

Abstract
The central nervous system (CNS) plays key role in the homeostatic regulation of body weight. Satiation and adiposity signals, providing acute and chronic information about available fuel, are produced in the periphery and act in the brain to influence energy intake and expenditure, resulting in the maintenance of stable adiposity. Diet-induced obesity (DIO) does not result from a failure of these central homeostatic circuits. Rather, the threshold for defended adiposity is increased in environments providing ubiquitous access to palatable, high-fat foods, making it difficult to achieve and maintain weight loss. Consequently, mechanisms by which nutritional environments interact with central homeostatic circuits to influence the threshold for defended adiposity represent critical targets for therapeutic intervention.
Energy balance relies on a tightly regulated homeostatic system matching energy intake with energy expenditure in order to maintain a stable body weight. In other words, body weight, and specifically body fat, is normally defended against acute perturbations. For example, animals that are calorically restricted to produce substantial weight loss quickly rebound when returned to ad-libitum feeding, recovering their initial body weight within days (Levin and Keesey, 1998; Woods et al., 2003). Analogously, animals chronically over-fed via an intra- gastric catheter quickly shed the excess weight gain by reducing their daily food intake when over-feeding stops (Hagan et al., 1999; Seeley et al., 1996a). Similarly, humans subjected to experimental weight loss or weight gain adjust their food intake and/or energy expenditure in order to defend baseline adiposity (Leibel et al., 1995; Roberts et al., 1990; Sims, 1976). Consequently, most adults, even those who are overweight or obese, maintain a relatively stable body weight when measured over long intervals. In a recent longitudinal study, for example, men with no history of obesity gained an average of only 0.18 kg·bw / year, and an age-matched obese group gained just 0.04 kg·bw / year (Van Wye et al., 2007), illustrating the remarkably precise nature of the homeostatic system regulating adiposity.
The CNS plays a key role to balance the energy equation. Environmental and internal signals, indicating energy needs and availability, are integrated in CNS circuits. These recruit appropriate effector systems to adjust behavioral and physiological responses, maintaining adiposity at a relatively constant level. Considerable progress has been made to elucidate the molecular and cellular pathways, primarily within the hypothalamus and hindbrain, comprising these circuits. An emergent principle of this homeostatic regulation is that acute satiation signals, arising in the gastrointestinal system (GI) and secreted phasically during meals, are integrated with more tonically-active adiposity signals to adjust nutrient intake and energy expenditure.
Satiation signals induce feelings of fullness and influence the decision to stop eating (Woods and D’Alessio, 2008). Many satiation signals are peptides secreted from enteroendocrine cells in the wall of the GI tract. They stimulate adjacent sensory nerves, forwarding information to the hindbrain to initiate appropriate reflexes, and influence the decision to keep eating or to end the meal. When administered peripherally at the start a meal, satiation signals result in less food being eaten (Fujimoto et al., 1993; Gibbs and Smith, 1977; Ruttimann et al., 2009). The response is dose-dependent and short-lived. Conversely, administering antagonists to satiation signals results in larger meals being eaten, indicating that endogenous satiation signals normally limit meal size (Hewson et al., 1988; Williams et al., 2009). Several satiation signals have been identified, including cholecystokinin (CCK), glucagon-like peptide (GLP-1), glucagon, oxyntomodulin, peptide tyrosine-tyrosine (PYY), apolipoprotein A-IV, enterostatin, amylin, and members of the bombesin family of peptides (Moran, 2004; Woods and D’Alessio, 2008). The various satiation signals respond with differing sensitivity to diverse physicochemical stimuli created by the meal, including individual nutrients, to limit meal size.
Whereas satiation signals reflect the nutrient and caloric content of a meal, adiposity signals reflect the amount of stored body fat. The most-studied adiposity signals are insulin and leptin. Insulin is secreted from the pancreatic β-cells, and leptin is secreted from adipocytes, both in direct proportion to total adiposity (Bagdade et al., 1967; Frederich et al., 1995; Polonsky et al., 1988; Schwartz et al., 1996), providing reliable indicators of stored energy. Insulin and leptin are transported from the circulation into the brain. In lean individuals, increased flux of insulin or leptin stimulates receptors in the hypothalamus and in other brain regions to reduce food intake (Brief and Davis, 1984; Halaas et al., 1995; Seeley et al., 1996b; Woods et al., 1979). Conversely, experimentally reducing adiposity signaling in the brain results in hyperphagia and weight gain (Brüning et al., 2000; Cohen et al., 2001; Obici et al., 2002). Unlike satiation signals, adiposity signals have long-lasting effects that span several meals or days, stabilizing adipose stores.
Satiation and adiposity signals interact synergistically to influence energy intake and expenditure. Leptin and insulin signaling provide a background tone that determines sensitivity to satiation signals. For example, individuals that diet and lose fat secrete less leptin and insulin, and consequently have reduced leptin and insulin signaling in the brain. This lowers sensitivity to satiation signals including CCK (Emond et al., 2001; Matson et al., 2000). The result is that larger-than-normal meals are eaten. Conversely, when individuals acutely gain excess fat, leptin and insulin signaling in the CNS is elevated, enhancing sensitivity to satiation signals (Riedy et al., 1995; Williams et al., 2006). Thus, adiposity and satiation signals interact in a manner that facilitates the defense of stable adipose stores.
The obesity epidemic
Despite this robust homeostatic system, the world is in the midst of an obesity crisis. The National Health Examination Studies and the National Health and Nutrition Examination Studies, conducted regularly beginning in 1960, indicate that body weight of adult Americans was relatively stable between 1960 and 1980, with ~25% of the population classified as overweight or obese (Kuczmarski et al., 1994). Since then, this proportion has dramatically increased, and the Centers for Disease Control now estimates that more than 68% of adults in the United States are overweight or obese. These sobering statistics beg the question: If the central homeostatic system governing energy balance is so precise, why are populations becoming obese?
Although there is clearly a heritable component to an individual’s susceptibility to becoming overweight, the short time-frame in which our collective waistlines have expanded implies that a change in gene frequencies is unlikely to be the primary causative factor. Rather, the increased incidence of obesity is thought to reflect an interaction between our genotype and an increasingly obesogenic environment. Among the numerous possible environmental factors that may contribute to obesity, ubiquitous access to low-cost, calorically-dense foods, and especially those high in fat content, is considered among the most important.
Individuals fed a diet containing a high proportion of fat as calories tend to become obese. We and others have often referred to this high-fat diet-induced obesity (often referred to as DIO) as a dysregulation or failure of energy homeostasis. However, this characterization obscures the fact that obese individuals typically maintain a relatively stable body weight, and robustly defend their higher level of adiposity. Similar to lean animals, for example, DIO rats that are calorically restricted to produce a substantial weight loss, quickly rebound when returned to ad-libitum feeding, and fully recover their initial body weight within the same time frame as lean controls (Levin and Dunn-Meynell, 2000; Woods et al., 2003) (Fig. 1). This defense of excess body fat is also familiar to humans, as illustrated by the discouraging phenomenon of yo-yo dieting, in which weight-loss achieved by vigilant diet and/or exercise is regained when the vigilance is softened (Kramer et al., 1989; Leibel et al., 1995). Weight loss achieved by pharmacological interventions is also susceptible to weight regain upon removal of the intervention. The important point is that the integrity of the homeostatic system appears to be maintained in DIO (Morton et al., 2006), and may in fact contribute to the maintenance of obesity.
Figure 1. Homeostatic regulation of energy balance is maintained in diet-induced obesity.
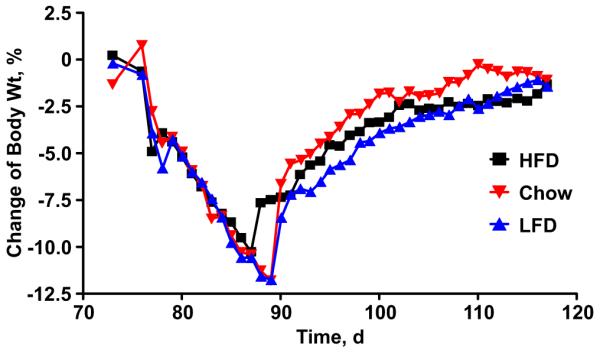
Both obese rats maintained on a high butter-fat diet (HFD, squares) and lean rats maintained on either low butter-fat diet (LFD) or standard chow (triangles), defend their body weight against acute perturbations. Rats were calorically restricted in order to produce a 12% weight loss. When returned to ad-lib feeding, all groups recovered to their initial body weight within a similar time-frame. Adapted from Woods, et al. 2003.
Consequently, DIO probably does not reflect a state of “dysregulated” energy balance. Rather, we suggest that the defended level of adiposity is a plastic phenotype that varies along a norm of reaction, within limits defined by genetic and/or epigenetic parameters, according to changing physiological and environmental conditions (e.g. Ghalambor et al., 2007). This concept is consistent with the observation that adipocytes themselves are also remarkably plastic in terms of the amount of stored fat they can accommodate (Sethi and Vidal-Puig, 2007), providing a buffer against acute changes in nutrient load. In a similar way, we suggest that plasticity in defended levels of adiposity can buffer against stochastic environmental conditions, including changes in the spatial or temporal availability of macronutrients. Given the historically transient availability of fat-rich food sources, individuals that defend higher levels of adiposity when faced with nutrient abundance should have greater fitness compared to conspecifics that exhibit less phenotypic plasticity (as in Neel, 1962). This would result in selection for pathways that couple the availability of ephemeral nutrients with hyperphagia and increased fat storage. Within this framework, it will be informative to ask how dietary fats might interact with central homeostatic circuits to shift the defended level of adiposity, as these mechanisms represent important nodes for therapeutic intervention. This is the topic of the current review.
