

Abstract
The locus coeruleus-noradrenergic systems supplies norepinephrine throughout the central nervous system. State-dependent neuronal discharge activity of locus coeruleus noradrenergic neurons has long-suggested a role of this system in the induction of an alert waking state. Work over the past two decades provides unambiguous evidence that the locus coeruleus, and likely other noradrenergic nuclei, exert potent wake-promoting actions via an activation of noradrenergic β- and α1-receptors located within multiple subcortical structures, including the general regions of the medial septal area, the medial preoptic area and, most recently, the lateral hypothalamus. Conversely, global blockade of β- and α1-receptors or suppression of norepinephrine release results in profound sedation. The wake-promoting action of central noradrenergic neurotransmission has clinical implications for treatment of sleep/arousal disorders, such as insomnia and narcolepsy, and clinical conditions associated with excessive arousal, such as post-traumatic stress disorder.
Keywords: arousal, locus coeruleus, noradrenaline, norepinephrine, psychostimulants, waking
Introduction
The regulation of sleep and waking serves a critical role in behavior and ultimately survival. Moreover, during waking, arousal levels fluctuate, ranging from sleepiness/sedation associated with a relative insensitivity to sensory stimuli to high-arousal stress or fear-related conditions associated with a hypersensitivity to sensory events (hyperalertness, hypervigilance). Fluctuations in sleep-wake/arousal state are accompanied by alterations in forebrain neuronal activity that are reflected in electroencephalographic (EEG) signals.1, 2 The formal examination of the neurobiology of arousal/waking dates back to the work of von Economo3, Bremer4 and Moruzzi and Magoun5, which identified a critical role of the brainstem in the induction and maintenance of arousal. Subsequent work has identified a large array of brainstem and basal forebrain neural systems that participate in the regulation of behavioral state. Included among these is the locus coeruleus (LC)-noradrenergic system.
The LC is a small pontine nucleus that provides the majority of brain norepinephrine (NE).6 NE acts at three major receptor families, α1, α2, and β, each comprised of multiple subtypes. α1- and β-receptors are thought to exist primarily postsynaptically whereas α2-receptors are present both pre- and postsynaptically. Early electrophysiological observations indicated that the LC-NE system may play a role in the regulation of behavioral state.6, 7 For example, LC neurons display higher discharge rates during waking than in sleep while within waking LC discharge rate is positively correlated with behavioral and EEG indices of arousal.8–10 Importantly, these alterations in LC discharge rate precede changes in behavioral state.8–10 Combined, these observations suggested a potentially causal role of the LC-noradrenergic system in the regulation of sleep-wake/arousal state. Results from studies conducted over the past two decades provide unambiguous evidence for a prominent wake/arousal-promoting role of the LC and other brain noradrenergic systems (e.g. A1, A2), as reviewed below.
Wake/Arousal-Promoting Actions of the Locus Coeruleus
Early lesion and pharmacological studies attempted to address whether there exists a causal relationship between LC neuronal activity and behavioral and EEG indices of waking/arousal (for review11, 12). In general, lesions of noradrenergic systems have had an inconsistent impact on indices of arousal (for review12). There is now strong reason to believe this limited action of noradrenergic lesions on behavioral state likely reflects the occurrence of time-dependent lesion-induced compensation within noradrenergic systems (for review12). Compensation is apparent within 7–10 days following a lesion and is observed at the level of release, receptor number, and second messenger sensitivity.12 For example, microdialysis measures of transmitter release unambiguously demonstrate that within 7–10 days following noradrenergic or dopaminergic lesions, extracellular levels of NE and dopamine are not significantly reduced unless tissue levels have been decreased by more than 90%.13–16 The vast majority of studies using catecholamine lesions generally do not achieve tissue depletion greater than 80–85%. Moreover, even when tissue levels have been reduced by at least 90%, the decrease in extracellular levels of these transmitters is substantially less than that observed in tissue measures (e.g. 60% reduction in extracellular levels vs. >90% reduction in tissue levels). Given lesions also increase postsynaptic receptor number and second messenger levels, the functional impairment in NE neurotransmission associated with these larger lesions (> 90% tissue depletion) is likely to be substantially less than predicted by the decrease in extracellular NE levels. Given these observations, it is not surprising that the time course of the sleep-wake effects of NE lesions is identical to that seen with lesion-induced compensation within catecholamine systems. For example, Lidbrink17, 18 reported that 6-OHDA lesions of the dorsal noradrenergic bundle initially increased slow-wave EEG activity, but by approximately 7 days following the lesion this effect was no longer apparent.
In contrast to lesions, acute suppression of LC-NE neurotransmission by systemic, ICV, or intra-brainstem administration of α2-receptor agonists, which in part activate presynaptic autoreceptors and thereby inhibit NE neurotransmission at postsynatpic noradrenergic receptors, elicit profound sedation.19, 20 Despite the consistency of these observations, the small size of the LC and the close proximity of this nucleus to other brainstem arousal-related nuclei preclude drawing unambiguous conclusions regarding the role of the LC vs. other brainstem nuclei in these drug-induced changes in arousal state.
