

Abstract
Polyelectrolyte multilayers (PEMs) fabricated from cationic polymers and DNA have been investigated broadly as materials for surface-mediated DNA delivery. One attractive aspect of this `multilayered' approach is the potential to exploit the presence of cationic polymer `layers' in these films to deliver DNA to cells more effectively. Past studies demonstrate that these films can promote transgene expression in vitro and in vivo, but significant questions remain regarding roles that the cationic polymers could play in promoting the internalization and processing of DNA. Here, we report physicochemical and in vitro cell-based characterization of DNA-containing PEMs fabricated using fluorescently end-labeled derivatives of a degradable polycation (polymer 1) used in past studies of surface-mediated transfection. This approach permitted simultaneous characterization of polymer and DNA in solution and in cells using fluorescence-based techniques, and provided information about the locations and behaviors of polymer 1 that could not be obtained using other methods. LSCM and flow cytometry experiments revealed that polymer 1 and DNA released from film-coated objects were both internalized extensively by cells, and that they were co-localized to a significant extent inside cells (e.g., ~58% of DNA was co-localized with polymer). Fluorescence anisotropy measurements of solutions containing partially eroded films were also consistent with the presence of aggregates of polymer 1 and DNA in solution (e.g., after release from surfaces, but prior to internalization by cells). Our results support the view that polymer 1 – which is incorporated into these materials as `layers' rather than as part of optimized, pre-formed `polyplexes' – can act to promote or enhance surface-mediated DNA delivery. More broadly, our results suggest opportunities to improve the delivery properties of DNA-containing PEMs by incorporation of additional `layers' of other conventional cationic polymers designed to address specific intracellular barriers to transfection, such as endosomal escape, more effectively.
Keywords: Thin Films, DNA Delivery, Cationic Polymers, Polyelectrolytes, Layer-by-Layer
Introduction
Materials and methods that can be used to localize the delivery of DNA to cells and tissues are important in a variety of fundamental and applied contexts, ranging from the development of new research tools to applications such as tissue engineering and the development of gene-based therapies. Localized delivery can be achieved using a range of methods, including the site-specific injection and catheter-based delivery of naked DNA or DNA-based nanoparticles (e.g., polyplexes or lipoplexes).1–5 Other materials-based approaches have focused on the immobilization of DNA on surfaces or encapsulation in polymers and coatings that can be used to promote local release from the surfaces of interventional devices, culture plates, porous matrices, etc.6–13 These and other physical/chemical approaches can be used to exert varying levels of spatial and/or temporal control over the delivery of DNA to cells and tissues both in vitro and in vivo.
Methods for the encapsulation, release, or delivery of naked (uncomplexed) DNA can be sufficient to promote useful levels of transgene expression in several different contexts.1,3,6,7,11 Delivery can often be facilitated and enhanced further, however, by complexation or co-incorporation of DNA with auxiliary polymer- and lipid-based agents that address specific cell-based barriers to internalization and processing.14–17 With respect to the development of surface-mediated approaches to delivery in particular, several groups have reported on methods for the direct adsorption or immobilization of well-defined, pre-formed lipoplexes and polyplexes on surfaces.8,10,13,18,19 As potential alternatives to these approaches, other groups have developed methods for the fabrication of thin, polyelectrolyte-based films (called `polyelectrolyte multilayers', or PEMs)20 composed of multiple different layers of DNA and cationic polymer.21 While this PEM-based approach can be used to promote surface-mediated cell transfection,22–35 these film-based assemblies are not generally fabricated using well-defined DNA `polyplexes', and thus several important questions exist regarding the mechanisms through which these multi-component materials function to promote transfection (and, ultimately, how they might be designed to do so more effectively). Here, we report physicochemical characterization and in vitro investigation of the intracellular trafficking of DNA and cationic polymer components released from a DNA-containing PEM used as a model in several past studies22,26,34,35 to promote surface-mediated transfection, with a particular focus on providing insight into roles that the cationic polymer components may play in promoting the internalization and processing of DNA.
The approach to the design of the PEMs reported here makes use of methods developed for the step-wise, layer-by-layer assembly of thin polymer films.20,36 These methods are entirely aqueous and take advantage of ionic interactions to drive the assembly of multilayered films by sequential (or `layer-by-layer') adsorption of a broad range of different cationic and anionic polymers. This iterative fabrication scheme results in the bottom-up assembly of nanostructured films, with each deposited layer serving as a platform for the subsequent deposition of an oppositely charged polymer layer. In the context of drug delivery, this approach confers several practical advantages,21,37–41 including (i) the potential for precise control over film thickness and drug loading (by controlling the number of layers deposited), (ii) the ability to deposit thin, conformal coatings on objects with complex surface structures, and (iii) the ability to incorporate natural polyelectrolytes (e.g. DNA, protein, etc.) directly into the structure of a film (i.e., as a `layer'). While incorporated small molecules can be released from PEMs by simple diffusion,37–40,42,43 films fabricated from larger biomacromolecular species can be designed to erode or disassemble44 using a broad range of natural or synthetic polyelectrolytes that contain hydrolytically,22,35,45–50 reductively,31,32,51–54 or enzymatically degradable bonds.24,28,55–60 Finally, in the context of DNA delivery considered here, we note that the inherent juxtaposition and commingling of DNA with cationic polymers create opportunities to release DNA as part of an aggregate or electrostatic complex that could promote the internalization and processing of DNA more effectively. A growing body of literature describing the design of PEMs for DNA delivery has recently been reviewed comprehensively;21 specific examples of particular relevance to the work reported here are described in further detail below.
As noted above, many fundamental questions remain regarding the roles that the cationic polymers in a PEM might play in promoting cell transfection. For example, in cases for which incorporated DNA is released directly into solution, is it released as naked (unbound) DNA, or is it released in a form that is bound to polymer? Several investigators have applied methods used conventionally for the characterization of pre-formed polyplexes to characterize the presence (or absence) of polymer/DNA aggregates in solutions containing partially eroded films. For example, Ji and coworkers used combinations of transmission electron microscopy (TEM), zeta-potential measurements, and dynamic light scattering (DLS) to identify aggregates with diameters on the order of several hundred nanometers following the erosion of enzymatically degradable poly(lysine)/DNA films.57,60 Our group has also used DLS and measurements of zeta-potential to characterize the sizes and charges of aggregates (ranging in size from ~100 – 600 nm) resulting from the erosion of DNA-containing films fabricated using hydrolytically degradable26 or `charge-shifting'61 cationic polymers. The results of these studies are consistent with the view that DNA can be released as part of a polymer aggregate (or, alternatively, that aggregates can be formed in solution once polymer and DNA are released). It is important to note, however, that these characterization methods do not provide direct information about the compositions of these aggregates (e.g., whether or not they are comprised of both DNA and polymer, and, if so, to what extnt). Several recent studies have sought to address some of these key issues by directly incorporating pre-formed polymer/DNA polyplexes as layers within PEMs (rather than individual layers of polymer or DNA).28,29,58 In an initial study, Meyer et al. used several different methods to characterize the structures and compositions of imbedded polyplexes during film assembly,58 but the results of these studies did not provide direct insight into the structures of aggregates that were released from the films or subsequently internalized by cells.28,29,58

Our group has reported extensively on the characterization and evaluation of PEMs fabricated using plasmid DNA and polymer 1.22,26,35,46,50,62–65 Polymer 1 is a hydrolytically degradable poly(β-amino ester) (PBAE),15,66 and can be used to fabricate thin films (~100 nm thick) that (i) promote the gradual release of DNA from the surfaces of film coated objects,26,46 and (ii) promote surface-mediated cell transfection when placed in contact with cells and tissues in vitro22,26 and in vivo.34,35 Several of these past studies have used films fabricated using fluorescently labeled plasmid DNA constructs to characterize the release and/or transfer of DNA from film-coated surfaces.22,35,50,64,67 Unfortunately, a more complete understanding of the locations and behaviors of polymer 1 in these past studies has been limited by the lack of fluorescently labeled derivatives of this cationic polymer (and, more generally, by difficulties associated with characterizing the locations and dynamics of unlabeled polymer 1 using other methods of analysis). This current investigation sought to characterize the erosion, release, and intracellular trafficking both of DNA and polymer 1 using PEMs fabricated with two new fluorescently end-labeled derivatives of polymer 1 (polymer 1TMR and polymer 1OG).68
This study is presented in three parts. In the first part, we demonstrate that fluorescently end-labeled derivatives of polymer 1 can be used to fabricate erodible, DNA-containing PEMs and that this approach permits simultaneous characterization of the release profiles of both the polymer and DNA components of these assemblies (e.g. by measurements of solution fluorescence). In the second part, we use laser scanning confocal microscopy (LSCM) and flow cytometry to characterize and quantify the internalization and intracellular trafficking of polymer 1 and DNA by cells. The results of these studies reveal significant levels of physical co-localization of polymer 1 and DNA in cells. Finally, we report on characterization of changes in the fluorescence anisotropy of polymers released from partially eroded films that provide additional evidence of interactions between polymer 1 and DNA in solution (e.g., subsequent to release from a film, but prior to internalization by cells). When combined, the results of this study shed new light on the behaviors of these DNA-containing materials and provide insight into the roles that polymer 1 could play in promoting surface-mediated cell transfection.