Reduced sensitivity to the adiposity signals insulin and leptin
One mechanism by which an ecological abundance of dietary fats might increase the defended level of adiposity is by reducing responsivity to the adiposity hormones leptin and insulin. Consistent with this, obese individuals maintain higher plasma leptin levels in proportion to their greater adiposity, suggesting reduced responsivity to its catabolic action (Frederich et al., 1995). Moreover, obesity is associated with reduced sensitivity to the behavioral and cellular actions of exogenously-administered leptin. Administering leptin directly into the CNS of DIO rats (Scarpace et al., 2001; Widdowson et al., 1997) and mice (El-Haschimi et al., 2000) does not reduce food intake, and likewise fails to activate the JAK/STAT (El-Haschimi et al., 2000; Munzberg et al., 2004) and S6K (Cota, 2008) signaling pathways that are downstream of the leptin receptor and important for its catabolic action. Similarly, DIO animals maintain higher plasma insulin levels in proportion to their level of body fat (Bagdade et al., 1967), but exhibit a blunted anorectic response to the administration of exogenous insulin into the brain (Arase et al., 1988; Chavez et al., 1995; Clegg et al., 2005). Activation of PI3K downstream of the insulin receptor is blunted in DIO (De Souza et al., 2005; Posey et al., 2009). Reduced leptin and insulin signaling in obese individuals would limit the capacity to increase sensitivity to satiation signals, thereby allowing larger meal size and facilitating the defense of greater adiposity (Fig. 2) (Myers et al., 2010). Although we hereafter follow convention by using the term leptin/ insulin resistance to describe these blunted behavioral and/or cellular responses to adiposity signals, we interpret this as a regulated process, not the dysfunctional phenomenon the word “resistance” might imply. Specific mechanisms tying the availability of HFD to the development and maintenance of leptin/ insulin resistance represent important therapeutic targets, and have been the focus of intense research by various groups.
Figure 2. Diet-induced leptin and insulin resistance facilitate defense of greater adiposity.
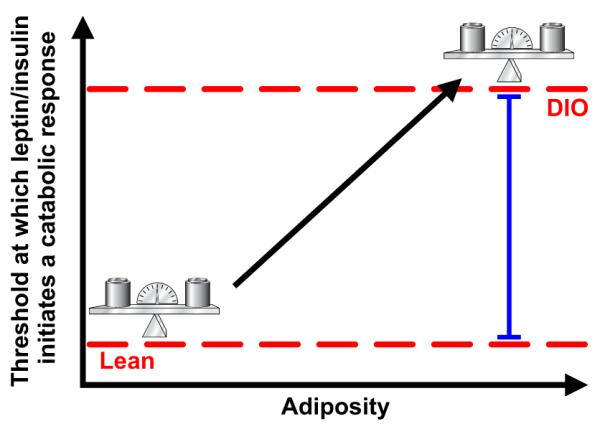
Plasma concentrations of the adiposity signals leptin and insulin increase in proportion to increased adiposity. However, the threshold plasma concentration at which CNS leptin or insulin action indicates appropriate fat storage (red dashed line) is increased, such that higher concentrations are needed to elicit an anorectic response, and greater adiposity is defended. Resistance to adiposity hormones can be conceptualized as the difference between these thresholds.
Molecular lipid sensors as modulators of leptin and insulin sensitivity
PKC-θ
The serine-threonine protein kinase C (PKC)- θ is a hypothalamic lipid-sensing protein thought to provide a mechanism linking dietary fats to the development of obesity. Members of the novel class of PKC isoforms, including PKC-θ, bind and are activated by intracellular diacylglycerols (DAGs), which accumulate when fatty acid delivery to the cell outpaces the rates of fat oxidation and triglyceride synthesis (Erion and Shulman, 2010), and this is associated with blunted insulin signaling in peripheral tissues (e.g. Griffin et al., 1999). PKC-θ is also expressed in the medial-basal hypothalamus (Benoit et al., 2009; Dewing et al., 2008), where accumulation of long-chain fatty acyl CoAs and DAGs is increased in rats chronically maintained on a diet high in saturated fats, rats receiving an acute gavage of saturated fats, and rats receiving infusions of palmitate directly into the brain (Benoit et al., 2009). In keeping with this, activation of hypothalamic PKC-θ is likewise increased in these treatments, and is associated with a blunted anorectic response to central insulin. Conversely, insulin sensitivity is restored to DIO rats by virally-mediated knockdown of PKC-θ in the medial-basal hypothalamus, implicating increased hypothalamic DAG accumulation and activation of PKC-θ in diet-induced CNS insulin resistance (Benoit et al., 2009).
Peroxisome-proliferator activated receptors (PPARs)
The peroxisome proliferator activated receptors (PPARs), another class of lipid sensors that may link maintenance on a HFD with obesity, are nuclear receptors activated by intracellular lipids to regulate gene expression. The PPARγ isoform, for example, is highly expressed in adipocytes, where it is a master regulator of adipogenesis (Patel et al., 2003; Rosen et al., 1999; Tontonoz et al., 1994) and lipogenesis (Lehrke and Lazar, 2005; Patel et al., 2003), thereby coupling the availability of fatty acids with increased fat synthesis and triglyceride storage. PPARγ is also expressed in key hypothalamic sites known to be important for the regulation of energy balance (Mouihate et al., 2004; Sarruf et al., 2008). We (Ryan et al., 2011) and others (Lu et al., 2011) have found that activation of CNS PPARγ, likely in the hypothalamus, leads to increased food intake and body fat. Reducing the activity of CNS PPARγ by local administration of a specific antagonist (Ryan et al., 2011), or with neuron- specific gene deletion (Lu et al., 2011), leads to acute and chronic reductions in HFD intake and blunts weight gain associated with HF-feeding. However, no differences are observed when rodents are maintained on a low-fat diet (Lu et al., 2011; Ryan et al., 2011; Sarruf et al., 2008), indicating that increased activation of CNS PPARγ under conditions of HF-feeding contributes to the development and maintenance of a higher body weight. CNS PPARγ also contributes to leptin resistance. Reducing the activity of CNS PPARγ in HFD-fed rats by central administration of its antagonist, or in mice by neuron-specific genetic knockdown of PPARγ, maintains the anorectic response to exogenous leptin in DIO animals (Ryan et al., 2011) and likewise accentuates leptin-induced STAT3 signaling (Lu et al., 2011). Together, these data indicate that activation of hypothalamic PPARγ by increased flux of fatty acids results in leptin resistance that facilitates the development and maintenance of DIO. However, important questions remain. With respect to PPARγ, the key hypothalamic target genes are unknown, and it is not clear which cellular and molecular mechanisms link the PPARγ and leptin signaling pathways. Moreover, other lipid-sensing PPAR isoforms are also expressed in hypothalamus where they may also modulate food intake and body weight (Chakravarthy et al., 2007; Moreno et al., 2004). Chakravarthy (2007) for example, reported that activation of hypothalamic PPARα, by endogenous products of fatty acid synthase, increases food intake. Less is known, however, about a potential role for PPARα in DIO. Future studies will be necessary to address these key questions.
The innate immune response
Recent attention has focused on the role of chronic, low-grade activation of the innate immune system in the pathogenesis of DIO and its associated co-morbidities. The toll-like receptor (TLR) 4, a type-I transmembrane receptor that recognizes lipopolysaccharide (LPS), a lipid derived from bacterial cell walls, is a key player in the innate immune response. Upon binding by LPS, TLR4 activates pathways either dependent or independent of the adaptor molecule, myeloid-differentiation primary-response gene 88 (MyD88). The MyD88-dependent pathway activates mitogen-activated protein kinases (MAPKs) and c-Jun N-terminal kinase (JNK), leading to activation of the transcription factor Activator protein-1 (AP-1). The MyD88- dependent pathway also activates inhibitor-of-κB kinase-β (IKKβ), leading to activation of the transcription factor nuclear-factor-κB (NF-κB). In this way, increased TLR4 activity leads to the increased production of several inflammatory cytokines, including interleukins -1β and -6 (IL-1β, IL-6) and tumor-necrosis factor-α (TNFα). The MyD88-independent pathway activates the transcription factor interferon-regulatory Factor 3 (IRF-3), increasing interferon-β (IFN- β) as well as other chemokines (Akira and Takeda, 2004; Kawai and Akira, 2010; Milanski et al., 2009; Selvarajoo, 2006). In response to infection, this coordinated response leads to fever and other appropriate physiological responses. Among these is reduced insulin sensitivity in muscle, liver and adipose tissue, associated with greater lipid and glucose availability to fuel energetically- demanding immune responses (Grimble, 2002; Wellen and Hotamisligil, 2005).
In addition to providing the non-specific response to pathogens, the innate immune system also exhibits low-level chronic activity associated with obesity and the metabolic syndrome. In both humans and rodents, plasma markers of inflammation are significantly associated with body mass index, serum triglycerides and systemic insulin resistance (Chan et al., 2002; Duncan and Schmidt, 2001; Pickup and Crook, 1998). Consistent with this, adipose tissue itself produces a wide range of pro-inflammatory cytokines and acute-phase reactants, including IL-1β, IL-6 and TNFα, contributing to increased systemic inflammation associated with increased adiposity in obese individuals (Hotamisligil, 2005; Rajala and Scherer, 2003; Uysal et al., 1997). It is now recognized that chronic low-grade systemic inflammation contributes to the increased risk of peripheral insulin resistance associated with obesity (Donath and Shoelson, 2011). One consequence has been attempts to use anti-inflammatory agents to treat type-2 diabetes mellitus (Fleischman et al., 2008; Goldfine et al., 2008; Larsen et al., 2007).