To more selectively manipulate LC neuronal discharge activity, Adams and Foote developed a combined recording/infusion probe that uses electrophysiological recordings to guide placement of an infusion needle within a 200–300 microns distance from the LC and to monitor the electrophysiological effects at LC neurons of peri-LC infusions of drugs that either activate or inhibit LC neuronal discharge rate.21, 22 With the consistent placement of the infusion needle in close proximity to the LC, much smaller infusion volumes (35–150 nl) can be used, greatly limiting the spread of infusate beyond the boundaries of the LC. Using this probe, we demonstrated that unilateral LC activation (via infusion of the cholinergic agonist, bethanechol) produced a bilateral activation of forebrain EEG in the halothane-anesthetized rat (Figure 1).22 A number of observations indicated that infusion-induced alterations in LC discharge were responsible for EEG activation, rather than drug action at a site distant to the LC. These include: 1) LC activation always preceded EEG activation by only a few seconds; 2) infusions only activated forebrain EEG if they were placed within a radius of approximately 500 μm of the center of the LC, a radius that rules out participation of a number of brainstem nuclei (Figure 1); 3) a similar latency between LC activation and EEG activation was observed regardless of whether infusions were placed lateral or medial to the LC; 4) infusions placed anterior or posterior to the LC did not alter forebrain EEG activity state; and 5) EEG activation was prevented by pretreatment with drugs that suppress NE neurotransmission (an α2-agonist and a β-antagonist).
Figure 1.

Effects of LC activation on cortical EEG (ECoG) activity state. For these studies, the LC was first located electrophysiologically using a combined recording-infusion probe in the halothane-anesthetized rat. Once located, a 100–150 nl infusion of the cholinergic agonist, bethanechol, was made adjacent to the LC to activate LC neurons while simultaneously recording LC discharge activity. The effect of infusion-induced activation of the LC on ECoG (and hippocampal EEG, not shown) was monitored. A: Schematic of the infusion/recording probe used to activate LC. Bethanechol was infused 300–400 μm lateral or medial to the recording electrode that was used to locate the main body of the LC. B: Photomicrograph of a peri-LC infusion site. In this example, an infusion lateral of the LC is depicted. N indicates position within the track created by the infusion needle where the infusate exited the needle. E indicates position of the recording electrode within the LC. C: Simultaneous effects of bethanechol infusion on LC activity (LC Trigger, bottom trace) and ECoG activity (top trace). Bethanechol infusion (indicated by horizontal bar) increased LC discharge rate approximately two-thirds of the way through the 60-second infusion. Several seconds following LC activation, EEG desynchronization is observed. EEG recordings were ipsilateral to the manipulated LC. However, identical effects were observed in the contralateral hemisphere. D: Power spectral analyses of 11-second pre-infusion and post-infusion ECoG epochs. Power of low-frequency activity is decreased following bethanechol-induced activation of ECoG. E: Schematic depicting bethanechol infusion sites that were effective or ineffective for activating forebrain EEG. Solid circles indicate sites at which bethanechol infusion activated EEG. Shaded boxes indicate sites at which bethanechol infusion had no obvious EEG effects. There is a radius of approximately 500 μm around LC within which infusions, placed either medially or laterally, activated forebrain EEG. Infusions placed immediately anterior to the LC were also ineffective (not shown). These mapping studies strongly argue against drug action at other arousal-related brainstem nuclei (e.g. cholinergic, serotonergic). Abbreviations: Me5, mesencephalic nucleus of the trigeminal nerve; Mo5, motor nucleus of the trigeminal nerve; Pr5, principle sensory nucleus of the trigeminal nerve; 4V, fourth ventricle (from22).
Additional studies in the lightly-anesthetized rat displaying spontaneously activated (desynchronized) forebrain EEG, demonstrated that bilateral suppression of LC neuronal discharge (via 35 nl infusions of an α2-receptor agonist) produced a robust increase in slow-wave EEG activity (Figure 2).23 In these studies, anesthesia level was adjusted to elicit sustained, spontaneous desynchronized forebrain EEG, indicative of a lightly anesthetized state. Under these conditions, EEG activity and LC neuronal discharge rates were monitored before and following suppression of LC neuronal firing produced by 35 nl infusions of the α2-agonist, clonidine. Bilateral, but not unilateral, clonidine infusions robustly suppressed desynchronized EEG activity and increased slow-wave activity. Importantly, these studies document that even minimal LC neuronal discharge activity within a single hemisphere (i.e. 5–10% of basal levels) is sufficient to maintain bilateral forebrain activation. This is likely an important factor for why LC/noradrenergic lesions, which almost certainly never destroy all LC NE neurons and thereby fail to reduce completely extracellular NE levels do not result in large alterations in time spent awake (see above).
