA. Continuous nerve cell renewal in the olfactory system. In: Jacobson M, editor. Handbook of Sensory Physiology. Springer; New York: 1978. pp. 55–83. [Google Scholar]
- Grimm I, Messemer N, Stanke M, Gachet C, Zimmermann H. Coordinate pathways for nucleotide and EGF signaling in cultured adult neural progenitor cells. J Cell Sci. 2009 doi: 10.1242/jcs.044891. [DOI] [PubMed] [Google Scholar]
- Guo Z, Packard A, Krolewski RC, Harris MT, Manglapus GL, Schwob JE. Expression of pax6 and sox2 in adult olfactory epithelium. J Comp Neurol. 2010;518:4395–4418. doi: 10.1002/cne.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel DE, Eipper BA, Ronnett GV. Neuropeptide Y functions as a neuroproliferative factor. Nature. 2001;410:940–944. doi: 10.1038/35073601. [DOI] [PubMed] [Google Scholar]
- Hassenklöver T, Schwartz P, Schild D, Manzini I. Purinergic signaling regulates cell proliferation of olfactory epithelium progenitors. Stem Cells. 2009;27:2022–2031. doi: 10.1002/stem.126. [DOI] [PubMed] [Google Scholar]
- Hegg CC, Greenwood D, Huang W, Han P, Lucero MT. Activation of purinergic receptor subtypes modulates odor sensitivity. J Neurosci. 2003;23:8291–8301. doi: 10.1523/JNEUROSCI.23-23-08291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegg CC, Irwin M, Lucero MT. Calcium store-mediated signaling in sustentacular cells of the mouse olfactory epithelium. Glia. 2009;57:634–644. doi: 10.1002/glia.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegg CC, Jia C, Chick WS, Restrepo D, Hansen A. Microvillous cells expressing IP3R3 in the olfactory epithelium of mice. Eur J Neurosci. 2010;32:1632–1645. doi: 10.1111/j.1460-9568.2010.07449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegg CC, Lucero MT. Purinergic receptor antagonists inhibit odorant-induced heat shock protein 25 induction in mouse olfactory epithelium. Glia. 2006;53:182–190. doi: 10.1002/glia.20258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb JD, Mumm JS, Calof AL. Apoptosis in the neuronal lineage of the mouse olfactory epithelium: regulation in vivo and in vitro. Dev Biol. 1995;172:307–323. doi: 10.1006/dbio.1995.0025. [DOI] [PubMed] [Google Scholar]
- Howell OW, Doyle K, Goodman JH, Scharfman HE, Herzog H, Pringle A, Beck-Sickinger AG, Gray WP. Neuropeptide Y stimulates neuronal precursor proliferation in the post-natal and adult dentate gyrus. J Neurochem. 2005;93:560–570. doi: 10.1111/j.1471-4159.2005.03057.x. [DOI] [PubMed] [Google Scholar]
- Howell OW, Scharfman HE, Herzog H, Sundstrom LE, Beck-Sickinger A, Gray WP. Neuropeptide Y is neuroproliferative for post-natal hippocampal precursor cells. J Neurochem. 2003;86:646–659. doi: 10.1046/j.1471-4159.2003.01895.x. [DOI] [PubMed] [Google Scholar]
- Hsu P, Yu F, Feron F, Pickles JO, Sneesby K, Mackay-Sim A. Basic fibroblast growth factor and fibroblast growth factor receptors in adult olfactory epithelium. Brain Res. 2001;896:188–197. doi: 10.1016/s0006-8993(01)02173-4. [DOI] [PubMed] [Google Scholar]
- Huard JM, Youngentob SL, Goldstein BJ, Luskin MB, Schwob JE. Adult olfactory epithelium contains multipotent progenitors that give rise to neurons and non-neural cells. J Comp Neurol. 1998;400:469–486. [PubMed] [Google Scholar]