Materials and Methods
Materials
Linear poly(ethylene imine) (LPEI, MW = 25,000) was purchased from Polysciences, Inc. (Warrington, PA). Poly(sodium 4-styrene sulfonate) (SPS) was purchased from Acros Organics (Geel, Belgium). Sodium acetate buffer was purchased from Accugene (Rockland, ME). Phosphate-buffered saline (PBS) was prepared by dilution of commercially available concentrate (EM Science, Gibbstown, NJ). Cell Scrub buffer was purchased from Gene Therapy Systems Inc. (San Diego, CA). Test-grade n-type silicon wafers were purchased from Silicon Inc. (Boise, ID). Glass microscope slides used as glass substrates in laser scanning confocal microscopy (LSCM) experiments were purchased from VWR International (West Chester, PA). 316 Stainless Steel woven wire cloth (mesh size – 120 × 120, wire diameter – 0.0036 in.) used as wire mesh substrates for flow cytometry experiments was purchased from McMaster-Carr (Santa Fe Springs, CA). Tetramethylrhodamine (TMR) cadaverine, Oregon Green (OG) 488 cadaverine, Hoechst 34580, Calcein AM, and Lysotracker Red were purchased from Invitrogen (Carlsbad, CA). Plasmid DNA (gWiz Luciferase) was obtained from a commercial supplier (Aldevron, Fargo, ND). Polymer 1, polymer 1TMR, and polymer 1OG were synthesized as previously described.68 For experiments requiring fluorescently labeled DNA, Cy5 Label-IT and TMR Label-IT nucleic acid kits were purchased from Mirus Bio Corporation (Madison, WI) and used according to the manufacturer's instructions. Glass inset dishes used for LSCM were purchased from MatTek (Ashland, MA). Deionized water (18 MΩ) was used to prepare all buffer and polymer solutions. All materials were used as received unless otherwise noted. LPEI and SPS solutions used to fabricate PEM base layers were filtered through a 0.2 μm nylon membrane syringe filter prior to use. Compressed air used to dry films and coated substrates was filtered through a 0.2 μm membrane syringe filter.
General Considerations
All substrates used for multilayer film fabrication, erosion experiments, and cell based experiments were cleaned with acetone, ethanol, methanol, and water and dried using 0.2 μm filtered compressed air prior to use. Silicon and glass surfaces were then activated by etching in an oxygen plasma for 5 minutes (Plasma Etch, Carson City, NV) prior to film fabrication. Optical thicknesses of films fabricated on silicon substrates were characterized using a Gaertner LSE ellipsometer (632.8 nm, incident angle = 70°). Data were processed using the Gaertner Ellipsometer Measurement program. Relative thicknesses were calculated assuming an average index of refraction of 1.577 for the multilayered films. Thickness was measured at five locations on the films and is presented as an average value (with standard deviation) determined from the measurement of three different films. All films were rinsed with water and dried using air passed through a 0.2 μm filter prior to measurement. UV/Vis absorbance values for PBS solutions used to characterize the release of DNA from films were recorded using a Beckman Coulter DU520 UV-Vis spectrophotometer (Fullerton, CA). Absorbance measurements of solutions of released DNA were recorded at a wavelength of 260 nm. Solution fluorescence measurements were made using a Jobin Yvon Fluoromax-3 fluorometer at an excitation wavelength of 543 nm and reporting the emission as the average of values collected at wavelengths from 580 – 590 nm. The pH of buffers used for erosion and hydrolysis experiments was measured using a pH meter. LSCM was performed using a Bio-Rad Radiance 2100 Rainbow laser scanning confocal microscope equipped with a multiphoton laser. Images were processed using the Bio-Rad LaserSharp 2000 processing kit and ImageJ software (NIH). Flow cytometry analysis of cells was conducted using a BD FACSCalibur Flow Cytometer (BD Bioscience, San Jose, CA).
Preparation of Polyelectrolyte Solutions
Solutions of polymer 1, polymer 1TMR, and polymer 1OG (5 mM with respect to the molecular weight of the repeat unit) were prepared in sodium acetate buffer (100 mM, pH 4.9), as described in past studies.22,46 Solutions of LPEI and SPS used for the fabrication of LPEI/SPS base layers (20 mM with respect to the molecular weight of the repeat unit) were prepared in 10 mM NaCl solution in 18 MΩ water. LPEI solutions contained 4 mM HCl to aid polymer solubility. Solutions of plasmid DNA were prepared at a concentration of 1 mg/mL in acetate buffer (100 mM, pH 4.9).
Fabrication of DNA-Containing PEMs
All polymer 1/DNA films were fabricated on planar silicon, planar glass, or stainless steel mesh substrates pre-coated with thin multilayered films composed of 10 layer pairs (referred to hereafter as `bilayers') of LPEI and SPS (terminated with SPS) to provide a suitably charged surface for the fabrication of polymer 1/DNA films, as reported previously.46 These LPEI/SPS precursor layers were fabricated using an automated dipping robot (Riegler & Kirstein GmbH, Potsdam, Germany). DNA-containing films fabricated using polymer 1 or fluorescently labeled polymer 1 were fabricated manually using an alternating dipping procedure similar to that described previously for the fabrication of polymer 1/DNA films.22,46 Briefly: 1) substrates were submerged in a solution of the cationic polymer for 5 minutes, 2) substrates were removed and submerged in two sequential 100 mM acetate buffer baths for 1 minute each, 3) substrates were removed and submerged in a DNA solution for 5 minutes, and 4) substrates were rinsed again as described in step 3. This procedure was repeated until 8 bilayers of cationic polymer and DNA were deposited. For films used to characterize stepwise film growth, substrates were removed, rinsed with water, and dried with 0.2 μm filtered air after every two deposition cycles for characterization by ellipsometry. Films used in erosion and release experiments and cell-based experiments were dried with filtered air and stored in a vacuum dessicator prior to use. All films were fabricated at room temperature.
Characterization of Film Release Profiles
Film-coated substrates were placed in a plastic UV-transparent cuvette containing 1 mL of PBS (pH = 7.4, 137 mM NaCl; a volume sufficient to completely immerse the film-coated portion of the substrates). Samples were then incubated at 37 °C and removed at predetermined time intervals to characterize the absorbance (at 260 nm, the absorbance maximum of DNA) and fluorescence (ex: 543 nm; emission: 580 – 590 nm) of the PBS (to characterize DNA release and the release of polymer 1FL, respectively) as a function of time. Film-coated substrates were then placed in a new cuvette containing fresh PBS and returned to the incubator.