Figure 2.

Effects of bilateral suppression of LC discharge in the lightly-anesthetized rat. A similar experimental approach was taken to that outlined in Figure 1, other than 1) animals were lightly anesthetized and 2) they received bilateral peri-LC clonidine infusions (35, 100 ng/35, 100 nl). The top panels (LC) depict oscilloscope traces of a multiunit LC recording before (Pre-Clonidine) and after (Post-Clonidine) a peri-LC clonidine infusion. Clonidine completely suppressed LC discharge. The effects of LC suppression on cortical EEG (ECoG, middle panels) and hippocampal EEG (HEEG, bottom panels) were characterized. Shown are 25-second raw ECoG/HEEG traces along with the results of power spectral analysis (graphs below raw traces). Power spectral analysis was calculated on 8-minute epochs that included the 25-second EEG segment shown. Prior to clonidine infusion, baseline EEG was stably activated as indicated by limited slow-wave activity in cortical EEG (ECoG, middle panels) and nearly pure theta activity in hippocampal EEG (HEEG, bottom panels). Peri-LC clonidine infusions that completely suppressed LC discharge bilaterally resulted in a profound increase in slow-wave activity in ECoG and HEEG recordings and a decrease in theta activity in HEEG. These effects appeared within seconds-minutes of LC suppression and persisted for the entire period in which LC neurons were completely inactive (> 60–90 minutes). Recovery from these EEG effects quickly followed the emergence of minimal LC discharge activity. Shading in the power spectral plots indicates the theta frequency band (2.3–6.9 Hz) in the HEEG power spectra. Mapping studies indicated an ~500–600μm radius around the LC for clonidine-induced increases in EEG indices of sedation (from23).
These observations indicate that LC neuronal activity is both sufficient and necessary for the maintenance of an activated forebrain in the anesthetized rat. More recent studies using optogenetic activation of the LC observed similar arousal-promoting actions in unanesthetized animals.24 These studies demonstrate that brief activation of the LC in the range of 3–5 Hz elicits increases in EEG/EMG measures of waking. This effect was most consistent when the duration of activation was at least 5–8 seconds, consistent with observations in anesthetized animals described above. Conversely, inhibition of the LC for 1-hour decreased time spent awake in the active period of the animal’s circadian cycle, while not altering time spent asleep. This is particularly notable given that 10-minute optogenetic inhibition of the LC only decreased extracellular levels in the prefrontal cortex by approximately 50%24, suggesting that perhaps not all LC discharge activity was suppressed. The above-reviewed observations in anesthetized animals demonstrated that only near-complete suppression of LC discharge (bilaterally) resulted in an increase in EEG indices of sedation. Thus, it seems likely that a more profound suppression of LC-NE neurotransmission would elicit greater actions on sleep-wake state than observed in these optogenetic studies.
Neurocircuitry and Receptor Mechanisms of Noradrenergic Dependent Waking: Wake-Promoting Actions of α1- and β-Receptors in the Medial Septal and Medial Preoptic Areas
Collectively the observations reviewed above provide strong evidence that the LC-NE system exerts wake-promoting actions. This raises the question of which LC terminal fields and which receptor post-synaptic receptors participate in these actions. Subcortically, the general regions of the medial septal area (MSA), the medial preoptic area (MPOA), and the substantia innominata (SI) are known to modulate forebrain EEG activity state 25–27 and each of these regions receives LC-noradrenergic input.28–30 A series of studies examined whether NE acts within these subcortical regions to promote waking. In these studies, the sleep-wake/arousal effects of intratissue infusion of NE, a noradrenergic α1-receptor agonist (phenylephrine) or a noradrenergic β-receptor agonist (isoproterenol) was examined in anesthetized and unanesthetized rats.31–34 In unanesthetized animals, the effects of wake-promoting actions of remote-controlled infusions of NE and NE agonists were examined in sleeping animals implanted with EEG/EMG electrodes. This approach permitted examination of the wake/arousal-promoting actions of the infusions from a stable and low-arousal state (i.e. sleeping) while avoiding infusion-related handling of the animals (see32).
Wake-promoting actions of NE β- and α1-receptors within the MSA and MPOA, but not SI
These studies demonstrated that intratissue infusion (150 nl-250 nl) of β- or α1-receptor agonists (isoproterenol or phenylephrine, respectively) into the MSA or MPOA (situated immediately caudal to the MSA; Figures 3–5) produce robust and dose-dependent increases in EEG activation in anesthetized animals and EEG/behavioral indices of waking in unanesthetized animals.31–36 Infusions immediately outside these regions were devoid of wake-promoting actions.31–34 In unanesthetized animals, the wake-promoting actions of β-and α1-receptors within both the MSA and MPOA were additive but not supra-additive/synergistic (i.e. greater than the sum of the individual effects of β- and α1-receptors; Figure 534).
Figure 3.