- Islam Z, Harkema JR, Pestka JJ. Satratoxin G from the black mold Stachybotrys chartarum evokes olfactory sensory neuron loss and inflammation in the murine nose and brain. Environ Health Perspect. 2006;114:1099–1107. doi: 10.1289/ehp.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Cussen AR, Hegg CC. ATP differentially upregulates fibroblast growth factor 2 and transforming growth factor alpha in neonatal and adult mice: effect on neuroproliferation. Neuroscience. 2011a;177:335–346. doi: 10.1016/j.neuroscience.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Doherty JD, Crudgington S, Hegg CC. Activation of purinergic receptors induces proliferation and neuronal differentiation in Swiss Webster mouse olfactory epithelium. Neuroscience. 2009;163:120–128. doi: 10.1016/j.neuroscience.2009.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Hegg CC. NPY mediates ATP-induced neuroproliferation in adult mouse olfactory epithelium. Neurobiol Dis. 2010;38:405–413. doi: 10.1016/j.nbd.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Roman C, Hegg CC. Nickel sulfate induces location-dependent atrophy of mouse olfactory epithelium: protective and proliferative role of purinergic receptor activation. Toxicol Sci. 2010;115:547–556. doi: 10.1093/toxsci/kfq071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Sangsiri S, Belock B, Iqbal T, Pestka JJ, Hegg CC. ATP mediates neuroprotective and neuroproliferative effects in mouse olfactory epithelium following exposure to satratoxin G in vitro and in vivo. Toxicol Sci. 2011b doi: 10.1093/toxsci/kfr213. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekar S, Jia C, Hegg CC. Purinergic receptor activation evokes neurotrophic factor neuropeptide Y release from neonatal mouse olfactory epithelial slices. J Neurosci Res. 2009;87:1424–1434. doi: 10.1002/jnr.21954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CT, Coulombe PA, Reed RR. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat Neurosci. 2007;10:720–726. doi: 10.1038/nn1882. [DOI] [PubMed] [Google Scholar]
- Lin JH, Takano T, Arcuino G, Wang X, Hu F, Darzynkiewicz Z, Nunes M, Goldman SA, Nedergaard M. Purinergic signaling regulates neural progenitor cell expansion and neurogenesis. Dev Biol. 2007;302:356–366. doi: 10.1016/j.ydbio.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglapus GL, Youngentob SL, Schwob JE. Expression patterns of basic helix-loop-helix transcription factors define subsets of olfactory progenitor cells. J Comp Neurol. 2004;479:216–233. doi: 10.1002/cne.20316. [DOI] [PubMed] [Google Scholar]
- Mishra SK, Braun N, Shukla V, Fullgrabe M, Schomerus C, Korf HW, Gachet C, Ikehara Y, Sevigny J, Robson SC, Zimmermann H. Extracellular nucleotide signaling in adult neural stem cells: synergism with growth factor-mediated cellular proliferation. Development. 2006;133:675–684. doi: 10.1242/dev.02233. [DOI] [PubMed] [Google Scholar]
- Miwa N, Storm DR. Odorant-induced activation of extracellular signal-regulated kinase/mitogen-activated protein kinase in the olfactory bulb promotes survival of newly formed granule cells. J Neurosci. 2005;25:5404–5412. doi: 10.1523/JNEUROSCI.1039-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani G, Tonelli S, Elsaesser R, Paysan J, Tirindelli R. Neuropeptide Y in the olfactory microvillar cells. Eur J Neurosci. 2006;24:20–24. doi: 10.1111/j.1460-9568.2006.04878.x. [DOI] [PubMed] [Google Scholar]
- Moon C, Liu BQ, Kim SY, Kim EJ, Park YJ, Yoo JY, Han HS, Bae YC, Ronnett GV. Leukemia inhibitory factor promotes olfactory sensory neuronal survival via phosphoinositide 3-kinase pathway activation and Bcl-2. J Neurosci Res. 2009;87:1098–1106. doi: 10.1002/jnr.21919. [DOI] [PubMed] [Google Scholar]