Characterization of Internalization of Polymer 1FL and DNA Using LSCM
COS-7 cells were grown in glass inset confocal microscopy dishes at an initial seeding of 1.5 × 105 cells/mL in 2 mL of growth medium [90% (v/v) Dulbecco's modified Eagle's medium, 10% (v/v) fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin]. Cells were allowed to grow overnight to approximately 80% confluence, and growth medium was replaced with 2 mL of fresh growth medium. Glass slides coated with multilayered films were placed manually into dishes and were supported above cells by the concentric ring inset present in the confocal microscopy dishes. Cells were incubated for 24 hours at 37 °C in a CO2 incubator. Substrates were then removed and analyzed immediately using a Bio-Rad Radiance 2100 MP Rainbow LSCM. Immediately prior to imaging, cells were stained with one or more (see text) of either Hoechst 34580 (nuclear stain), Calcein AM (live cell stain), or Lysotracker Red (a stain for acidic intracellular vesicles) according to the manufacturer's protocols. LSCM images were acquired using a 60×/1.4 NA oil immersion objective. Hoechst 34580 was excited using a 780 nm (multiphoton laser). Calcein AM and Oregon Green 488 probes were excited using a 488 nm laser. Lysotracker Red® and tetramethylrhodamine probes were excited using a 543 nm laser. Probes were excited sequentially from shortest wavelength excitation to longest wavelength excitation (i.e. UV excitation, then blue excitation, and then green excitation). Fluorescence emission signals were collected for three individual channels using direct scanning mode (N = 1, scan speed = 50 lps) and merged to create three-color images.
Characterization of Intracellular Co-Localization
Experiments designed to characterize the extents to which polymer 1FL, DNA, and/or Lysotracker Red® signal was colocalized in cells were performed using two different methods: (1) software analysis of polymer1OG staining and DNATMR or Lysotracker Red staining was performed using the JACoP ImageJ plugin69 (http://imagej.nih.gov/ij/plugins/track/jacop.html), or, alternatively, (2) processing of polymer 1OG staining and DNATMR or Lysotracker Red® staining using the `Feature Edges' ImageJ plugin (http://www.imagescience.org/meijering/software/featurej) to define locations of particles followed by manual counting of individual and colocalized particles in merged images (see text). A threshold was applied to all images equally prior to conducting JACoP analyses to eliminate background signal. For analyses using `FeatureJ Edges', a default smoothing factor of 1 was used to define the shapes and locations of particles.
Characterization of Internalization of Polymer 1FL and DNA Using Flow Cytometry
COS-7 cells were grown in 12 well plates at an initial seeding density of 1.25 × 105 cells/mL in 1 mL of growth medium [90% (v/v) Dulbecco's modified Eagle's medium, 10% (v/v) fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin]. Cells were allowed to grow overnight to approximately 80% confluence, and growth medium was replaced with 1 mL of fresh growth medium. Eight bilayer polymer 1TMR/DNA films were fabricated on stainless steel mesh substrates using polymer 1TMR and Cy5-labeled DNA. Coated mesh substrates were then placed in direct contact with cells. Populations of cells to be used as no-polymer/no-film controls were treated directly with aliquots containing 20 μg of naked Cy5-labeled DNA. All experiments, including cell controls, were performed in triplicate. After 24 hours, substrates were removed and cells were washed using Cell Scrub buffer according to the manufacturer's protocol. Cells were then detached using 0.5 mL trypsin and collected as a pellet in a 1.5 mL micro-centrifuge tube at 3000 rpm. The cells were then re-suspended in a 0.1 % (w/v) bovine serum albumin (BSA) solution in PBS, re-collected by centrifugation, and re-suspended in a 0.1% BSA solution. For each sample, a total of 30,000 cells were then analyzed immediately using a BD FACSCalibur flow cytometer. TMR-associated cell fluorescence was detected by excitation using a 488 nm laser and collecting emission using a 585 nm (± 21 nm) detector. Cy5 fluorescence was detected by excitation using a 635 nm laser and collecting emission using a 670 nm long pass filter. Cell fluorescence data was analyzed using the WinMDI v. 2.9 Flow Cytometry Application (facs.scripps.edu/software). Cells positive for polymer 1TMR and Cy5-labeled DNA were determined by gating all cells into positive and negative regions as determined by flow cytometry data collected for cells grown in the absence of film coated substrates. Results are expressed as the percentage of cells positive for TMR or Cy5 relative to all cells observed. The mean DNA fluorescence intensity was taken as the geometric mean Cy5 fluorescence intensity of all cells and was normalized using the mean Cy5 fluorescence intensity of untreated negative control cells.
Characterization of Released Polymer Using Measurements of Fluorescence Anisotropy
Fluorescence anisotropy was measured using a Jobin Yvon Fluoromax-3 fluorometer exciting at 543 nm, and anisotropy values are reported as the average of values at 580–590 nm according to the equation below,
where <r> is the fluorescence anisotropy, I∥ is the emission when the excitation and emission polarizers are both aligned vertically, and I⊥ is the emission when the excitation polarizer is aligned vertically and the emission polarizer is aligned horizontally.70 G is a grating correction factor determined by where the parallel configuration has both polarizers aligned horizontally and the perpendicular configuration has the excitation polarizer aligned horizontally and the emission polarizer aligned vertically. For all experiments, the erosion of polymer 1TMR/DNA films used to collect samples for these fluorescence anisotropy measurements was conducted using the general procedure described above. Anisotropy values and error bars are presented as the average and standard deviation of anisotropy measurements for six polymer 1TMR/DNA films and three polymer-only (DNA-free) control solutions consisting of 40μg polymer 1TMR in 1 mL of PBS.
Results and Discussion
Characterization of the Fabrication & Erosion of Films Assembled Using Fluorescent Polymer 1
We recently reported two different synthetic approaches to the synthesis of fluorescently labeled derivatives of polymer 1.65,68 The first approach involved random incorporation of fluorescently labeled repeat units into the backbone of polymer 1 during polymerization (e.g., by introduction of a fluorescently-labeled co-monomer during synthesis).65 The second approach makes use of methods for post-polymerization modification by the reaction of end-functionalized samples of polymer 1 with primary amine-containing fluorophores.68 Relative to the first approach, this second approach has several advantages, including: (i) the ability to generate fluorescently labeled derivatives of polymer 1 with molecular weights and polydispersity indices (PDIs) that are identical (or very similar) to those of samples of unlabeled polymer 1, (ii) access to end-functionalized polymers containing a well-defined number of fluorescent labels (reducing the likelihood of physicochemical changes in polymer behavior caused by random labeling),and (iii) the `modular' nature of this approach provides a straightforward route to families of otherwise similar polymers bearing end groups with different spectral properties (because fluorescent end groups can be added in the final synthetic step rather than during polymerization). Our past studies demonstrate that this second approach can be used to synthesize end-labeled polymers suitable for the layer-by-layer fabrication and characterization of PEMs assembled using a model anionic polymer [poly(sodium styrene sulfonate) (SPS)].68
To facilitate physical characterization and cell-based trafficking experiments using the DNA-containing films described here, we synthesized two end-labeled polymers (polymer 1TMR and polymer 1OG; see structures above) bearing TMR and Oregon Green fluorescent end groups. All films used specifically for characterization of film growth and erosion were fabricated using aqueous solutions of polymer 1TMR; films used for cell-based experiments discussed below were fabricated using either polymer 1TMR or polymer 1OG. All films used in this study were fabricated using plasmid DNA encoding luciferase (as opposed to plasmid encoding EGFP, as used in our past studies on cell transfection22,26) to simplify fluorescence-based characterization of the intracellular trafficking in subsequent cell-based experiments.
We performed an initial series of experiments using films fabricated on reflective silicon substrates to characterize the growth profiles of films fabricated with the fluorescently labeled polymer. Figure 1A shows a plot of optical film thickness versus the number of layer pairs (or `bilayers') of polymer and DNA deposited for films fabricated using plasmid DNA and either polymer 1 (closed squares) or polymer 1TMR (closed circles). Films having these general compositions are referred to hereafter as `polymer 1/DNA films' and `polymer 1TMR/DNA films', respectively. Inspection of these data reveals that the two different films exhibit similar linear growth profiles and reach thicknesses of ~150 nm after the deposition of eight bilayers. The growth profile for the polymer 1/DNA films is similar to those reported for these films in our past studies.46 The similarity of the growth profiles for the polymer 1TMR/DNA films to those of the films fabricated using unlabeled polymer demonstrates that the incorporation of fluorescent end-labels does not influence film growth significantly. Additional characterization of polymer 1OG/DNA films using fluorescence microscopy revealed the presence of uniform green and red fluorescence across the surfaces of these substrates and confirmed the presence of fluorescently labeled polymer and DNA, respectively, in the films (e.g., see Figure S1 of the Supporting Information).