NE acts within the general regions of the MSA and MPOA to promote waking. A) Schematics indicate boundaries within the general regions of the MSA (Top Row) and MPOA (Bottom Row) within which NE and NE α1- and β-receptor agonist infusions promote waking in the sleeping rat. These studies identify a nearly continuous portion of the medial basal forebrain where NE acts to promote waking (dotted lines) that extends in the anterior-posterior dimension from the anterior MSA to the posterior MPOA. The region termed the MSA encompasses the medial septum, the vertical limb of the diagonal band of Broca, the posterior portions of the shell region of the nucleus accumbens. The region termed the MPOA encompasses preoptic area of the hypothalamus and portions of the bed nucleus of the stria terminalis (BST). Previous mapping studies suggest that the shell accumbens or BST are not prominently involved in NE-induced waking. Infusions outside the MSA and MPOA are generally ineffective at increasing waking. Panels are arranged anterior-posterior with the anterior-most panel shown in the upper left and the posterior-most panel shown in the bottom right position. Photomicrographs depict intra-tissue infusion sites from experiments involving NE agonist infusions into the MSA and MPOA. In these photomicrographs, large arrows indicate the most ventral extent of the infusion needle. Small arrow indicates the lateral ventricle. Note minimal tissue damage associated with these infusions. B: Effects of NE infusions into the MPOA on EEG and EMG indices of sleep-wake state. Shown are 10-minute traces of cortical EEG (ECoG) and EMG recorded immediately prior to (top traces, PRE NE) and 10-minute following (bottom traces, POST NE) NE infusion into MPOA. Prior to the infusion, the animal spent the majority of time in slow-wave sleep (resting with large amplitude, slow-wave activity present in ECoG and low-amplitude activity present in EMG). The most striking post-infusion changes are the decrease in large-amplitude, slow-wave ECoG activity and the increase in EMG amplitude, indicative of alert, active waking. Depending on dose, this wake-promoting effect is observed for > 90 minutes. Similar effects are observed with NE agonist infusions into the MSA. Abbreviations: AC, anterior commissure; CC, corpus callosum; CP, caudate-putamen; GP, globus pallidus; I, internal capsule; LS, lateral septum; LV, lateral ventricle; M, midline, MS, medial septum; NA, nucleus accumbens; SI, substantia innominata (from33).
Figure 5.

Additive wake-promoting effects of α1- and β-agonist receptor stimulation within the MSA and MPOA. Panels depict the wake-promoting effects of vehicle, 10 nmol of the α1-agonist, phenylephrine (PHEN), 4 nmol of the β-agonist, isoproterenol (ISO), and combined PHEN + ISO (Combined) when infused into the MSA (Top Panel) and MPOA (Bottom Panel). For both regions, a relatively low dose of each drug was used that elicited only a mild wake-promoting action when administered alone. In the combined treatment group, additive wake-promoting effects of ISO and PHEN were observed. Symbols represent means (± SEM) of time (seconds) spent awake per 30-min testing epoch. PRE1 and PRE2 represent 30-minute pre-infusion epochs occurring immediately prior to the infusion. POST1-POST3 represent 30-minute post-infusions epochs, beginning immediately following the infusions. *P<0.05, **P<0.01 compared to PRE1; +P<0.05 compared to Combined (from34).
There are three identified β-receptor subtypes: β1-β3.12 Intra-tissue infusions of the β2-agonist, clenbuterol, into either the MSA or the MPOA produced dose-dependent increases in time spent awake similar to that seen with the non-selective β-agonist, isoproterenol37. In addition to its β2-agonist action, clenbuterol also possesses weak β1-antagonistic properties38. However, infusion of a selective β1-antagonist into the MSA or MPOA had no impact on sleep-wake state37, indicating that clenbuterol-induced waking results from activation of β2-receptors and not inhibition of β1-receptors.
The SI, situated immediately lateral to both the MSA and MPOA (see Figure 3), provides a potent activating influence on forebrain EEG.27, 39 These SI projections act in parallel with efferents from the MSA to regulate the activity state of the neocortex and hippocampus, respectively. Thus, it was posited initially that NE-driven activation of hippocampal and cortical EEG involved the simultaneous actions of NE within the MSA and SI, respectively. However, infusion of NE, phenylephrine (α1-agonist), isoproterenol (βagonist), or the indirect noradrenergic agonist, amphetamine, directly into the SI failed to impact EEG/EMG or behavioral indices of waking/arousal (Figure 4).31–33, 40 In contrast, under identical experimental conditions, intra-SI infusions of the excitatory amino acid, glutamate, produced a robust activation of forebrain EEG.27, 31 Across all drug-infusion studies, the only condition in which a noradrenergic drug was observed to promote waking when infused into SI was with the highest concentration of NE examined (15 μg/250 nl) which produced a moderate increase in waking (Figure 4).33, 41 However, in this case, the latency to waking was substantially longer and the time spent awake substantially reduced relative to infusion into the MPOA (Figure 4)33, suggesting that at high concentrations NE diffuses from the SI to the MPOA where it acts to increase waking.