Consumption of a HFD also initiates a pro-inflammatory response in the hypothalamus. DeSouza (2005) evaluated the effect of consuming a HFD on hypothalamic gene expression. mRNAs for immune-related proteins were the largest group modulated by diet, and this was confirmed by increased protein expression of IL-1β, IL-6 and TNFα locally in the hypothalamus. Moreover, HF-fed rats had increased activation of two inflammatory pathways upstream of cytokine production, JNK and NF-κB. Consistent with other reports (Arase et al., 1988; Chavez et al., 1995; Clegg et al., 2005), HF-fed rats were resistant to the anorectic effects of central insulin. Importantly, this was reversed by the central administration of a JNK inhibitor, implicating hypothalamic inflammatory pathways in the pathogenesis of obesity and central insulin resistance (De Souza et al., 2005).
The mechanisms by which consumption of HFD leads to increased hypothalamic inflammation are not completely understood, but may rely on activation of TLR4. In addition to recognizing LPS, TLR4 can be bound by saturated fatty acids and invoke an immune response (Fessler et al., 2009, Lee et al., 2001; Shi et al., 2006; but see Erridge and Samani, 2009), positioning TLR4 as a physiological sensor of fatty acids. Activation of TLR4 by fatty acids has been implicated as a key mechanism linking DIO to increased plasma concentration of inflammatory markers and insulin resistance in peripheral tissues (Shi et al., 2006; Tsukumo et al., 2007).
Milanski (2009) and colleagues tested the hypothesis that hypothalamic TLR4 mediates hypothalamic inflammation in animals maintained on a HFD, resulting in leptin resistance. They observed that both HF-feeding and direct hypothalamic infusion of long-chain saturated fatty acids increased hypothalamic cytokine expression and reduced the anorectic response to leptin. Direct TLR4 loss-of-function inhibited HFD-induced inflammation and weight gain, and restored leptin sensitivity. Similarly, Kleinridders (2009) observed that specifically blocking MyD88- dependent TLR4 activity, by deleting MyD88 selectively in the CNS, leads to reduced HFD intake and DIO, and restores leptin sensitivity in DIO mice. Although MyD88- TLR4 inflammatory signaling can signal through both JNK/ AP1 and IKKβ/ NF-κB, the effect of CNS MyD88 to mediate HFD-induced obesity relies on IKKβ signaling, and JNK activity remained unaltered in these mice (Kleinridders et al., 2009).
Consistent with the possibility that hypothalamic inflammation contributes to the development of DIO, Zhang (2008) reported that activation of hypothalamic IKKβ/ NF-κB increases in obesity, both in leptin-deficient ob/ob mice fed a chow diet and in DIO animals. Moreover, acute infusions of glucose or oleic acid into the brain of fasted mice also increased hypothalamic IKKβ/ NF-κB activity, suggesting that an over-supply of nutrients locally in the brain can drive this inflammatory response. To specifically test the role of hypothalamic IKKβ/ NF-κB in DIO, the authors reduced IKKβ activity with virally-mediated or constitutive knockdown. Both manipulations reduced HFD intake and weight gain in DIO animals, whereas increasing hypothalamic NF-κB activity had the opposite effect. Moreover, increased hypothalamic NF-κB activity reduced the anorectic action of insulin and leptin, as well as activation of their downstream-signaling pathways, PI3K and STAT3 (Zhang et al., 2008). Providing a critical link, Posey (2009) found that HF-feeding increases the local abundance of saturated long-chain acyl- CoAs in the hypothalamus, thereby directly connecting the consumption of HFD with increased accumulation of intracellular lipid and activation of inflammatory pathways in the hypothalamus. HF-feeding was also associated with increased inflammatory signaling via IKKβ in this study, and it reduced the anorectic response to insulin. Hypothalamic insulin sensitivity was restored by intracerebroventricular administration of an IKK inhibitor. Importantly, the ability of dietary fat to induce this inflammatory pathway did not depend on increased total caloric intake, as rats pair-fed the HFD, to the amount of calories consumed by controls fed a low-fat diet, also had increased activation of hypothalamic IKKβ despite similar caloric intake (Posey et al., 2009).
One potential link between hypothalamic inflammation and leptin/ insulin resistance is the protein-tyrosine phosphatase-1B (PTP1B). PTP1B dephosphorylates the insulin- and leptin- associated Janus kinase (JAK), thereby acting as a negative regulator of both insulin and leptin signaling (Bourdeau et al., 2005; Dubé and Tremblay, 2005). PTP1B is expressed in hypothalamus, where it is increased in HF-fed animals (White et al., 2009; Zabolotny et al., 2008). Genetic knockdown of PTP1B in all neurons (Bence et al., 2006), or specifically in neurons expressing the anorexogenic neuropeptide precursor, proopiomelanocortin (POMC) (Banno et al., 2010), improves leptin sensitivity and protects against DIO. The mechanism by which PTP1B is increased by HFD is unclear, but may involve inflammatory signaling pathways. Hypothalamic PTP1B mRNA and protein are increased during acute TNFα injection. This may depend in part on activation of NF-κB downstream of TNFα, as PTP1B appears to be an NFκB target gene. Its promoter contains a putative NF-κB binding site, and consistent with this, treatment with TNFα increases the recruitment of the NFκB subunit p65 to the PTP1B promoter (Zabolotny et al., 2008). However, although central administration of TNFα induces leptin and insulin resistance, associated with activation of IKKβ, this is not blunted by knockdown of PTP1B expression with antisense oligonucleotides (Picardi et al., 2010). Consequently, it remains uncertain whether PTP1B is a critical mediator of HFD-induced leptin and/or insulin resistance downstream of NF-κB or other inflammatory signaling pathways.
Another potential molecular link between hypothalamic inflammation and leptin/ insulin resistance is provided by suppressor-of-cytokine signaling (SOCS) proteins. SOCS proteins were originally identified as anti-inflammatory, negative regulators of cytokine receptor signaling that inhibit the JAK-STAT pathway. Activation of STAT transcription factors downstream of cytokine receptors is a major source of SOCS expression, resulting in negative feedback that restrains initial pro-inflammatory responses (Howard and Flier, 2006; Myers et al., 2008; Thaler and Schwartz, 2010). The leptin receptor is a member of the class-I cytokine receptor family, which includes the IL-6 receptor and other cytokine receptors (Baumann et al., 1996; Tartaglia et al., 1995). Signaling through the leptin receptor drives increased SOCS-3 expression (Bjorbaek et al., 1999). In turn, SOCS-3 is a negative regulator of leptin signaling and is thought to contribute to HFD-induced leptin resistance. Mice deficient in SOCS-3 (Howard et al., 2004) or with brain-specific SOCS-3 deletion (Mori et al., 2004) are more sensitive to the anorectic effects of leptin and are resistant to DIO. Hypothalamic SOCS-3 is increased in DIO, likely the result of both increased signaling at cytokine receptors in response to low-grade inflammation, and increased leptin receptor signaling in response to rising leptin levels. Consistent with this, SOCS-3 was found to be a critical mediator of hypothalamic leptin/ insulin resistance downstream of the IKKβ/ NF-κB pathway (Zhang et al., 2008). However, the mechanism by which IKKβ was linked to SOCS-3 in that study is unclear, because no differences in cytokine expression were observed.
Open questions regarding the role of the innate immune response
Role of microglia
Despite a growing consensus regarding the important role of hypothalamic inflammation in the etiology of DIO, several open questions remain. First, it is not known which cell types are involved in the initiation of HFD-induced inflammatory responses in hypothalamus (Thaler and Schwartz, 2010). An important feature of inflamed tissues is macrophage infiltration. Obesity- associated macrophage infiltration of adipose tissue has been described in both humans and rodents and contributes to the emergence and maintenance of low-grade peripheral inflammation in obesity (Kanda et al., 2006; Weisberg et al., 2003; Xu et al., 2003). Perhaps analogously, microglial infiltration of the arcuate nucleus and median eminance, but not other brain regions, is reportedly increased in HFD-fed rats. Moreover the cellular distribution of TLR4 in hypothalamus occurs primarily on microglia (Milanski et al., 2009), suggesting that hypothalamic microglial cells may be key to initiating the innate-immune response to increased abundance of saturated fats. Therefore, although evidence suggests that IKKβ/ NF-κB in specific neuronal populations is critical to HFD-induced inflammation and participates in the etiology of HFD-induced leptin/ insulin resistance (Zhang et al., 2008), the role of microglia to initiate this neuronal inflammation remains unclear. Intriguingly, cultured hypothalamic neurons are resistant to inflammation induced by saturated fatty acids (Choi et al., 2010), supporting the possibility that communication between microglia and neurons is necessary for the initiation of HFD-induced hypothalamic inflammation.