Figure 1.
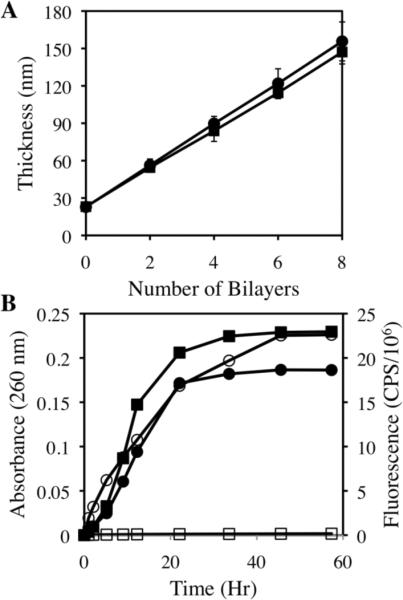
(A) Plot of ellipsometric thickness versus number of polymer 1/DNA bilayers (closed squares) and polymer 1FL/DNA bilayers (closed circles) deposited on silicon substrates. (B) Plot of solution absorbance at 260 nm (closed symbols) and solution fluorescence (Ex: 543 nm Em: 580 – 590 nm, open symbols) versus time for polymer 1/DNA films (squares) and polymer 1TMR/DNA films (circles) eroded in a solution of PBS buffer at pH 7.4 and 37 °C. Solution absorbance and fluorescence correspond to DNA and polymer 1 or polymer 1TMR release, respectively. The absorbance of polymer 1 and polymer 1TMR is negligible at 260 nm. Substrates used in these experiments were precoated with 10 bilayers of LPEI/SPS (~20 nm thick) prior to fabrication of polymer 1-containing films (see text).
Figure 1B shows both DNA and polymer 1 release profiles for polymer 1/DNA and polymer 1TMR/DNA films (eight bilayers thick) incubated in PBS at 37 °C. For these experiments, DNA release profiles were characterized by measuring increases in solution absorbance (at 260 nm, the maximum absorbance of DNA) over time, as described in our past studies.26,46 Inspection of these absorbance data (closed symbols) reveals that both films release DNA into solution gradually over a period of ~30 hours. These release profiles are generally consistent with those of our past reports on DNA-containing films fabricated using unlabeled polymer 1.46
We used fluorometry to characterize the release profiles of polymer 1 during these film erosion experiments (excitation at 543 nm and emission from 580 – 590 nm, the maximum emission for TMR). Inspection of the fluorescence data arising from the incubation of polymer 1TMR/DNA films (Figure 1B, open circles) reveals that polymer 1TMR is released gradually into solution over ~40 hours. (No increases in fluorescence were observed during the incubation of polymer 1/DNA (unlabeled) films; Figure 1B, open squares). These results are important in two primary contexts. First, these data provide the first direct observation and confirmation of the release of polymer 1 into solution during the erosion of a polymer 1/DNA film. Second, these data demonstrate that both polymer 1 and DNA are released into solution simultaneously, and on the same approximate time scale. While these data cannot be used to determine the extent to which polymer 1 and DNA may (or may not) be associated with each other once they are released, these data are consistent with the potential presence of such aggregates in solution. We return to these observations and considerations again in the discussion below.
Characterization of Internalization of Polymer 1 and DNA by Cells
We next performed a series of different in vitro cell-based experiments to characterize the extents to which the DNA and/or polymer released from these films are internalized by cells. For initial LSCM-based characterization experiments, we used films eight bilayers thick fabricated on the surfaces of planar glass substrates. These films were fabricated using polymer 1OG and TMR-labeled DNA to facilitate the characterization of the locations of both polymer and DNA in film-treated cells (and to accommodate the use of other fluorescent probes and cell stains in subsequent experiments). These LSCM experiments were conducted by placing film-coated substrates in close proximity to, but not in direct contact with, cells growing on the bottom of a confocal inset microscopy dish (e.g., film-coated substrates were maintained ~1 mm above cells by resting them on the elevated edges of the inset portion of the confocal dishes).
Figure 2 shows representative LSCM images of COS-7 cells incubated for one day in the presence of glass slides coated with polymer 1OG/DNATMR films. Panels A and B are low magnification images showing the locations of DNATMR and polymer 1OG, respectively, in a larger population of ~60 cells (both images are of the same population of cells; cell nuclei are stained blue to indicate approximate locations of cells; cell membranes not shown). These images reveal significant punctate red and green fluorescence in the vicinity of nearly every cell in this image. The images in panels C and D show low and high magnification images of cells (treated with a polymer 1TMR/DNA film) that were stained with the live-cell stain Calcein AM prior to imaging to facilitate delineation of cell shapes and borders. These confocal microscopy images indicate that areas of orange punctate fluorescence (a result of overlap of red polymer-associated fluorescence and green cell-associated fluorescence in these images) are located inside cells and provide evidence that polymer 1 is indeed internalized by nearly all cells. The punctate nature of the orange fluorescence in these images, combined with the apparent trafficking of many of these structures to locations proximal to cell nuclei, suggests that polymer 1 is internalized by endocytosis and likely resides to a significant extent in endosomal or lysosomal vesicles (see additional discussion below).
Figure 2.
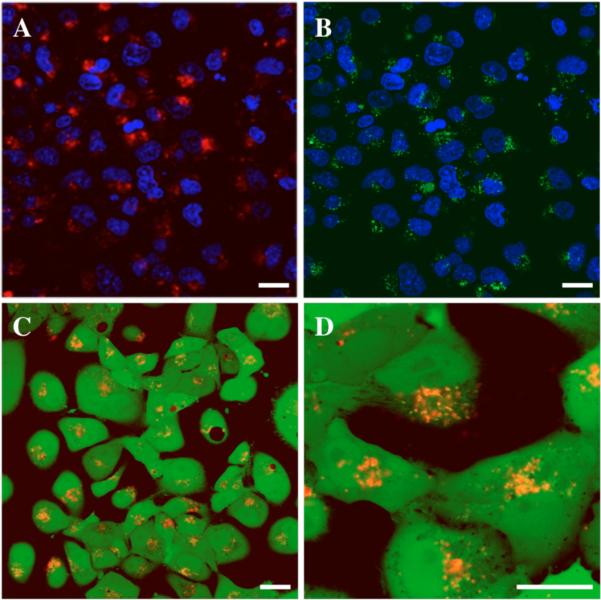
LSCM images of COS-7 cells incubated for one day in the presence of glass slides coated with either (A and B) a polymer 1OG/DNATMR film or (C and D) a polymer 1TMR/DNA film. Panels A and B show low magnification images of the same population of cells with nuclei stained blue to indicate approximate cell locations (cell membranes not shown for clarity). Panels A and B show the locations of DNATMR (red) and Polymer 1OG (green), respectively. Panels C and D show low and high magnification images of film-treated cells, respectively, with orange fluorescence corresponding to polymer 1TMR colocalized with the live cell stain Calcein AM staining (green) used prior to imaging to aid in identifying cell shapes and borders. Scale bars = 50 μm.
Zhang and co-workers have reported characterization of the internalization of DNA by HeLa cells growing directly on enzymatically degradable poly(lysine)/hyaluronic acid PEMs containing embedded layers of fluorescently labeled plasmid DNA pre-complexed with either poly(lysine), β-cyclodextrin, or a poly(lysine)-β-cyclodextrin conjugate.28 Although LSCM was used to characterize the internalization and trafficking of DNA, this past study permitted only indirect conclusions regarding internalization of the cationic components of these materials. To the best of our knowledge, the results shown in Figure 2 provide the first direct (albeit preliminary) demonstration that the cationic polymer components of a degraded DNA-containing PEM can be internalized by cells.