Figure 4.

NE and α1- and β-agonist infusions within the MSA and MPOA, but not SI, promote waking. Shown are the effects of NE, the α1-agonist, phenylephrine (PHEN), and the β-agonist, isoproterenol (ISO) infused into MPOA (top and middle panels) or MSA (bottom panel). Symbols represent mean (± SEM) of time (seconds) spent awake in 30-minute (1800-seconds) epochs. PRE1 and PRE2 represent 30-min pre-infusion epochs occurring immediately prior to the infusion. POST1-POST3 represent 30-minute post-infusions epochs, beginning immediately following the infusions. Top Panel: Effects of unilateral infusion of vehicle, 4 nmol NE, or 16 nmol NE, infused into either the MPOA or SI on time spent awake. Middle Panel: Effects of unilateral infusion of vehicle, PHEN (40 nmol) or ISO (15 nmol) infused into either the MPOA or SI on time spent awake. NE, PHEN and ISO increase time spent awake when infused into the MPOA, but not into the SI. The only exception to this was observed with the high dose of NE. In this case the latency to waking was longer, the magnitude of waking smaller and the duration shorter than that observed with infusions into the MPOA. The wake-promoting effects of intra-MPOA infusion of all of these compounds are dose-dependent. Larger effects are observed with bilateral infusions. Similar wake-promoting effects of β and α1-receptor activation are observed in MSA. Bottom Panel: Effects of vehicle and PHEN (10 nmol, 50 nmol) infusion into the MSA on time spent awake. PHEN exerts dose dependent wake-promoting effects when infused into the MSA. Similar effects were observed with infusion of the β-agonist, ISO. For all panels, a lack of visible error bars indicates the magnitude of the SEM fell within the range corresponding to the dimensions of the symbol. There were no significant differences between any of the groups during the pre-infusion epochs. *P<0.05, **P<0.01 compared to PRE-1 (from31–33, 40}).
Collectively, these observations demonstrate NE acts at β- and α1-receptors within the MSA and MPOA, but not SI, to promote waking. Limited studies reported minimal sleep-wake actions of the α1-agonist, methoxamine, when infused into the MPOA.42, 43 Although the basis for this apparent discrepancy is unclear, possible contributing factors include an elevated baseline level of waking, which would obscure the acute wake-promoting actions of an infusion, and more extensive needle/cannula-related tissue damage within the MPOA in these latter studies.
Neurocircuitry within the MSA and MPOA involved in NE-induced waking
The terms MSA and MPOA refer to relatively large and heterogeneous structures, each containing multiple subnuclei. The anatomical resolution of previous mapping studies (approximately 500 μm) limits definitive identification of the specific subnuclei involved in the wake-promoting actions of NE within these general regions. Distinct populations of sleep-active and wake-active neurons are found in the general regions of the MSA and MPOA. Within the MPOA, sleep-active neurons are found within both the median preoptic nucleus (MnPN) and the ventrolateral preoptic nucleus (VLPO).44, 45 Results from previous NE/NE-agonist mapping studies suggest that proximity to the VLPO does not correlate with the wake-promoting effects of intra-MPOA α1-agonist infusion.33 On the other hand, the MnPN is well within the general area targeted in previous intra-MPOA infusion studies.
Neither α1- or β-agonists affect sleep-active neurons within MPOA and MSA.46 In contrast, α1-agonist administration, but not β-agonist, increases the activity of a subpopulation of waking-active neurons46. Thus, α1-receptor-dependent waking may involve an activation of waking-active neurons in the MSA and MPOA. Within the SI and MPOA, sleep-active neurons express α2-receptors and α2-receptor stimulation was observed to inhibit sleep-active neurons within the MSA and MPOA.46, 47, 48 Thus, the wake-promoting actions of NE may also involve α2-receptor-dependent inhibition of sleep-active neurons.
Within the MSA, evidence indicates that the activity of GABAergic and cholinergic neurons combine to influence hippocampal EEG activity.49 To date, the extent to which α1- and β-receptors are differentially distributed across neurochemically-defined (i.e. cholinergic vs. GABAergic vs. galaninergic) or behaviorally-defined (i.e. sleep-active vs. waking-active) neuronal subpopulations within both the MSA and MPOA is unknown. If the wake-promoting effects of α1- and β-receptors involve actions on the same cell, at least two mechanisms exist through which these receptors could interact to exert additive arousal-modulating actions. First, these receptors could exert independent and additive effects on membrane potential via parallel actions on second messenger systems (e.g. phosphoinositol and cAMP). Additionally, evidence indicates that α1-receptors can potentiate β-receptor-mediated cAMP production.50 However, this latter mechanism would not explain why βantagonists fail to exert sedative effects alone but potentiate the sedative effects of α1-antagonists, as described below (see Synergistic Actions of β- and α1-Receptors in the Maintenance of Alert Waking).