Importance of various components of the inflammatory cascade
It also remains unclear which specific components of the inflammatory cascade are critical to the development of HFD-induced insulin/ leptin-resistance. The innate immune response to HFD includes increased levels of several cytokines and pro-inflammatory signaling pathways (De Souza et al., 2005), making it difficult to identify the relative importance of individual components of this response. As discussed above, initial studies reported that chronic maintenance on a HFD increases the hypothalamic expression of multiple pro- inflammatory cytokines including IL-1β, IL-6 and TNFα, and that both pro-inflammatory cytokine expression and DIO are blunted by loss-of-function of TLR4 or JNK, suggesting that cytokine signaling is a key link between inflammation and diet-induced leptin/ insulin resistance (De Souza et al., 2005; Milanski et al., 2009). In contrast, other studies observed no change (Kleinridders et al., 2009; Posey et al., 2009; Zhang et al., 2008) or variable increases (Oh-I et al., 2010) in cytokine expression with obesity, despite that activation of hypothalamic IKKβ was a critical mediator of DIO in those studies. These data therefore implicate non-cytokine signaling downstream of NF-κB. Intriguingly, ceramide biosynthesis, which is initiated downstream of IKKβ, is essential for TLR4-dependent insulin resistance in muscle (Holland et al., 2011). Moreover, following an acute intravenous infusion of lipids, hypothalamic ceramide concentration is increased in wild-type, but not in TLR4-defective mice, and this could be blocked with systemic administration of the IKK inhibitor, sodium salicylate (Holland et al., 2011). Collectively, these data suggest the hypothesis that hypothalamic ceramides might participate in IKKβ-dependent leptin/ insulin resistance in the hypothalamus. Additional studies are needed to directly test this possibility.
Paradoxical role of cytokine signaling in the regulation of energy balance
The role of pro-inflammatory cytokine signaling is counterintuitive because these same cytokines are also important central mediators of illness-induced anorexia (Asarian and Langhans, 2005; Laye et al., 2000; Plata-Salaman et al., 1996). It is not clear how a robust, acute, pro-inflammatory response to pathogens can elicit reductions in food intake and body weight, whereas the chronic low-grade pro-inflammatory response to fats elicits the opposite response. Thaler and Schwartz (2010) proposed that this might result from differences in the affected neuronal populations or in the affected cell types, where pathogen-provoked immune responses are initiated by microglia and HFD-induced immune responses are initiated directly in the neurons (or vice-versa). Another possibility is that HFD-induced obesity results from anti- inflammatory counter-regulatory responses, including SOCS-3, rather than reflecting the inflammation itself. In the case of pathogen-provoked inflammation, anorectic responses to robust cytokine signaling would result in sickness-induced anorexia that overwhelms any influence of counter-regulatory responses. On the other hand, in the case of diet-induced inflammation, although weak cytokine signaling may be insufficient to drive sickness-anorexia, chronic anti-inflammatory counter-regulatory responses would provoke hypothalamic leptin/ insulin resistance that leads to higher levels of defended body weight. In this way the scale and timeframe of the inflammatory response would predict the outcome with respect to adiposity.
Endoplasmic reticulum stress
The endoplasmic reticulum (ER) may have a key role as a nutrient sensor (Hotamisligil, 2010) mediating the interaction between abundance of dietary fat and the defense of a higher body weight. The ER forms a network of membranes and tubules for folding, storage, and transport of proteins, assisted by molecular chaperone proteins. Increased demand on the ER results in an imbalance between protein load and folding capacity, a condition known as ER stress. ER stress is sensed by the luminal domains of the transmembrane proteins Inositol- Requiring-1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) to activate a complex response known as the unfolded protein response (UPR) (Hotamisligil, 2006; Marciniak and Ron, 2006; Schroder and Kaufman, 2005). The UPR restores the match between ER folding capacity and needs (Bernales et al., 2006). First, the UPR reduces protein load in the ER by lowering protein synthesis and increasing clearance. Second, the increase in UPR target genes, including processing enzymes and molecular chaperones, results in an increased capacity of the ER to handle unfolded proteins. Finally, if homeostasis cannot be re- established by the first two responses, the cell undergoes apoptosis (Bernales et al., 2006; Ron and Walter, 2007). ER stress has been reported in the liver (Ozcan et al., 2004) and adipose tissue (Boden et al., 2008) of obese or diabetic individuals, and contributes to impaired glucose homeostasis in DIO (Ozcan et al., 2006).
Hypothalamic ER stress is increased in obesity and may contribute to insulin/ leptin resistance in DIO individuals. Activated PERK is increased in the hypothalamus of DIO (Ozcan et al., 2009; Zhang et al., 2008), but not diet-resistant (Ozcan et al., 2009) mice, and this could be blocked by central administration of the chemical chaperone, taurine-conjugated ursodeoxycholic acid (TUDCA) (Zhang et al., 2008). Moreover, acute treatment with the ER stress-inducing agent tunicamycin directly into the brain increases hypothalamic expression of the orexigenic neuropeptides agouti-related peptide (AgRP) and neuropeptide-Y (NPY), and reduces leptin-induced activation of STAT3 (Ozcan et al., 2009). To further investigate the role of ER stress in DIO, mice with a brain-specific knockout of X-box-binding-protein-1 (XBP1) were challenged with HFD. Because XBP1, which is activated by IRE1, is a master regulator of ER folding capacity (Ozcan et al., 2009; Ron and Walter, 2007), its deletion reduces ER function and thereby reduces the threshold for induction of the UPR. As anticipated, XNKO mice were more susceptible to DIO and diet-induced leptin resistance compared to control mice (Ozcan et al., 2009), suggesting that ER stress can act as a link between increased dietary fat consumption, resistance to leptin signaling, and eventually to the maintenance and defense of greater adiposity.
While these data suggest that the UPR may play a role to couple obesity and/or nutrient- induced ER stress with leptin resistance and obesity, important questions remain. First, it will be important to determine the mechanisms by which ER stress is increased in DIO. Increased activation of the UPR may be a consequence of obesity itself, occurring only after chronic HF- feeding and possibly contributing to the maintenance of obesity, or may result from increased availability of nutrients preceding weight gain, and thereby contributing to the onset of obesity. Second, it will be important to understand the downstream mechanisms by which ER stress contributes to leptin and insulin resistance. It appears that inflammatory signaling through IKKβ/ NF-κB may be both upstream (Ozcan et al., 2009) and downstream (Ozcan et al., 2009; Purkayastha et al., 2011; Zhang et al., 2008) of the UPR. Intracerebroventricular administration of TUDCA blunts HFD-induced increases in NF-κB activity, whereas knockdown of brain IKKβ blunts HFD-induced increases in UPR signaling (Ozcan et al., 2009). Although it is not clear whether ER stress is initially a consequence or cause of obesity-associated inflammation, these data suggest that obesity may create a situation whereby inflammation and ER stress cooperate in a feed-forward manner, resulting in the maintenance of CNS leptin/ insulin resistance in the presence of HFD.
It is important to note that physiological activation of the UPR to maintain homeostasis in the ER, as opposed to pathophysiological activation of the UPR leading to apoptosis and cell death, may be difficult to discriminate on the basis of gene expression and protein phosphorylation. In light of the available data, it remains unclear whether homeostatic or apoptotic UPR pathways (or both) are critical to the observed effects on leptin/ insulin resistance. Specifically, it will be important to determine whether ER-stress-invoked apoptosis occurs in key neuroanatomical substrates for leptin and insulin action and, if so, whether this contributes to the observed outcomes. Recent evidence suggests that palmitate induces ER stress and increases apoptosis in a neuronal cell model (Mayer and Belsham, 2010) and that DIO is associated with increased apoptosis and reduced synaptic plasticity in the arcuate and lateral hypothalamic nuclei (Moraes et al., 2009). However, the extent to which apoptosis results from ER stress in vivo remains undetermined.
Perspectives on hypothalamic leptin and insulin resistance in DIO animals
To date, most studies investigating mechanisms that link the consumption of HFD with the defense of greater body adiposity have focused on molecular aspects of hypothalamic leptin/ insulin resistance. However, the extent to which reduced responsivity to leptin and insulin is critical to the initiation versus the maintenance of HFD-induced obesity remains largely untested (Myers et al., 2010). When an individual is in a stable environment with abundant food, its homeostatic control system for body adiposity helps it maintain and defend a particular level of body fat as discussed above. If the environment is changed in such a way that the average fat consumption is increased (i.e., the individual is put on a HFD, whether voluntarily or not), the level of fat maintained is often increased and may lead to obesity. In this situation, the observed changes in leptin and insulin sensitivity may in fact be secondary, as opposed to causal, to the entire process. Nonetheless, they may also be necessary for the obesity to develop since experimental elimination of key mediators, including those discussed above, precludes the development of DIO.
Studies investigating mechanisms of diet-induced cellular hormone resistance typically use a DIO rodent model in which animals have been maintained on HFD for many weeks. We therefore know a good deal about how prolonged exposure to HFD, with its accompanying obesity, modulates cellular signaling pathways. In contrast, we know much less about the time course over which these changes develop and what the precipitating factors might be. In light of this, future studies that rigorously describe a time course for 1) the development of insulin and leptin resistance at the level of food intake and body weight responses, 2) changes in the response of receptor signaling pathways, and 3) the activation of lipid-sensing pathways in the hypothalamus, including inflammation and ER stress, will be informative and should be a priority for future research.
Hedonic attractiveness of fats
Despite recent focus on, and the clear importance of, central resistance to peripheral adiposity signals, this is unlikely to be the only contributor to defense of greater body adiposity in environments characterized by ubiquitous access to high-fat foods. Many factors can regulate food intake and adiposity, and the amount of body fat that is ultimately defended likely represents the integrated effect of diverse factors. Among these is the hedonic attractiveness of fat; i.e. its pleasurable properties beyond those derived from caloric content alone. In other words, one reason that we consume fatty foods is simply because we enjoy eating them! The pleasurable aspects of fat consumption derive from both unlearned attraction to orosensory factors and learned preferences based on postoral factors (Ackroff and Sclafani, 2010). Both humans (Drewnowski, 1997; Drewnowski et al., 1992) and rodents (Imaizumi et al., 2000; Takeda et al., 2000; Yoneda et al., 2007a; Yoneda et al., 2007b) have a hedonic preference for fat that increases with increasing fat concentration (Figlewicz and Benoit, 2009; Manabe et al., 2010). Moreover, some fats elicit a conditioned place preference in rodents (Figlewicz, 2004; Imaizumi et al., 2000), and act in a concentration-dependent manner as a reinforcer under a progressive ratio schedule operant task (Yoneda et al., 2007b), implying that at least some dietary fats have both rewarding and reinforcing properties.