To provide additional insight and characterize the internalization of polymer 1 and DNA more quantitatively (and in significantly larger populations of cells), we also characterized film-treated cells using flow cytometry. These experiments were conducted using eight-bilayer films fabricated on stainless steel mesh substrates using polymer 1TMR and DNA labeled with Cy-5 (20% of DNA was labeled; the remainder was unlabeled). Cells were incubated in the presence of film-coated mesh substrates for 24 hours prior to trypsinization and analysis. Figure 3A shows a plot of the percentages of film-treated cells that were positive for either polymer 1TMR or Cy-5 DNA; data shown are for three different populations of cells treated with either (1) no substrates (negative cell control), (2) film-coated substrates, or (3) a solution containing a 20 μg bolus of naked Cy-5 DNA (an amount of DNA determined to be similar to the amount of DNA released during the erosion of a film-coated substrate). Figure 3B presents the results of these same experiments as the average mean fluorescence intensity of Cy-5 DNA associated with the cells (rather than as the percentage of fluorescence-positive cells).
Figure 3.

Results of flow cytometry data evaluating association of polymer 1TMR and DNACy5 with COS-7 cells. (A) Percentage of cells positive for polymer 1TMR or DNACy5 (grey and white bars, respectively) and (B) relative cellular uptake of DNACy5 as determined by the normalized mean DNA fluorescence intensity. COS-7 cells were treated with a polymer 1TMR/DNACy5 films on mesh substrates, 20 μg of naked DNACy5, or left untreated (negative cell control) for 24 hours prior to harvesting and analysis by flow cytometry (see text for details). Values and error bars are reported as the average and standard deviation, respectively, for three samples analyzing ~30,000 cells each.
The data in Figure 3A demonstrate that nearly all cells incubated in the presence of film-coated substrates are positive for both polymer (~95% of cells; grey bar) and DNA (~99% of cells; white bar). These results also reveal that ~99% of cells in control populations treated with the bolus of naked DNA are also associated with DNA. Inspection of the mean fluorescence intensity values for these populations shown in Figure 3B, however, shows that cells incubated in the presence of film-coated substrates are associated with significantly higher amounts of DNA (~3.5-fold more) as compared to cells incubated with DNA alone. This result is consistent with the view that the presence of cationic polymer 1 could promote more effective internalization of DNA (e.g., by formation of polymer 1/DNA aggregates that stimulate or facilitate endocytosis, etc.). It is, of course, also possible that the close proximity of the film-coated substrates to many of the cells in these experiments could lead to higher local concentrations of DNA that also promote more effective internalization. Although these current experiments cannot discount the contributions of such substrate/proximity effects, we note that such effects would be much less likely for these more open, coated mesh substrates than for film-coated solid substrates (such as film-coated glass slides used above and in our past studies on surface-mediated transfection22). More broadly, however, we note that such substrate/proximity effects or influences would, of course, be desirable in the practical context of promoting more efficient surface-mediated DNA delivery. Further evidence for the role that polymer 1 could play in promoting internalization of DNA is provided by the results of additional confocal experiments described below.
Characterization of Co-Localization of Polymer 1 and DNA in Cells Using LSCM
We performed a series of additional LSCM experiments to determine the extents to which internalized polymer and internalized DNA are co-localized inside cells. Figure 4 shows a set of high-magnification LSCM images of cells incubated in the presence of glass substrates coated with a polymer 1OG/DNATMR film for 22 hours. Panels A and B show the locations of DNATMR (red) and polymer 1OG (green), respectively, in cells (the locations of cell membranes/borders were determined from corresponding bright field images and are indicated for several cells as dashed lines in these images). Panel C shows a merged image of the images in panels A and B, and also includes the locations of cell nuclei (blue stain). The presence of yellow punctate structures where green and red fluorescence signals (in panels A and B) of similar intensities overlap suggests that polymer 1 and DNA are co-localized within these cells to a significant extent. Closer evaluation of the image in panel C also reveals areas of punctate polymer-associated green fluorescence and DNA-associated red fluorescence that do not appear to overlap by visual inspection.
Figure 4.

High magnification LSCM images of the same population of COS-7 cells incubated for 22 hours in the presence of a glass slide coated with a polymer 1OG/DNATMR film. Panels A and B show the locations of DNATMR (red) and polymer 1OG (green), respectively. Panel C shows the merge of panels A and B, with the locations of cell nuclei included as indicated by blue staining. Yellow indicates the colocalization of polymer 1OG and DNATMR with similar fluorescence intensities. Panel D shows the merge of panels A and B after being processed using ImageJ software to identify the locations of polymer 1OG and DNATMR particles (see text for details). For all images, the locations of cell membranes/borders were determined using a corresponding bright field image and are indicated here for several cells as white dashed lines. These images and others (see Figure S2 in Supporting Information) were used in combination to quantify the extents of polymer 1OG and DNATMR colocalization reported in Table I (see text for details). Scale bar = 50 μm.
To provide more quantitative insight into the extents to which the red and green fluorescence signals in this image were co-localized, we analyzed a series of LSCM images using two different methods. The results of these analyses are summarized in Table 1. These images were processed using features of the ImageJ processing program (see Materials and Methods for additional details) to identify the locations of red and green features in these images (e.g., see Figure 4D; additional representative images used for these analyses are included in Figure S2 of the Supporting Information). The extent to which these different red and green signals overlapped was then first determined by visual inspection and manual counting. This method of analysis revealed that ~58% of the punctate DNA-associated (red) features overlap, at least to some extent, with areas of green polymer-associated fluorescence. Automated pixel-analysis of the same processed images using a co-localization algorithm associated with ImageJ69 returned a similar value (~54% co-localization of signal). These results demonstrate that polymer 1 and DNA are co-localized to a significant degree in these images (e.g., slightly more than half of the fluorescent DNA-associated features exist in the same locations as polymer 1OG in these cells).
Table 1.
Characterization of the extents of overlap of Polymer 1OG-associated and DNATMR-associated fluorescence in cells incubated with polymer 1OG/DNATMR filmsa
| Analysis Method | DNA Colocalized with Polymer 1 Average ± σ (%) | Polymer 1 Colocalized with DNA Average ± σ (%) |
|---|---|---|
| Manual Counting | 57.5 ± 10.0 | 39.2 ± 6.8 |
| JaCOP Analysis | 54.0 ± 3.5 | 35.4 ± 2.5 |
Values were calculated using separate manual counting and automated software processes and are reported as the average extent of colocalization of polymer 1OG and DNATMR with standard deviation for measurements of three separate regions of cells (see text for details).
As mentioned above, inspection of the images in Figure 4 also reveals areas of red and green fluorescence that do not overlap, either by visual inspection or when the image processing steps described above were used. (In contrast to the results above, for example, we observed only ~35–40% of the punctate areas of polymer-associated fluorescence to overlap with areas of red DNA-associated fluorescence; see Table 1, Column 2). There are several possible reasons for this, including, foremost, the possibility that these signals identify the locations of free (unbound) DNA and free (unbound) polymer. We note, however, that only 50% of the DNA used to fabricate the films used in these experiments was fluorescently labeled – the remaining 50% of the plasmid in the dipping solutions used to fabricate these films was not labeled, and the locations of this unlabeled DNA would thus not be apparent in these images. It is therefore also possible that at least some of the areas that appear as green (identifying the apparent locations of polymer only) could also represent the locations of aggregates of polymer with unlabeled DNA. In view of this possibility, we note that the values calculated above for the percentage of polymer 1-associated structures that overlap with DNA (~35–40%) are likely to be underestimates. Values representing the extent to which areas of DNA fluorescence overlap with polymer-associated fluorescence (~54–58%) are likely more representative of the actual extents of polymer/DNA colocalization, because every chain of polymer 1OG used to fabricate these films should, in principle, be fluorescently end-labeled.68
The results of additional confocal microscopy-based trafficking experiments using polymer 1OG/DNA films and cells stained with a fluorescently-labeled organelle stain specific for acidic intracellular vesicles (Lysotracker Red) revealed that a significant amount of internalized polymer was located within cell endosomes or lysosomes (data not shown). The results of these confocal microscopy experiments were more difficult to quantify (they ranged from ~50% to ~85% co-localization, depending on methods used). Qualitatively, however, these results suggest that endosomal escape, which has been identified as an important intracellular barrier to transfection in numerous other studies of non-viral DNA delivery agents, likely represents an important barrier to transfection using this PEM-based approach as well (our results above, collectively, suggest that internalization efficiency is high and, thus, less likely to be rate-limiting). This view is also consistent with observations arising from our past studies characterizing levels of surface-mediated cell transfection mediated by objects coated with otherwise identical films fabricated using polymer 1 and plasmid DNA encoding pEGFP.22,26 Whereas our current results demonstrate that polymer and DNA are internalized by nearly all cells, those past studies revealed levels of cell transfection that were significantly lower (e.g., ranging from ~5 to ~40% of EGFP-positive cells, as determined by fluorescence microscopy).