In addition to hippocampal projections, the MSA also sends direct projections to the neocortex.51–53 Therefore, NE could simultaneously modulate hippocampal and cortical EEG activity state via simultaneous actions on both of these ascending MSA projections paths. Alternatively, NE could influence EEG and behavioral activity state via alterations in efferent pathways that project to subcortical arousal-related regions, such as the hypothalamus, thalamus, or midbrain. Such actions could involve direct projections from the MSA to these regions54, 55 or indirect via hippocampal efferents.56, 57
Contrasting actions of MSA/MPOA β-receptor blockade in anesthetized vs. unanesthetized animals
As reviewed above, stimulation of β-receptors within the MSA is sufficient to increase EEG and behavioral indices of waking/arousal. Moreover, in the lightly-anesthetized animal, bilateral blockade of MSA β-receptors decreases EEG activation elicited either through LC activation or in the lightly-anesthetized preparation.58 These observations indicate that in the anesthetized animal MSA β-receptors are necessary for forebrain EEG activation. Whether this is true of MPOA β-receptors remains to be determined. In contrast, in the unanesthetized rat, neither bilateral blockade of MSA β-receptors58 or blockade of MPOA β-receptors35 alters EEG or behavioral indices of arousal. Nonetheless, global suppression of LC-NE neurotransmission, using ICV or intrabrainstem-administered α2-agonists, potently decreases EEG/behavioral indices of arousal.59, 60 The lack of sedative effects of bilateral MSA/MPOA β-receptor blockade likely reflects the wake-promoting actions of: 1) α1-receptors within these regions; and/or 2) α1- and β-receptors located outside these regions. In sum, in contrast to that observed in the presence of anesthesia, the wake-promoting actions of MSA β-receptors are not necessary for behavioral and EEG indices of alert waking even though stimulation of these β-receptors is sufficient to promote alert waking.
Origin and organization of the noradrenergic innervation of the MSA, MPOA and SI
The above-described observations indicate that LC neurons exert a robust excitatory influence on forebrain activity state, actions that likely involve NE signaling within the MSA and MPOA. However, there are a number of additional noradrenergic nuclei that could also provide input to these regions and thus participate in the modulation of behavioral state. Recent retrograde tracing studies indicate that although the LC provides the majority of noradrenergic input to the MSA, MPOA and SI (~50%), the A1/C1 (~25%) and A2/C2 (~25–40%) cell groups also provide notable contributions.30 These observations suggest a likely arousal-promoting role of the A1/C1 and the A2/C2 noradrenergic/adrenergic nuclei.
In the case of the LC, basal forebrain-projecting noradrenergic neurons are largely distributed uniformly within the LC.30 This contrasts with a rough topographic distribution of LC neurons projecting to neocortex (with the exception of the prefrontal cortex), hippocampus, cerebellum, and spinal cord.61 Additional observations indicate that individual LC neurons display a high degree of collateralization across these three arousal-related basal forebrain regions, ensuring that alterations in LC discharge are relayed simultaneously to these regions.30 In sum, these observations indicate that LC efferents exert coordinated actions across multiple anatomically-distinct, yet functionally-related (i.e. arousal-related), basal forebrain fields.
As mentioned, unlike the MSA and MPOA, the SI is relatively insensitive to the wake-promoting actions of NE and NE agonists.33, 40 Nonetheless, in vitro, NE depolarizes cholinergic basal forebrain neurons, indicating a neuromodulatory role of NE within this region.62 To date, the behavioral/cognitive functions of NE within the SI remain to be fully elucidated. However, evidence indicates a role of the SI in state-dependent attentional processes (for review63). Moreover, noradrenergic α1-receptors within the SI appear to participate in the priming effect of systemic epinephrine administration on the cerebral auditory evoked potential.64 Thus, NE likely acts within the SI to modulate state-dependent behavioral/cognitive processes, such as attention. The high degree of collateralization across the MSA, MPOA and SI permits the LC to simultaneously modulate behavioral state while modulating SI-dependent behavioral and physiological processes.
Additional Regions Involved in NE-Dependent Waking
In addition to the MSA and MPOA, the lateral hypothalamus (LH) has been implicated in the regulation of sleep-wake state and state-dependent processes.65, 66 In particular, the neuropeptide family, hypocretin (HCRT; orexin), exerts potent wake-promoting actions.67, 68 HCRT-synthesizing neurons are located solely in the perifornical region of the LH, an area that receives a moderately dense noradrenergic innervation most of which arises from outside the LC.69, 70 Recently completed studies demonstrate additive wake-promoting actions of α1- and β-receptor agonists (phenylephrine and isoproterenol, respectively) infused into the LH (Figure 6).71 Surprisingly, this NE-dependent waking was not associated with an activation of HCRT neurons as measured by expression of the immediate-early gene, c-fos.71 This latter observation is consistent with in vitro electrophysiological studies demonstrating an inhibitory action of NE on HCRT neuronal discharge under certain conditions72, 73 (but see74). Thus, the wake-promoting actions of NE within the LH may be independent of the HCRT system. However, this is clearly a question that needs further study.