The important question concerns the link between consuming the HFD and initiating the progression toward obesity and its associated co-morbidities. A corollary question is whether, when an individual is placed on a HFD, the ensuing obesity is secondary to modulation of the homeostatic control system, of the hedonic/reward system, or both. It is clear that hedonics plays a role, for when an animal is allowed to voluntarily ingest the HFD, and body weight becomes stable at an elevated level, homeostatic peptides in the hypothalamus are present at the same levels as those in stable animals fed a low-fat diet; i.e., in this condition the homeostatic system is not trying to reduce body weight. In contrast, if animals are force-fed the HFD by having it put directly into their stomachs each day, they can be made just as obese as individuals eating the food, but homeostatic neuropeptide levels indicate that the individual is too heavy; i.e., orexigenic peptides are decreased and anorexic peptides are increased relative to controls (Hagan et al., 1999; Seeley et al., 1996a). Further, once force-feeding ends, the animals eat little or no food and body weight declines. The difference seems to be whether the animals experience the hedonically pleasant aspects of the diet; i.e., whether they voluntarily taste, chew and swallow it. The important point is that although both involuntarily overfed and voluntarily overfed individuals presumably share degrees of dysfunction of lipid-sensing enzyme cascades, inflammatory cytokines in the plasma and CNS, ER folding problems, and so on (although this remains to be tested directly), one is actively attempting to lose weight while the other is content to remain in the obese state.
The animal freely consuming the HFD activates its hedonic/reward circuits with every bite. These include corticolimbic circuits that link the prefrontal cortex, the amygdala, the ventral tegmental area, the nucleus accumbens, and the ventral pallidum with the medial forebrain bundle. This network, via axonal circuits, connects the hindbrain and midbrain to key hypothalamic nuclei involved with homeostatic controls over energy balance (Cota, 2006; Figlewicz and Benoit, 2009; Saper et al., 2002). What is important is that there is considerable crosstalk between the homeostatic and non-homeostatic systems, mediated by in part by direct actions of insulin and leptin at their receptors within the corticolimbic system, as well as by secondary hypothalamic effector systems (Davis et al., 2011a; Davis et al., 2011b; Figlewicz, 2004; Figlewicz et al., 2001; Hommel et al., 2006). These interactions are congruent with a highly integrated system governing food intake. For example, when an animal is food-deprived, several changes occur. In the homeostatic circuits, the reduced levels of insulin and leptin act to blunt the action of satiation signals such that when food is encountered, the animal will eat larger meals. In the reward circuits, hedonically pleasing sensations of food become exaggerated (Hommel et al., 2006). In a word, food tastes better, and this results in more food being eaten, even in the face of inhibitory signals from the gut and adipose tissue.
When a HFD is suddenly made available to an individual, sensory properties of the food (e.g., odor, taste, mouth feel) are intrinsically pleasant and lead to more food being consumed. As this continues, excess calories accumulate as increased adiposity, and increased circulating fatty acids lead to increased lipid flux into the brain, insulin/ leptin resistance, an inflammatory condition, and so on. Stated another way, the increased palatability of the diet initiates a vicious cycle in which hedonics cause more food to be eaten than is necessary to meet energy needs, and the increased calories in turn initiate events that lead to insulin/ leptin resistance and a consequent tendency to eat even more food (Fig. 4). In this context, it is interesting to consider how cellular leptin/ insulin resistance in key reward pathways (Matheny et al., 2011) might modulate the hedonic response to foods. In the same way that reduced leptin and insulin signaling in the fasted animal exaggerates the hedonic pleasing sensations of food, so too might blunted leptin and insulin signaling in DIO exaggerate the pleasurable aspects of eating, increase the amount of food eaten, and thereby contribute to this viscous cycle.
Figure 4. Vicious cycle facilitating defense of greater adiposity with high-fat feeding.
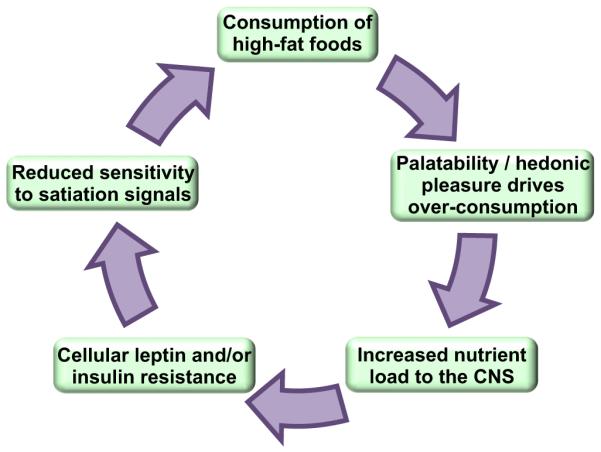
Consumption of high-fat foods acts on central reward pathways to elicit further intake beyond what is necessary to maintain homeostasis. This facilitates increased nutrient flux to central 27-Ryan et al. homeostatic circuits, which leads to cellular resistance to leptin and insulin. Blunted leptin and insulin signaling results in reduced sensitivity to satiation signals, and even more food is consumed. In this way, a feed-forward cycle is created, leading to hyperphagia and obesity. It is critical to understand the mechanisms by which dietary fats and/or their metabolites interact with central homeostatic and reward circuitry, as these represents key targets for therapeutic intervention.
Therapeutic implications
Despite great progress to elucidate central homeostatic pathways regulating adiposity, and despite hopeful perspectives that accompany nearly every new finding, safe and effective therapies for the treatment of obesity remain elusive. Most individuals eventually regain adipose tissue that is lost by caloric restriction, likely because the integrity of the homeostatic system is maintained in obesity and thereby drives behavioral and physiological responses to return adipose stores to the pre-restricted (higher) steady-state. Evidence suggests that consumption of hedonically pleasing, calorically-dense foods, and particularly those high in fat content plays a role to mediate this upward shift in the defended level of adiposity. Consequently, therapeutic interventions are needed that target the lipid-sensing and hedonic mechanisms facilitating this interaction between the nutritional environment and central homeostatic circuits. In this way, therapies may be developed that maintain the defense of a healthy level of adiposity in the face of an obesogenic environment. Furthermore, the most successful therapies are likely to be those that target these interactions at multiple nodes to over-ride the redundancies in the system.
The most effective currently-available therapy for obesity is bariatric surgery. In contrast to individuals that lose a significant percentage of body fat by caloric restriction only to regain the weight when the restriction is relieved, individuals that lose weight with bariatric surgery maintain this lesser adiposity for many years. This is associated with remarkable improvements in glucose control and reversal of type-2 diabetes and other co-morbidities (Sjostrom et al., 2004). Thus it appears that bariatric surgery may shift the defended level of adiposity, even in the face of a modern environment characterized by ubiquitous access to low-cost, HF-foods. Despite its efficacy, however, bariatric surgery is not a practical solution for the large numbers of individuals now facing obesity and its consequences. Considerable effort is underway to understand the mechanisms responsible for changes in defended adiposity following these procedures, in order to target affected pathways in a less-invasive manner.
Several weight-loss surgeries are currently performed, including Roux-en-y gastric bypass (RYGB), vertical sleeve gastrectomy (VSG), gastric banding (GB), and ileal transposition, and these encompass varying combinations of gastric restriction and re-routing of the flow of nutrients through the gut. Contrary to the widely held belief that a small stomach drives the reduced food intake, evidence suggests that the therapeutic benefit of bariatric surgeries may not depend on the restrictive component of these procedures (Melissas et al., 2007; Stefater et al., 2010). For example, rats that have recovered from VSG and were subsequently calorically-deprived to produce approximately 30% weight loss can and do overeat and recover their defended levels of adiposity within the same time frame as sham- operated controls. This outcome indicates that VSG rats could consume more calories but choose not to (Stefater et al., 2010b), and that their post-operative food intake reflects a new level of defended adiposity.
Consequently it will be critical to understand how bariatric surgeries modulate the interactions among HFD, leptin/ insulin sensitivity, the various lipid-sensing pathways described above, and the hedonic response to eating. Recent data from animal models indicate that changes in leptin sensitivity may not be a critical factor leading to weight loss following surgery. In one study (Stefater et al., 2010) VSG did not improve leptin sensitivity of DIO rats beyond that achieved by pair-fed controls, and moreover VSG, RYGB, and GB are all effective in obese Zucker rats, which lack a functional leptin receptor (Lopez et al., 2009; Xu et al., 2002). On the other hand, the importance of CNS insulin sensitivity in these models has not yet been tested, and induction of hypothalamic inflammation or other CNS lipid-sensing pathways is unknown. Various satiation signals are increased in both humans and rodents following RYGB or VSG (Chambers et al., 2011; Chelikani et al., 2010; Harvey, 2010; Peterli et al., 2009; Shin et al., 2010b). The importance of these changes to facilitate observed metabolic improvements will require further investigation, and would be definitively addressed by testing the effects of surgery in various mouse knockout models. Several studies indicate that the hedonic experience of food is altered in both humans, who report a reduced drive to eat sweet and/or fatty foods following bariatric surgery (Olbers et al., 2006; Thirlby et al., 2006), and rats, which shift their preference from higher to lower concentrations of oil or sucrose (Shin et al., 2010a). Thus it seems that bariatric surgeries can interfere with the relationship between physiology and the modern nutritional environment in several ways, providing a permanent combination therapy that leads to the defense of lesser adiposity.