We note here that polymer 1 was selected for use in this and several past studies largely on the basis of the levels of control that it provides over film growth and film erosion.22,26,46 Polymer 1 has been demonstrated to be an effective transfection agent when used to form welldefined polyplexes,66 but the structure of this polymer has not been optimized or designed rationally to promote efficient endosomal escape or navigate other intracellular barriers. In this particular context, our results demonstrating that polymer and DNA are co-localized inside cells are important, because they suggest new opportunities to design films capable of promoting more efficient surface-mediated transfection by either (i) further modifying the structure of polymer 1 to incorporate additional functionality, or (ii) exploiting the layer-by-layer nature of the assembly process use here to deposit additional layers of other polymers [e.g., branched poly(ethyleneimine), etc.] that have been used in other studies15,16 to address particular intracellular barriers more effectively. It is important to note that if polymer 1 were not observed to be internalized or co-localized with DNA in the experiments above, these potential molecular-level design alternatives would likely be less effective (or would at least be significantly less attractive) as options for further optimization.
Characterization of Interactions Between Polymer 1FL and DNA in Solution
Finally, we mention that while the results above reveal that polymer 1 and DNA are substantially co-localized inside cells, these data do not permit specific conclusions about the manner in which co-localization occurs. For example, our observations are consistent with the direct internalization of polymer 1/DNA aggregates, but we cannot exclude the alternative possibility that polymer 1 and DNA may be internalized separately (e.g., by endocytosis) and then trafficked actively to the same locations inside cells (e.g., into lysosomes, etc.). The release results shown in Figure 1B demonstrate that both polymer 1 and DNA are released simultaneously during film erosion. Dynamic light scattering (DLS) analysis of solutions of partially eroded polymer 1/DNA films arising from otherwise identical experiments revealed the presence of large aggregates in solution with sizes as small as ~30 – 300 nm in size (data not shown). These results are consistent with those of our past studies.26 However, as discussed in the Introduction, DLS does do not provide direct insight into the compositions of these aggregates (e.g., whether or not they are comprised of polymer 1 alone, DNA alone, or mixtures of polymer 1 and DNA). In a final set of experiments, we sought to determine whether our new fluorescently-labeled derivatives of polymer 1 could be used to provide additional evidence of polymer 1/DNA aggregates in solution (e.g., after film erosion, but prior to internalization by cells) using measurements of fluorescence anisotropy.
Changes in fluorescence anisotropy are used in many applications to characterize interactions between different molecules in solution (e.g., protein-DNA interactions, antigen-antibody binding, small molecule-protein binding, and peptide-protein interactions).70 This method is based, in general, on two observations. The first is that the polarization of light emitted from a fluorescent species excited using polarized light is related to the rate at which that species rotates or tumbles in solution (e.g., depolarization occurs more rapidly for small molecules than for large molecules that exhibit slower rates of rotational diffusion). The second observation is that when a fluorescent species binds to another molecule, the increased size of the resulting complex results in a change in the rate of rotational diffusion (as a result of an increase in the mass of the complex) and, thus, a reduction in the extent to which fluorescence is depolarized. This decrease in the depolarization of fluorescence can be measured using fluorometry and expressed as an increase in fluorescence anisotropy (see Materials and Methods for additional details).
Two recent studies suggest that fluorescence anisotropy can be used to characterize the formation and disruption of polycation/DNA polyplexes formed in solution.71,72 We therefore sought to determine whether observations of changes in the fluorescence anisotropy of solutions used to erode films fabricated using DNA and fluorescently-labeled polymer 1FL could provide evidence of the presence (or absence) of complexes formed by these two species in solution. We chose to use fluorescently labeled polymer 1 in these experiments, as opposed to fluorescently labeled DNA, because polymer 1 is substantially smaller than the plasmid DNA used here and should therefore, in principle, yield larger changes in anisotropy upon binding. A series of initial experiments to measure fluorescence anisotropy in solutions of defined mixtures of polymer 1FL and DNA (i.e., preformed soluble interpolyelectrolyte complexes) suggested that changes in fluorescence anisotropy could be used to provide qualitative insight into the existence or absence of physical interactions between these species (data not shown).
Figure 5 shows a plot of fluorescence anisotropy versus time for PBS solutions collected at various time points during the erosion of polymer 1TMR/DNA films eight bilayers thick (a plot of measurements recorded for solutions of polymer 1TMR alone is also included for comparison). Two key observations arise from these results. First, fluorescence anisotropy is measured at all time points in this experiment to be higher than that of solutions of free, unbound polymer 1TMR. Second, the magnitude of the fluorescence anisotropy changes significantly over a period of ~45 hours (for example, there is an initial maximum after the first hour of erosion, a minimum observed at eight hours, and a subsequent gradual increase in anisotropy over the remaining ~35 hours of the experiment). The time-dependent nature of these changes in fluorescence anisotropy appears to be general and was observed over many different experimental trials.
Figure 5.
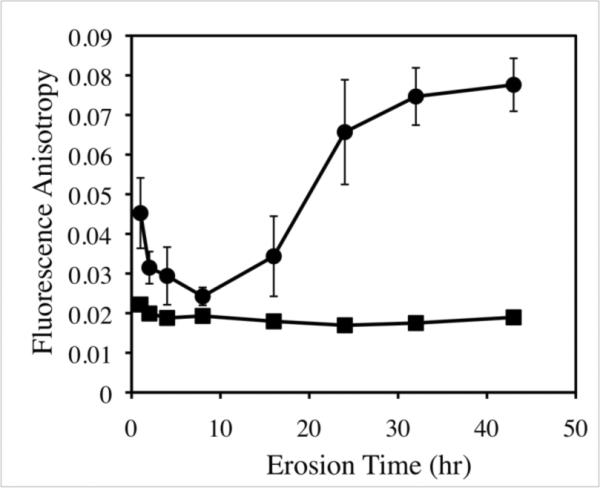
Plot of fluorescence anisotropy versus time measured for polymer 1TMR released from polymer 1TMR/DNA films incubated in PBS buffer at 37 °C (circles) and a solution of 40 μg polymer 1TMR alone (squares) incubated in PBS buffer at 37 °C in the absence of DNA (see text for details). Fluorescence anisotropy values of solutions for polymer 1TMR/DNA film erosion are representative of polymer 1TMR released over discrete time intervals (i.e. 0 – 1 hours, 1 – 2 hours, 2 – 4 hours, etc.; see text for details). The 40 μg samples of polymer 1TMR were incubated in solution for the entirety of the experiment in order to provide fluorescence anisotropy values that are reflective of polymer 1TMR released from films (since the polymer within films can continually degrade due to hydrolysis).
The results shown in Figure 5 are consistent with the presence of aggregates of polymer 1TMR and DNA in these solutions, but they also suggest that the compositions and/or sizes of these aggregates can change significantly over time. In this context, we note that these data represent average values of fluorescence anisotropy arising from the release of polymer 1TMR and DNA into solution at different, discrete points in the erosion process, and that any aggregates of polymer 1TMR present in solution could arise in at least three ways: (i) from polymer 1TMR/DNA 1TMR/DNA complexes released directly from the films (as pre-existing aggregates), (ii) from the aggregation of polymer 1TMR and DNA films after the release of each of these components individually (i.e., aggregates that form in solution), or (iii) from the aggregation of free polymer 1TMR with existing aggregates in solution (e.g., to form larger complexes).