Figure 6.
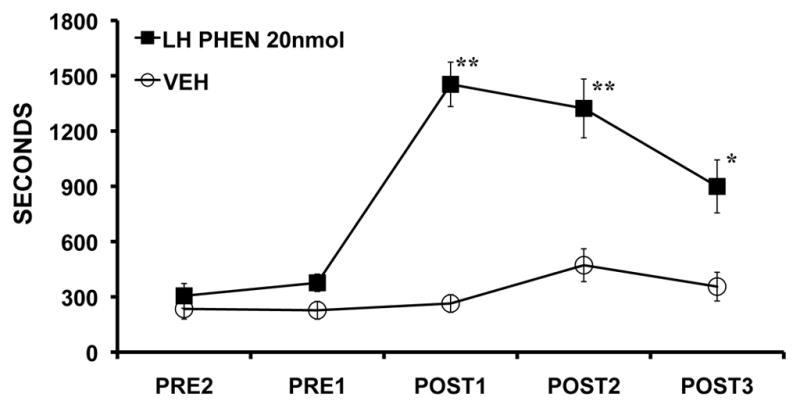
NE acts within the LH to promote alert waking. Shown are the effects of bilateral intra-LH infusion of the α1-agonist, phenylephrine (PHEN; 20 nmol/250 nl) on time spent awake as determined from EEG/EMG indices. At this dose, PHEN increased time spent awake for a sustained period. Unilateral infusions also produced significant increases in waking, but of a somewhat lesser intensity. Intra-LH β-agonist infusion produced smaller magnitude, though significant, wake-promoting effects (data not shown). PRE1 and PRE2 represent 30-minute pre-infusion epochs occurring immediately prior to the infusion. POST1-POST3 represent 30-minute post-infusions epochs, beginning immediately following the infusions. Symbols represent means (± SEM) of time (seconds) spent awake per 30-min testing epoch. *P<0.05, **P<0.01 compared to PRE1 (from71).
Finally, there are numerous additional sites in which NE may act to modulate behavioral state and state-dependent processes. For example, within neocortex and thalamus, NE exerts a depolarizing action, eliciting a transition from a burst-mode of firing associated with sleep to a single-spike mode of firing associated with waking.75, 76 Moreover, a variety of additional brainstem-originating monoaminergic systems are known to modulate sleep-wake/arousal state including, serotonergic, dopaminergic, cholinergic and histaminergic systems.11, 25, 77, 78 All of these systems are highly interconnected and in many cases have been demonstrated to exert reciprocally activating effects.79–81 To date, the full extent to which NE acts on these other neurotransmitter systems to modulate behavioral state has not been examined systematically. Nonetheless, dorsal pontine tegmental β-receptor activation has been demonstrated to inhibit REM sleep.82
Combined, these observations indicate that noradrenergic regulation of behavioral state involves coordinated actions of NE, arising from the LC and other noradrenergic nuclei (i.e. A1, A2), across a distributed network of cortical and subcortical structures.
Synergistic Actions of β- and α1-Receptors in the Maintenance of Alert Waking
Substantial evidence indicates that stimulation of either α1- or β-receptors within any one of a number of brain regions is sufficient to induce the alert waking state. This high degree of redundancy suggests that α1-/β-receptor action in any given region is unlikely to be necessary for the maintenance of alert waking, consistent with observations reviewed above regarding a lack of effect of β-antagonist infusions into the MSA on indices of arousal in unanesthetized animals. To initially address the degree to which α1- and/or β-receptors, globally within the brain, are necessary for the maintenance of alert waking, the EEG effects of β-receptor blockade (ICV timolol), α1-receptor blockade (intraperitoneal prazosin), or combined β- and α1-receptor blockade were examined in rats under conditions associated with high arousal levels (a brightly-lit novel environment). Combined blockade of β- and α1-receptors elicited a profound increase in large-amplitude, slow-wave activity in cortical EEG indicative of sedation (Figure 7).83 This increase in slow-wave activity was in contrast to the minimal EEG effects of β-antagonist treatment alone or an increase in sleep-spindles seen with α1-receptor blockade (Figure 7; see also84). Therefore, under these conditions, combined blockade of β- and α1-receptors produces synergistic sedative effects.
Figure 7.