The key point here is that successful therapeutic intervention for the large population facing the social, financial and health consequences of obesity will depend on understanding both 1) how consumption of calorically dense high-fat diets increases the defended level of adiposity and 2) how effective therapies manage to disrupt CNS mechanisms underlying this change in defended body weight, including HFD-induced leptin/ insulin resistance. Such an endeavor will ultimately require a systems approach involving a variety of disparate disciplines and expertise.
Figure 3. Nutrient-sensing mechanisms contributing to diet-induced leptin and insulin resistance.

Excess lipid flux into the central nervous system leads to activation of nutrient- sensing pathways in the hypothalamus including: PKCθ, PPARγ, ER stress, and inflammatory mediators. Activation of these pathways results in blunted cellular responses to circulating leptin and insulin, including changes in transcriptional activity and/or neuronal firing. Consequently, higher plasma concentrations of leptin and insulin are needed to induce downstream effectors. Body fat stores are increased until this new threshold is reached.
Acknowledgements
We thank Glenn Doerman for drawing the figures. The authors were supported by the National Institute of Diabetes and Digestive and Kidney Diseases (F32DK082173 to K.K.R., R01DK17844 to S.C.W., R01DK07305 to R.J.S.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackroff K, Sclafani A. Oral and postoral determinants of dietary fat appetite. In: JP M, J l.C., editors. Fat Detection: Taste, Texture, and Post Ingestive Effects. CRC Press; Boca Raton: 2010. Chapter 12. [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Arase K, Fisler JS, Shargill NS, York DA, Bray GA. Intracerebroventricular infusions of 3-OHB and insulin in a rat model of dietary obesity. Am J Physiol. 1988;255:R974–R981. doi: 10.1152/ajpregu.1988.255.6.R974. [DOI] [PubMed] [Google Scholar]
- Asarian L, Langhans W. Current perspectives on behavioural and cellular mechanisms of illness anorexia. Int Rev Psychiatry. 2005;17:451–459. doi: 10.1080/02646830500381450. [DOI] [PubMed] [Google Scholar]
- Bagdade JD, Bierman EL, Porte D., Jr The significance of basal insulin levels in the evaluation of the insulin response to glucose in diabetic and nondiabetic subjects. J Clin Invest. 1967;46:1549–1557. doi: 10.1172/JCI105646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, Bence KK. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. The J Clin Invest. 2010;120:720–734. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim H, Lai CF, Tartaglia LA. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proceedings of the National Academy of Science, USA. 1996;93:8374–8378. doi: 10.1073/pnas.93.16.8374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, Lefevre A-L, Cruciani-Guglielmacci C.l., Magnan C, Yu F, et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. The Journal of Clinical Investigation. 2009;119:2577–2589. doi: 10.1172/JCI36714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S.n., Papa FR, Walter P. Intracellular Signaling by the Unfolded Protein Response. Annual Review of Cell and Developmental Biology. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in Endoplasmic Reticulum Stress Related Proteins and Genes in Adipose Tissue of Obese, Insulin-Resistant Individuals. Diabetes. 2008;57:2438–2444. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau A, Dubé N, Tremblay ML. Cytoplasmic protein tyrosine phosphatases, regulation and function: the roles of PTP1B and TC-PTP. Current Opinion in Cell Biology. 2005;17:203–209. doi: 10.1016/j.ceb.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Brief DJ, Davis JD. Reduction of food intake and body weight by chronic intraventricular insulin infusion. Brain Res Bull. 1984;12:571–575. doi: 10.1016/0361-9230(84)90174-6. [DOI] [PubMed] [Google Scholar]
- Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- Chakravarthy MV, Zhu Y, Lopez M, Yin L, Wozniak DF, Coleman T, Hu Z, Wolfgang M, Vidal-Puig A, Lane MD, Semenkovich CF. Brain fatty acid synthase activates PPARα to maintain energy homeostasis. The Journal of Clinical Investigation. 2007;117:2539–2552. doi: 10.1172/JCI31183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AP, Jessen L, Ryan KK, Sisley S, Wilson-Perez HE, Stefater MA, Gaitonde S, Sorrell JE, Toure M, Berger J, et al. Weight-independent changes in blood glucose homeostasis after roux-en-Y gastric bypass and vertical sleeve gastrectomy surgeries in rats. Gastroenterology. 2011;141:950–958. doi: 10.1053/j.gastro.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JCN, Cheung JCK, Stehouwer CDA, Emeis JJ, Tong PCY, Ko GTC, Yudkin JS. The central roles of obesity-associated dyslipidaemia, endothelial activation and cytokines in the Metabolic Syndrome: an analysis by structural equation modeling. Int J Obes Relat Metab Disord. 2002;26:994–1008. doi: 10.1038/sj.ijo.0802017. [DOI] [PubMed] [Google Scholar]
- Chavez M, Kaiyala K, Madden LJ, Schwartz MW, Woods SC. Intraventricular insulin and the level of maintained body weight in rats. Behavioral Neuroscience. 1995;109:528–531. doi: 10.1037//0735-7044.109.3.528. [DOI] [PubMed] [Google Scholar]
- Chelikani P, Shah I, Taqi E, Sigalet D, Koopmans H. Comparison of the Effects of Roux-en-Y Gastric Bypass and Ileal Transposition Surgeries on Food Intake, Body Weight, and Circulating Peptide YY Concentrations in Rats. Obesity Surgery. 2010;20:1281–1288. doi: 10.1007/s11695-010-0139-6. [DOI] [PubMed] [Google Scholar]
- Choi SJ, Kim F, Schwartz MW, Wisse BE. Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. American Journal of Physiology - Endocrinology and Metabolism. 2010;298:E1122–E1130. doi: 10.1152/ajpendo.00006.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg DJ, Benoit SC, Reed JA, Woods SC, Dunn-Meynell A, Levin BE. Reduced anorexic effects of insulin in obesity-prone rats fed a moderate-fat diet. Am J Physiol Regul Integr Comp Physiol. 2005;288:R981–986. doi: 10.1152/ajpregu.00675.2004. [DOI] [PubMed] [Google Scholar]
- Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. Journal of Clinical Investigation. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Barrera JG, Seeley RJ. Leptin in energy balance and reward: two faces of the same coin? Neuron. 2006;51:678–680. doi: 10.1016/j.neuron.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Cota D, Matter EK, Woods SC, Seeley RJ. The role of hypothalamic mTORC1 and diet-induced obesity. Journal of Neuroscience. 2008;28:1389–1408. doi: 10.1523/JNEUROSCI.1389-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JF, Choi DL, Schurdak JD, Fitzgerald MF, Clegg DJ, Lipton JW, Figlewicz DP, Benoit SC. Leptin Regulates Energy Balance and Motivation Through Action at Distinct Neural Circuits. Biological Psychiatry. 2011a;69:668–674. doi: 10.1016/j.biopsych.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JF, Choi DL, Shurdak JD, Krause EG, Fitzgerald MF, Lipton JW, Sakai RR, Benoit SC. Central melanocortins modulate mesocorticolimbic activity and food seeking behavior in the rat. Physiology & Behavior. 2011b;102:491–495. doi: 10.1016/j.physbeh.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- Dewing P, Christensen A, Bondar G, Micevych P. Protein Kinase C Signaling in the Hypothalamic Arcuate Nucleus Regulates Sexual Receptivity in Female Rats. Endocrinology. 2008;149:5934–5942. doi: 10.1210/en.2008-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Drewnowski A. Why do we like fat? Journal of the American Dietetic Association. 1997;97(Suppl 7):S58–S62. doi: 10.1016/s0002-8223(97)00732-3. [DOI] [PubMed] [Google Scholar]
- Drewnowski A, Kurth C, Holden-Wiltse J, Saari J. Food preferences in human obesity: Carbohydrates versus fats. Appetite. 1992;18:207–221. doi: 10.1016/0195-6663(92)90198-f. [DOI] [PubMed] [Google Scholar]
- Dubé N, Tremblay ML. Involvement of the small protein tyrosine phosphatases TC-PTP and PTP1B in signal transduction and diseases: From diabetes, obesity to cell cycle, and cancer. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2005;1754:108–117. doi: 10.1016/j.bbapap.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Duncan BB, Schmidt MIS. Chronic activation of the innate immune system may underlie the metabolic syndrome. Sao Paulo Medical Journal. 2001;119:122–127. doi: 10.1590/S1516-31802001000300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. Journal of Clinical Investigation. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emond M, Ladenheim EE, Schwartz GJ, Moran TH. Leptin amplifies the feeding inhibition and neural activation arising from a gastric nutrient preload. Physiology and Behavior. 2001;72:123–128. doi: 10.1016/s0031-9384(00)00393-0. [DOI] [PubMed] [Google Scholar]
- Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Samani NJ. Saturated Fatty Acids Do Not Directly Stimulate Toll-Like Receptor Signaling. Arterioscler Thromb Vasc Biol. 2009;29:1944–1949. doi: 10.1161/ATVBAHA.109.194050. [DOI] [PubMed] [Google Scholar]
- Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Current Opinion in Lipidology. 2009;20:379–385. doi: 10.1097/MOL.0b013e32832fa5c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP. Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behavioral Neuroscience. 2004;118:479–487. doi: 10.1037/0735-7044.118.3.479. [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2009;296:R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ. Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav. 2001;73:229–234. doi: 10.1016/s0031-9384(01)00486-3. [DOI] [PubMed] [Google Scholar]
- Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate Improves Glycemia and Inflammatory Parameters in Obese Young Adults. Diabetes Care. 2008;31:289–294. doi: 10.2337/dc07-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nature Medicine. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Machidori H, Iwakiri R, Yamamoto K, Fujisaki J, Sakata T, Tso P. Effect of intravenous administration of apolipoprotein A-IV on patterns of feeding, drinking and ambulatory activity in rats. Brain research. 1993;608:233–237. doi: 10.1016/0006-8993(93)91463-3. [DOI] [PubMed] [Google Scholar]
- Ghalambor CK, McKay JK, Carroll SP, Reznick DN. Adaptive versus non-adaptive phenotypic plasticity and the potential for contemporary adaptation in new environments. Functional Ecology. 2007;21:394–407. [Google Scholar]
- Gibbs J, Smith GP. Cholecystokinin and satiety in rats and rhesus monkeys. American Journal of Clinical Nutrition. 1977;30:758–761. doi: 10.1093/ajcn/30.5.758. [DOI] [PubMed] [Google Scholar]
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. Use of Salsalate to Target Inflammation in the Treatment of Insulin Resistance and Type 2 Diabetes. Clinical and Translational Science. 2008;1:36–43. doi: 10.1111/j.1752-8062.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- Grimble RF. Inflammatory status and insulin resistance. Current opinion in Clinical Nutrition and Metabolic Care. 2002;5:551–559. doi: 10.1097/00075197-200209000-00015. [DOI] [PubMed] [Google Scholar]
- Hagan M, Rushing P, Schwartz M, Yagaloff K, Burn P, Woods S, Seeley R. Role of the CNS melanocortin system in the response to overfeeding. Journal of Neuroscience. 1999;19:2362–2367. doi: 10.1523/JNEUROSCI.19-06-02362.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffel M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Harvey EJ. Hormone changes affecting energy homeostasis after metabolic surgery. The Mount Sinai journal of medicine. 2010;77:446–465. doi: 10.1002/msj.20203. [DOI] [PubMed] [Google Scholar]
- Hewson G, Leighton GE, Hill RG, Hughes J. The cholecystokinin receptor antagonist L364,718 increases food intake in the rat by attenuation of endogenous cholecystokinin. British Journal of Pharmacology. 1988;93:79–84. doi: 10.1111/j.1476-5381.1988.tb11407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang L-P, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. The Journal of Clinical Investigation. 2011;121:1858–1870. doi: 10.1172/JCI43378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Role of Endoplasmic Reticulum Stress and c-Jun NH2-Terminal Kinase Pathways in Inflammation and Origin of Obesity and Diabetes. Diabetes. 2005;54:S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends in Endocrinology & Metabolism. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Imaizumi M, Takeda M, Fushiki T. Effects of oil intake in the conditioned place preference test in mice. Brain Res. 2000;870:150–156. doi: 10.1016/s0006-8993(00)02416-1. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K.-i., Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Brüning JC. MyD88 Signaling in the CNS Is Required for Development of Fatty Acid-Induced Leptin Resistance and Diet-Induced Obesity. Cell Metabolism. 2009;10:249–259. doi: 10.1016/j.cmet.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer FM, Jeffery RW, Forster JL, Snell MK. Long-term follow-up of behavioral treatment for obesity: Patterns of weight regain among men and women. International Journal of Obesity. 1989;13:123–136. [PubMed] [Google Scholar]
- Kuczmarski R, Flegal K, Campbell S, Johnson C. Increasing prevalence of overweight among US adults. The national health and nutrition examination surveys, 1960 to 1991. JAMA. 1994;272:205–211. doi: 10.1001/jama.272.3.205. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, VÃ,lund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. New England Journal of Medicine. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- Laye S, Gheusi G, Cremona S, Combe C, Kelley K, Dantzer R, Parnet P. Endogenous brain IL-1 mediates LPS-induced anorexia and hypothalamic cytokine expression. Am J Physiol Regul Integr Comp Physiol. 2000;279:R93–98. doi: 10.1152/ajpregu.2000.279.1.R93. [DOI] [PubMed] [Google Scholar]
- Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4. Journal of Biological Chemistry. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- Lehrke M, Lazar MA. The Many Faces of PPARγ. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332:621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA. Defense of body weight against chronic caloric restriction in obesity-prone and -resistant rats. Am J Physiol Regul Integr Comp Physiol. 2000;278:R231–237. doi: 10.1152/ajpregu.2000.278.1.R231. [DOI] [PubMed] [Google Scholar]
- Levin BE, Keesey RE. Defense of differing body weight set points in diet-induced obese and resistant rats. American Journal of Physiology. 1998;274:R412–R419. doi: 10.1152/ajpregu.1998.274.2.R412. [DOI] [PubMed] [Google Scholar]
- Lopez PP, Nicholson SE, Burkhardt GE, Johnson RA, Johnson FK. Development of a Sleeve Gastrectomy Weight Loss Model in Obese Zucker Rats. Journal of Surgical Research. 2009;157:243. doi: 10.1016/j.jss.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Sarruf DA, Talukdar S, Sharma S, Li P, Bandyopadhyay G, Nalbandian S, Fan W, Gayen JR, Mahata SK, et al. Brain PPAR-γ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat Med. 2011;17:618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe Y, Shigenobu M, Fushiki T. Preference for high fat foods in animals. In: Montmayeur JP l.C.J., editor. Fat Detection: Taste, Texture, and Post Ingestive Effects. CRC Press; Boca Raton: 2010. Chapter 10. [PubMed] [Google Scholar]
- Marciniak SJ, Ron D. Endoplasmic Reticulum Stress Signaling in Disease. Physiological Reviews. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- Matheny M, Shapiro A, Tümer N, Scarpace PJ. Region-specific diet-induced and leptin-induced cellular leptin resistance includes the ventral tegmental area in rats. Neuropharmacology. 2011;60:480–487. doi: 10.1016/j.neuropharm.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson CA, Reid DF, Cannon TA, Ritter RC. Cholecystokinin and leptin act synergistically to reduce body weight. American Journal of Physiology. 2000;278:R882–R890. doi: 10.1152/ajpregu.2000.278.4.R882. [DOI] [PubMed] [Google Scholar]
- Mayer CM, Belsham DD. Palmitate Attenuates Insulin Signaling and Induces Endoplasmic Reticulum Stress and Apoptosis in Hypothalamic Neurons: Rescue of Resistance and Apoptosis through Adenosine 5′ Monophosphate-Activated Protein Kinase Activation. Endocrinology. 2010;151:576–585. doi: 10.1210/en.2009-1122. [DOI] [PubMed] [Google Scholar]
- Melissas J, Koukouraki S, Askoxylakis J, Stathaki M, Daskalakis M, Perisinakis K, Karkavitsas N. Sleeve gastrectomy: a restrictive procedure? Obes Surg. 2007;17:57–62. doi: 10.1007/s11695-007-9006-5. [DOI] [PubMed] [Google Scholar]
- Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DML, Anhe G, Amaral ME, Takahashi HK, et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. The Journal of Neuroscience. 2009;29:359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, Romanatto T, Carvalheira JB, Oliveira ALR, Saad MJ, Velloso LA. High-Fat Diet Induces Apoptosis of Hypothalamic Neurons. PLoS ONE. 2009;4:e5045. doi: 10.1371/journal.pone.0005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran TH. Gut peptides in the control of food intake: 30 years of ideas. Physiol Behav. 2004;82:175–180. doi: 10.1016/j.physbeh.2004.04.048. [DOI] [PubMed] [Google Scholar]
- Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid x receptors in the adult rat CNS. Neuroscience. 2004;123:131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Mouihate A, Boisse L, Pittman QJ. A Novel Antipyretic Action of 15-Deoxy-Δ12,14-Prostaglandin J2 in the Rat Brain. J. Neurosci. 2004;24:1312–1318. doi: 10.1523/JNEUROSCI.3145-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Munzberg H. Mechanisms of Leptin Action and Leptin Resistance. Annual Review of Physiology. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr., Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress?”. American Journal Human Genetics. 1962;14:353–362. [PMC free article] [PubMed] [Google Scholar]
- Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. 2002;5:566–572. doi: 10.1038/nn0602-861. [DOI] [PubMed] [Google Scholar]
- Oh-I S, Thaler JP, Ogimoto K, Wisse BE, Morton GJ, Schwartz MW. Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. American Journal of Physiology - Endocrinology and Metabolism. 2010;299:E47–E53. doi: 10.1152/ajpendo.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olbers T, Bjorkman S, Lindroos A, Maleckas A, Lonn L, Sjostrom L, Lonroth H. Body composition, dietary intake, and energy expenditure after laparoscopic Roux-en-Y gastric bypass and laparoscopic vertical banded gastroplasty: a randomized clinical trial. Ann Surg. 2006;244:715–722. doi: 10.1097/01.sla.0000218085.25902.f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic Reticulum Stress Plays a Central Role in Development of Leptin Resistance. Cell Metabolism. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee A-H, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NG, Holder JC, Smith SA, Kumar S, Eggo MC. Differential Regulation of Lipogenesis and Leptin Production by Independent Signaling Pathways and Rosiglitazone During Human Adipocyte Differentiation. Diabetes. 2003;52:43–50. doi: 10.2337/diabetes.52.1.43. [DOI] [PubMed] [Google Scholar]