The data in Figure 5 are consistent with the presence of macromolecular aggregates that contain both polymer 1 and DNA (the absence of large, time-dependent changes in the fluorescence anisotropy of solutions of polymer 1TMR alone suggests that polymer 1 itself does not self-aggregate substantially). We caution here, however, that DNA is not the only anionic polymer species present in these films, and that our past AFM, SEM, and confocal microscopy studies have demonstrated that these films can undergo significant physical rearrangements62,63,65 It is possible that the presence and possible release of SPS (an anionic polymer used to fabricate the underlying `base layers' on which these polymer 1/DNA films are deposited; see materials and methods) could also account for observations of aggregation and time-dependent differences in average fluorescence anisotropy values. Additional studies will be required to determine the extent to which fluorescence anisotropy can be used as a tool to characterize quantitatively the formation and/or relative compositions of polymer/DNA aggregates in solution. In the context of this current study, however, the relative differences in fluorescence anisotropy shown in Figure 5 are consistent with the view that polymer 1 and DNA can be released from these PEMs in forms that are at least partially aggregated with each other (or that can subsequently aggregate or bind to each other).
Summary and Conclusions
In summary, we have demonstrated that new fluorescently end-labeled derivates of polymer 1 can be used to provide insights into the behaviors of a class of erodible, DNA-containing PEMs used in several past studies to promote the surface-mediated delivery of DNA to cells. The availability fluorescently-labeled derivatives of polymer 1 permits characterization of film erosion and cellular internalization of film components using fluorescence-based techniques that provide information about the locations and behaviors of polymer 1 that cannot be readily obtained using other methods. Our results demonstrate for the first time that both polymer 1 and DNA are released simultaneously during film erosion, and that both of these film components are internalized readily by cells growing in vitro in the vicinity of substrates coated with these materials. Additional characterization by LSCM revealed significant levels of co-localization of polymer 1 and DNA in cells and suggested that these materials are sequestered, to a significant extent, within endosomes and lysosomes. Finally, the results of fluorescence anisotropy measurements of solutions containing partially eroded films provided additional preliminary evidence consistent with the presence of aggregates of polymer 1 and DNA (e.g., after release from film-coated surfaces, but prior to internalization by cells).
The results of this study, when combined, shed new light on the behaviors of the cationic polymer components of these DNA-containing assemblies and provide important insight into the roles that polymer 1 may play in promoting surface-mediated cell transfection. The results of our past studies have demonstrated that polymer 1/DNA films can promote significant levels of surface-mediated cell transfection in vitro and in vivo. The results of this current study provide additional support for the broader view that polymer 1 – which is incorporated into these PEM-based films as `layers' during layer-by-layer assembly, rather than as a component of pre-formed `polyplexes' – can act to promote or enhance the internalization and processing of DNA by cells. In this context, the observation that polymer and DNA are substantially co-localized inside cells is particularly important, because it suggests new opportunities to improve the gene delivery properties of these materials in future experiments – for example by modifying the structure of polymer 1 or by co-incorporation of additional `layers' of other cationic polymers (e.g., PEI, etc.) that are known to address other important intracellular barriers to transfection (such as endosomal escape) more effectively. These molecular-level design alternatives would likely prove less effective as a means of promoting more efficient surface-mediated transfection if polymer 1 were not bound to and/or co-localized with DNA after film erosion. Studies to evaluate the potential of these new design approaches are currently underway.
Supplementary Material
Acknowledgment
Financial support for this work was provided by the National Institutes of Health (R01 EB006820). We are grateful to Lance Rodenkirch and Michael Hendrickson at the W. M. Keck Center for Biological Imaging for technical support of instrumentation and numerous helpful discussions associated with LSCM experiments. We also are grateful to Ryan Flessner and Christopher Jewell for many useful discussions and technical assistance.
Footnotes
Supporting Information Available. Additional fluorescence and confocal microscopy images used to characterize film morphology and composition and quantify aspects of intracellular trafficking of polymer 1 and DNA. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 2.Nishikawa M, Huang L. Hum. Gene Ther. 2001;12:861–870. doi: 10.1089/104303401750195836. [DOI] [PubMed] [Google Scholar]
- 3.Losordo DW, Vale PR, Hendel RC, Milliken CE, Fortuin FD, Cummings N, Schatz RA, Asahara T, Isner JM, Kuntz RE. Circulation. 2002;105:2012–2018. doi: 10.1161/01.cir.0000015982.70785.b7. [DOI] [PubMed] [Google Scholar]
- 4.Niidome T, Huang L. Gene Ther. 2002;9:1647–1652. doi: 10.1038/sj.gt.3301923. [DOI] [PubMed] [Google Scholar]
- 5.Sharif F, Daly K, Crowley J, O'Brien T. Cardiovasc. Res. 2004;64:208–216. doi: 10.1016/j.cardiores.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Shea LD, Smiley E, Bonadio J, Mooney DJ. Nat. Biotechnol. 1999;17:551–554. doi: 10.1038/9853. [DOI] [PubMed] [Google Scholar]
- 7.Klugherz BD, Jones PL, Cui XM, Chen WL, Meneveau NF, DeFelice S, Connolly J, Wilensky RL, Levy RJ. Nat. Biotechnol. 2000;18:1181–1184. doi: 10.1038/81176. [DOI] [PubMed] [Google Scholar]
- 8.Bielinska AU, Yen A, Wu HL, Zahos KM, Sun R, Weiner ND, Baker JR, Roessler BJ. Biomaterials. 2000;21:877–887. doi: 10.1016/s0142-9612(99)00229-x. [DOI] [PubMed] [Google Scholar]
- 9.Ziauddin J, Sabatini DM. Nature. 2001;411:107–110. doi: 10.1038/35075114. [DOI] [PubMed] [Google Scholar]
- 10.Segura T, Shea LD. Bioconjugate Chem. 2002;13:621–629. doi: 10.1021/bc015575f. [DOI] [PubMed] [Google Scholar]
- 11.Pannier AK, Shea LD. Mol. Ther. 2004;10:19–26. doi: 10.1016/j.ymthe.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 12.De Laporte L, Shea LD. Adv. Drug Delivery Rev. 2007;59:292–307. doi: 10.1016/j.addr.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jewell CM, Lynn DM. Curr. Opin. Colloid Interface Sci. 2008;13:395–402. doi: 10.1016/j.cocis.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabanov AV, Felgner PL, Seymour LW. Self-assembling complexes for gene delivery : from laboratory to clinical trial. Wiley; Chichester; New York: 1998. [Google Scholar]
- 15.Amiji M. Polymeric Gene Delivery: Principles and Applications. CRC Press; New York, New York: 2004. [Google Scholar]
- 16.Pack DW, Hoffman AS, Pun S, Stayton PS. Nat. Rev. Drug Discovery. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 17.Wasungu L, Hoekstra D. J. Controlled Release. 2006;116:255–264. doi: 10.1016/j.jconrel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 18.Luo D, Saltzman WM. Nat. Biotechnol. 2000;18:893–895. doi: 10.1038/78523. [DOI] [PubMed] [Google Scholar]