- Neary JT, Kang Y, Shi YF. Cell cycle regulation of astrocytes by extracellular nucleotides and fibroblast growth factor-2. Purinergic Signal. 2005;1:329–336. doi: 10.1007/s11302-005-8075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary JT, Rathbone MP, Cattabeni F, Abbracchio MP, Burnstock G. Trophic actions of extracellular nucleotides and nucleosides on glial and neuronal cells. Trends Neurosci. 1996;19:13–18. doi: 10.1016/0166-2236(96)81861-3. [DOI] [PubMed] [Google Scholar]
- Neary JT, Shi YF, Kang Y, Tran MD. Opposing effects of P2X(7) and P2Y purine/pyrimidine-preferring receptors on proliferation of astrocytes induced by fibroblast growth factor-2: implications for CNS development, injury, and repair. J Neurosci Res. 2008;86:3096–3105. doi: 10.1002/jnr.21765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary JT, Zimmermann H. Trophic functions of nucleotides in the central nervous system. Trends Neurosci. 2009;32:189–198. doi: 10.1016/j.tins.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Newman MP, Feron F, Mackay-Sim A. Growth factor regulation of neurogenesis in adult olfactory epithelium. Neuroscience. 2000;99:343–350. doi: 10.1016/s0306-4522(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Doi K, Ochi N, Katsunuma S, Nibu K. Effect of intranasal administration of basic fibroblast growth factor on olfactory epithelium. Neuroreport. 2009;20:764–769. doi: 10.1097/WNR.0b013e32832b169e. [DOI] [PubMed] [Google Scholar]
- Schwob JE, Szumowski KE, Stasky AA. Olfactory sensory neurons are trophically dependent on the olfactory bulb for their prolonged survival. J Neurosci. 1992;12:3896–3919. doi: 10.1523/JNEUROSCI.12-10-03896.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinogami M, Ishibashi T. Mitogen-activated protein kinase phosphatase-1 in olfactory neural regeneration. Neuroreport. 2000;11:3743–3746. doi: 10.1097/00001756-200011270-00030. [DOI] [PubMed] [Google Scholar]
- Simpson PJ, Wang E, Moon C, Matarazzo V, Cohen DR, Liebl DJ, Ronnett GV. Neurotrophin-3 signaling maintains maturational homeostasis between neuronal populations in the olfactory epithelium. Mol Cell Neurosci. 2003;24:858–874. doi: 10.1016/j.mcn.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Sultan-Styne K, Toledo R, Walker C, Kallkopf A, Ribak CE, Guthrie KM. Long-term survival of olfactory sensory neurons after target depletion. J Comp Neurol. 2009;515:696–710. doi: 10.1002/cne.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiriet N, Agasse F, Nicoleau C, Guegan C, Vallette F, Cadet JL, Jaber M, Malva JO, Coronas V. NPY promotes chemokinesis and neurogenesis in the rat subventricular zone. J Neurochem. 2011;116:1018–1027. doi: 10.1111/j.1471-4159.2010.07154.x. [DOI] [PubMed] [Google Scholar]
- Watt WC, Sakano H, Lee ZY, Reusch JE, Trinh K, Storm DR. Odorant stimulation enhances survival of olfactory sensory neurons via MAPK and CREB. Neuron. 2004;41:955–967. doi: 10.1016/s0896-6273(04)00075-3. [DOI] [PubMed] [Google Scholar]
- Watt WC, Storm DR. Odorants stimulate the ERK/mitogen-activated protein kinase pathway and activate cAMP-response element-mediated transcription in olfactory sensory neurons. Journal of Biological Chemistry. 2001;276:2047–2052. doi: 10.1074/jbc.M006703200. [DOI] [PubMed] [Google Scholar]
- Young JT. Histopathologic examination of the rat nasal cavity. Fundam Appl Toxicol. 1981;1:309–312. doi: 10.1016/s0272-0590(81)80037-1. [DOI] [PubMed] [Google Scholar]