- Peterli R, Wolnerhanssen B, Peters T, Devaux N, Kern B, Christoffel-Courtin C, Drewe J, von Flue M, Beglinger C. Improvement in glucose metabolism after bariatric surgery: comparison of laparoscopic Roux-en-Y gastric bypass and laparoscopic sleeve gastrectomy: a prospective randomized trial. Ann Surg. 2009;250:234–241. doi: 10.1097/SLA.0b013e3181ae32e3. [DOI] [PubMed] [Google Scholar]
- Picardi PK, Caricilli AM, Abreu L.L.F.d., Carvalheira JBC, Velloso LA, Saad MJA. Modulation of hypothalamic PTP1B in the TNF-α-induced insulin and leptin resistance. FEBS Letters. 2010;584:3179–3184. doi: 10.1016/j.febslet.2010.05.064. [DOI] [PubMed] [Google Scholar]
- Pickup JC, Crook MA. Is Type II diabetes mellitus a disease of the innate immune system? Diabetologia. 1998;41:1241–1248. doi: 10.1007/s001250051058. [DOI] [PubMed] [Google Scholar]
- Plata-Salaman CR, Sonti G, Borkoski JP, Wilson CD, French-Mullen JMH. Anorexia induced by chronic central administration of cytokines at estimated pathophysiological concentrations. Physiology and Behavior. 1996;60:867–875. [PubMed] [Google Scholar]
- Polonsky KS, Given BD, Hirsch L, Shapiro ET, Tillil H, Beebe C, Galloway JA, Frank BH, Karrison T, Van-Cauter E. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest. 1988;81:435–441. doi: 10.1172/JCI113338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. American Journal of Physiology - Endocrinology and Metabolism. 2009;296:E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proceedings of the National Academy of Sciences. 2011;108:2939–2944. doi: 10.1073/pnas.1006875108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- Riedy CA, Chavez M, Figlewicz DP, Woods SC. Central insulin enhances sensitivity to cholecystokinin. Physiology & Behavior. 1995;58:755–760. doi: 10.1016/0031-9384(95)00108-u. [DOI] [PubMed] [Google Scholar]
- Roberts SB, Young VR, Fuss P, Fiatarone MA, Richard B, Rasmussen H, Wagner D, Joseph L, Holehouse E, Evans WJ. Energy expenditure and subsequent nutrient intakes in overfed young men. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 1990;259:R461–R469. doi: 10.1152/ajpregu.1990.259.3.R461. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPARγ Is Required for the Differentiation of Adipose Tissue In Vivo and In Vitro. Molecular Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- Ruttimann EB, Arnold M, Hillebrand JJ, Geary N, Langhans W. Intrameal hepaticportal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology. 2009;150:1174–1181. doi: 10.1210/en.2008-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-γ in the regulation of energy balance. Nat Med. 2011;17:623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK. The Need to Feed: Homeostatic and Hedonic Control of Eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, Schwartz MW. Expression of PPARγ in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology. 2008;150:707–712. doi: 10.1210/en.2008-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpace PJ, Matheny M, Tumer N. Hypothalamic leptin resistance is associated with impaired leptin signal transduction in aged obese rats. Neuroscience. 2001;104:1111–1117. doi: 10.1016/s0306-4522(01)00142-7. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual Review of Biochemistry. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Peskind E, Raskind M, Nicolson M, Moore J, Morawiecki A, Boyko EJ, Porte DJ. Cerebrospinal fluid leptin levels: Relationship to plasma levels and to adiposity in humans. Nature Medicine. 1996;2:589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, Matson CA, Chavez M, Woods SC, Schwartz MW. Behavioral, endocrine and hypothalamic responses to involuntary overfeeding. American Journal of Physiology. 1996a;271:R819–R823. doi: 10.1152/ajpregu.1996.271.3.R819. [DOI] [PubMed] [Google Scholar]
- Seeley RJ, van Dijk G, Campfield LA, Smith FJ, Burn P, Nelligan JA, Bell SM, Baskin DG, Woods SC, Schwartz MW. Intraventricular leptin reduces food intake and body weight of lean rats but not obese Zucker rats. Horm Metab Res. 1996b;28:664–668. doi: 10.1055/s-2007-979874. [DOI] [PubMed] [Google Scholar]
- Selvarajoo K. Discovering differential activation machinery of the Toll-like receptor 4 signaling pathways in MyD88 knockouts. FEBS Letters. 2006;580:1457–1464. doi: 10.1016/j.febslet.2006.01.046. [DOI] [PubMed] [Google Scholar]
- Sethi JK, Vidal-Puig AJ. Thematic review series: Adipocyte Biology. Adipose tissue function and plasticity orchestrate nutritional adaptation. Journal of Lipid Research. 2007;48:1253–1262. doi: 10.1194/jlr.R700005-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AC, Zheng H, Pistell PJ, Berthoud HR. Roux-en-Y gastric bypass surgery changes food reward in rats. Int J Obes. 2010a;35:642–651. doi: 10.1038/ijo.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AC, Zheng H, Townsend RL, Sigalet DL, Berthoud HR. Meal-induced hormone responses in a rat model of Roux-en-Y gastric bypass surgery. Endocrinology. 2010b;151:1588–1597. doi: 10.1210/en.2009-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims EAH. Experimental obesity, diet-induced thermogenesis and their clinical implications. Clin Endocrinol Metab. 1976;5:377–395. doi: 10.1016/s0300-595x(76)80027-8. [DOI] [PubMed] [Google Scholar]
- Sjostrom L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, Dahlgren S, Larsson B, Narbro K, Sjostrom CD, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. 2004;351:2683–2693. doi: 10.1056/NEJMoa035622. [DOI] [PubMed] [Google Scholar]
- Stefater MA, Perez-Tilve D, Chambers AP, Wilson-Perez HE, Sandoval DA, Berger J, Toure M, Tschop M, Woods SC, Seeley RJ. Sleeve Gastrectomy Induces Loss of Weight and Fat Mass in Obese Rats, but Does Not Affect Leptin Sensitivity. Gastroenterology. 2010;138:2426–2436. doi: 10.1053/j.gastro.2010.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Imaizumi M, Fushiki T. Preference for vegetable oils in the two-bottle choice test in mice. Life Sciences. 2000;67:197–204. doi: 10.1016/s0024-3205(00)00614-7. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Thaler JP, Schwartz MW. Minireview: Inflammation and Obesity Pathogenesis: The Hypothalamus Heats Up. Endocrinology. 2010;151:4109–4115. doi: 10.1210/en.2010-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirlby RC, Bahiraei F, Randall J, Drewnoski A. Effect of Roux-en-Y gastric bypass on satiety and food likes: the role of genetics. J Gastrointest Surg. 2006;10:270–277. doi: 10.1016/j.gassur.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Tsukumo DML, Carvalho-Filho MA, Carvalheira JBC, Prada PCO, Hirabara SM, Schenka AA, Araðjo EP, Vassallo J, Curi R, Velloso L.c.A., Saad MJA. Loss-of-Function Mutation in Toll-Like Receptor 4 Prevents Diet-Induced Obesity and Insulin Resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-[alpha] function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- Van Wye G, Dubin JA, Blair SN, DiPietro L. Adult Obesity Does Not Predict 6-Year Weight Gain in Men: The Aerobics Center Longitudinal Study. Obesity. 2007;15:1571–1577. doi: 10.1038/oby.2007.186. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil G.k.S. Inflammation, stress, and diabetes. The Journal of Clinical Investigation. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. American Journal of Physiology - Endocrinology and Metabolism. 2009;296:E291–E299. doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdowson PS, Upton R, Buckingham R, Arch J, Williams G. Inhibition of food response to intracerebroventricular injection of leptin is attenuated in rats with diet-induced obesity. Diabetes. 1997;46:1782–1785. doi: 10.2337/diab.46.11.1782. [DOI] [PubMed] [Google Scholar]
- Williams DL, Baskin DG, Schwartz MW. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes. 2006;55:3387–3393. doi: 10.2337/db06-0558. [DOI] [PubMed] [Google Scholar]
- Williams DL, Baskin DG, Schwartz MW. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology. 2009;150:1680–1687. doi: 10.1210/en.2008-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, D’Alessio DA. Central control of body weight and appetite. J Clin Endocrinol Metab. 2008;93:S37–50. doi: 10.1210/jc.2008-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Lotter EC, McKay LD, Porte D., Jr Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature. 1979;282:503–505. doi: 10.1038/282503a0. [DOI] [PubMed] [Google Scholar]
- Woods SC, Seeley RJ, Rushing PA, D’Alessio DA, Tso P. A controlled high-fat diet induces an obese syndrome in rats. Journal of Nutrition. 2003;133:1081–1087. doi: 10.1093/jn/133.4.1081. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ohinata K, Meguid MM, Marx W, Tada T, Chen C, Quinn R, Inui A. Gastric bypass model in the obese rat to study metabolic mechanisms of weight loss. J Surg Res. 2002;107:56–63. doi: 10.1006/jsre.2002.6508. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Saitou K, Mizushige T, Matsumura S, Manabe Y, Tsuzuki S, Inoue K, Fushiki T. The palatability of corn oil and linoleic acid to mice as measured by short-term two-bottle choice and licking tests. Physiology & Behavior. 2007a;91:304–309. doi: 10.1016/j.physbeh.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Taka Y, Okamura M, Mizushige T, Matsumura S, Manabe Y, Tsuzuki S, Inoue K, Fushiki T. Reinforcing effect for corn oil stimulus was concentration dependent in an operant task in mice. Life Sciences. 2007b;81:1585–1592. doi: 10.1016/j.lfs.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Kim Y-B, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine Phosphatase 1B Expression Is Induced by Inflammation in Vivo. Journal of Biological Chemistry. 2008;283:14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER Stress Link Overnutrition to Energy Imbalance and Obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]