- 19.Saul JM, Linnes MP, Ratner BD, Giachelli CM, Pun SH. Biomaterials. 2007;28:4705–4716. doi: 10.1016/j.biomaterials.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 20.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 21.Jewell CM, Lynn DM. Adv. Drug Delivery Rev. 2008;60:979–999. doi: 10.1016/j.addr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jewell CM, Zhang JT, Fredin NJ, Lynn DM. J. Controlled Release. 2005;106:214–223. doi: 10.1016/j.jconrel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Yamauchi F, Kato K, Iwata H. Langmuir. 2005;21:8360–8367. doi: 10.1021/la0505059. [DOI] [PubMed] [Google Scholar]
- 24.Jessel N, Oulad-Abdeighani M, Meyer F, Lavalle P, Haikel Y, Schaaf P, Voegel JC. Proc. Natl. Acad. Sci. U. S. A. 2006;103:8618–8621. doi: 10.1073/pnas.0508246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reibetanz U, Claus C, Typlt E, Hofmann J, Donath E. Macromol. Biosci. 2006;6:153–160. doi: 10.1002/mabi.200500163. [DOI] [PubMed] [Google Scholar]
- 26.Jewell CM, Zhang JT, Fredin NJ, Wolff MR, Hacker TA, Lynn DM. Biomacromolecules. 2006;7:2483–2491. doi: 10.1021/bm0604808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimitrova M, Arntz Y, Lavalle P, Meyer F, Wolf M, Schuster C, Haikel Y, Voegel JC, Ogier J. Adv. Funct. Mater. 2007;17:233–245. [Google Scholar]
- 28.Zhang X, Sharma KK, Boeglin M, Ogier J, Mainard D, Voegel JC, Mely Y, Benkirane-Jessel N. Nano Lett. 2008;8:2432–2436. doi: 10.1021/nl801379y. [DOI] [PubMed] [Google Scholar]
- 29.Meyer F, Dimitrova M, Jedrzejenska J, Arntz Y, Schaaf P, Frisch B, Voegel JC, Ogier J. Biomaterials. 2008;29:618–624. doi: 10.1016/j.biomaterials.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 30.Blacklock J, You YZ, Zhou QH, Mao GZ, Oupicky D. Biomaterials. 2009;30:939–950. doi: 10.1016/j.biomaterials.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blacklock J, Vetter A, Lankenau A, Oupicky D, Mohwald H. Biomaterials. 2010;31:7167–7174. doi: 10.1016/j.biomaterials.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blacklock J, Sievers TK, Handa H, You YZ, Oupicky D, Mao GZ, Mohwald H. J. Phys. Chem. B. 2010;114:5283–5291. doi: 10.1021/jp100486h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Oulad-Abdelghani M, Zelkin AN, Wang YJ, Haikel Y, Mainard D, Voegel JC, Caruso F, Benkirane-Jessel N. Biomaterials. 2010;31:1699–1706. doi: 10.1016/j.biomaterials.2009.11.032. [DOI] [PubMed] [Google Scholar]
- 34.Demuth PC, Su X, Samuel RE, Hammond PT, Irvine DJ. Adv. Mater. 2010;22:4851–4856. doi: 10.1002/adma.201001525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saurer EM, Yamanouchi D, Liu B, Lynn DM. Biomaterials. 2011;32:610–618. doi: 10.1016/j.biomaterials.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertrand P, Jonas A, Laschewsky A, Legras R. Macromol. Rapid Commun. 2000;21:319–348. [Google Scholar]
- 37.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv. Mater. 2006;18:3203–3224. [Google Scholar]
- 38.Boudou T, Crouzier T, Ren KF, Blin G, Picart C. Adv. Mater. 2010;22:441–467. doi: 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- 39.Zelikin AN. ACS Nano. 2010;4:2494–2509. doi: 10.1021/nn100634r. [DOI] [PubMed] [Google Scholar]
- 40.Becker AL, Johnston APR, Caruso F. Small. 2010;6:1836–1852. doi: 10.1002/smll.201000379. [DOI] [PubMed] [Google Scholar]
- 41.Lavalle P, Voegel JC, Vautier D, Senger B, Schaaf P, Ball V. Adv. Mater. 2011;23:1191–1221. doi: 10.1002/adma.201003309. [DOI] [PubMed] [Google Scholar]
- 42.Qiu XP, Leporatti S, Donath E, Mohwald H. Langmuir. 2001;17:5375–5380. [Google Scholar]
- 43.Chung AJ, Rubner MF. Langmuir. 2002;18:1176–1183. [Google Scholar]
- 44.Lynn DM. Adv. Mater. 2007;19:4118–4130. [Google Scholar]
- 45.Vazquez E, Dewitt DM, Hammond PT, Lynn DM. J. Am. Chem. Soc. 2002;124:13992–13993. doi: 10.1021/ja026405w. [DOI] [PubMed] [Google Scholar]
- 46.Zhang JT, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 47.Wood KC, Boedicker JQ, Lynn DM, Hammond PT. Langmuir. 2005;21:1603–1609. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 48.Macdonald M, Rodriguez NM, Smith R, Hammond PT. J. Controlled Release. 2008;131:228–234. doi: 10.1016/j.jconrel.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Su XF, Kim BS, Kim SR, Hammond PT, Irvine DJ. ACS Nano. 2009;3:3719–3729. doi: 10.1021/nn900928u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saurer EM, Flessner RM, Sullivan SP, Prausnitz MR, Lynn DM. Biomacromolecules. 2010;11:3136–3143. doi: 10.1021/bm1009443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li BY, Haynie DT. Biomacromolecules. 2004;5:1667–1670. doi: 10.1021/bm0496155. [DOI] [PubMed] [Google Scholar]
- 52.Zelikin AN, Li Q, Caruso F. Angew. Chem., Int. Ed. 2006;45:7743–7745. doi: 10.1002/anie.200602779. [DOI] [PubMed] [Google Scholar]
- 53.Blacklock J, Handa H, Manickam DS, Mao GZ, Mukhopadhyay A, Oupicky D. Biomaterials. 2007;28:117–124. doi: 10.1016/j.biomaterials.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 54.Chen J, Huang SW, Lin WH, Zhuo RX. Small. 2007;3:636–643. doi: 10.1002/smll.200600301. [DOI] [PubMed] [Google Scholar]
- 55.Serizawa T, Yamaguchi M, Akashi M. Angew. Chem., Int. Ed. 2003;42:1115–1118. doi: 10.1002/anie.200390293. [DOI] [PubMed] [Google Scholar]
- 56.Picart C, Schneider A, Etienne O, Mutterer J, Schaaf P, Egles C, Jessel N, Voegel JC. Adv. Funct. Mater. 2005;15:1771–1780. [Google Scholar]
- 57.Ren KF, Ji J, Shen JC. Biomaterials. 2006;27:1152–1159. doi: 10.1016/j.biomaterials.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 58.Meyer F, Ball V, Schaaf P, Voegel JC, Ogier J. Biochim. Biophys. Acta, Biomembr. 2006;1758:419–422. doi: 10.1016/j.bbamem.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 59.Dimitrova M, Affolter C, Meyer F, Nguyen I, Richard DG, Schuster C, Bartenschlager R, Voegel JC, Ogier J, Baumert TF. Proc. Natl. Acad. Sci. U. S. A. 2008;105:16320–16325. doi: 10.1073/pnas.0800156105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang XF, Sun JK, Ji JA. React. Funct. Polym. 2011;71:254–260. [Google Scholar]
- 61.Zhang JT, Lynn DM. Adv. Mater. 2007;19:4218–4223. [Google Scholar]
- 62.Fredin NJ, Zhang JT, Lynn DM. Langmuir. 2005;21:5803–5811. doi: 10.1021/la050596+. [DOI] [PubMed] [Google Scholar]
- 63.Fredin NJ, Zhang JT, Lynn DM. Langmuir. 2007;23:2273–2276. doi: 10.1021/la0624182. [DOI] [PubMed] [Google Scholar]
- 64.Saurer EM, Jewell CM, Kuchenreuther JM, Lynn DM. Acta Biomater. 2009;5:913–924. doi: 10.1016/j.actbio.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fredin NJ, Flessner RM, Jewell CM, Bechler SL, Buck ME, Lynn DM. Microsc. Res. Tech. 2010;73:834–844. doi: 10.1002/jemt.20830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lynn DM, Langer R. J. Am. Chem. Soc. 2000;122:10761–10768. [Google Scholar]
- 67.Zhang JT, Montanez SI, Jewell CM, Lynn DM. Langmuir. 2007;23:11139–11146. doi: 10.1021/la702021s. [DOI] [PubMed] [Google Scholar]
- 68.Bechler SL, Lynn DM. J. Polym. Sci., Part A: Polym. Chem. 2011;49:1572–1581. doi: 10.1002/pola.24578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bolte S, Cordelieres FP. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 70.Jameson DM, Ross JA. Chem. Rev. 2010;110:2685–2708. doi: 10.1021/cr900267p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ko YT, Bickel U, Huang JY. Oligonucleotides. 2011;21:109–114. doi: 10.1089/oli.2010.0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshihara C, Shew CY, Ito T, Koyama Y. Biophys. J. 2010;98:1257–1266. doi: 10.1016/j.bpj.2009.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.