Synergistic sedative effects of α1- and β-receptor blockade globally within the brain. Shown are the effects of the β-antagonist, timolol (ICV), the α1-antagonist, prazosin (IP) and combined antagonist treatment on cortical EEG in animals exposed to an arousing brightly-lit novel environment. Animals were treated 30-minutes prior to testing with: 1) ICV vehicle + IP saline (VEH/VEH); 2) 150 μg ICV timolol + IP saline (TIM/VEH); 3) ICV vehicle + IP 500 μg/kg prazosin (VEH/PRAZ,), and; 4) combined timolol + prazosin (TIM/PRAZ). In this figure, EEG traces are from the second 5-min epoch of exposure to the novel environment. Vehicle-treated controls displayed behavioral and EEG indices of alert waking throughout most of the recording session. This was reflected in sustained EEG desynchronization (low-amplitude, high-frequency). β-receptor blockade alone (TIM/VEH) had no effects on EEG activity. α1-receptor blockade alone (VEH/PRAZ) increased the frequency and duration of sleep spindles (high-voltage spindles). In contrast to that observed with β-receptor blockade alone, in the presence of α1-receptor blockade, β-receptor blockade produced substantial increases in large-amplitude, slow-wave activity. Power spectral analysis quantified and confirmed these qualitative observations (data not shown; from62).
Interestingly, the sedative action of combined β-/α1-receptor blockade was not observed during the first 5-minutes of testing. Thus, under certain conditions of elevated arousal, combined actions of α1- and β-receptors are not necessary for the maintenance of EEG indices of arousal. These observations could suggest that the combined blockade of α1- and β-receptors increases the rate of habituation to an arousing, novel environment. Alternatively, initial exposure to the novel environment may involve actions of additional arousal-promoting systems that are capable of compensating for the loss of noradrenergic signaling.
Finally, it is of interest that although combined α1- and β-receptor blockade elicits profound sedation (i.e. nearly continuous slow-wave EEG activity), this treatment did not appear to induce sleep. Specifically, the animals maintained sufficient postural tone to remain in an upright position, albeit with minimal movement. Thus, in the absence of α1-/β-receptor action, actions of post-synaptic α2-receptors or other neurotransmitter systems maintain a certain degree of arousal.
Collectively, these observations demonstrate potent and additive wake-promoting actions of NE α1- and β-receptors that are necessary for the maintenance of alert waking. Much of the sedative actions of α2-agonists can be ascribed to autoreceptor-dependent suppression of post-synaptic α1- and β-receptors. The sedative actions of peri-LC α2-agonist infusion are consistent with this view. However, limited observations suggest that NE may inhibit sleep-active neurons via activation of α2-receptors in the MSA and MPOA.46–48
Potential Sleep-Promoting Actions of α1-Receptors within the MPOA
In contrast to the robust wake-promoting effects of NE agonists infused into the MPOA and MSA, 6-OHDA lesions of the ventral noradrenergic bundle (VNAB) have been reported to increase time spent awake.85 This has been interpreted as suggesting a potential sleep-promoting action of NE, particularly within the MPOA.86 However, as noted above, 6-OHDA lesion of the VNAB will not eliminate all VNAB noradrenergic input to the MPOA. Moreover there are multiple sources of the noradrenergic innervation of the MPOA (LC, A1, A2), the axons of which travel through both the dorsal and VNAB (see30). Combined, VNAB lesions will spare a prominent proportion of NE afferents into the MPOA while eliciting compensatory responses at the level of release and postsynaptic receptor function.87–89 Given this, we posit that the wake-promoting effects of an incomplete VNAB lesion reflects lesion-induced upregulation of noradrenergic neurotransmission within the MPOA and/or other arousal-related structures, as has been described elsewhere (for review, see12).
Lesions of the VNAB also disrupted the wake-promoting actions of NE when infused into the MPOA during the day and resulted in an NE-induced decrease in time spent awake when infused into the MPOA during the night.90 Consistent with this latter observation, infusion of an α1-antagonist into the MPOA increased time spent awake.43 Combined, these observations further suggest possible arousal-attenuating actions of MPOA α1-receptors. Currently, it is not clear how to reconcile the robust wake-promoting effects of intra-MPOA NE, α1- and β-agonist infusions reported by others33–35 with these more recent observations. However, there are a number of issues to consider. First, these effects appear relatively modest compared to the wake-promoting effects of NE agonist infusions. Second, baseline arousal level in these latter studies appears higher and more labile than in previous studies utilizing NE agonists. Alterations in baseline arousal level could influence the arousal-modulating actions of NE. Third, when performing intra-tissue infusions, it is critical to minimize tissue damage. However, the limited available evidence indicates substantial tissue damage may have occurred within the MPOA in these more recent studies (see91), which could alter the sensitivity of this region to NE-selective drugs. Fourth, given variability in baseline waking between groups observed in these studies, it would be useful to examine the effects of α1-antagonist infusions using a within-subject design, similar to studies with α1-agonists.
Behavioral State Modulatory actions of Norepinephrine Occur in Conjunction with other Behavioral Actions
Evidence reviewed above indicates potent arousal-promoting actions of the LC-NE and presumably other noradrenergic systems. However, this is not to say that the sole function of the central noradrenergic systems is the regulation of arousal. Indeed, NE-induced arousal occurs in tandem with a large variety of modulatory actions of NE on physiological and behavioral processes, including endocrine regulation, perception, motor function, attention and memory, and decision and action (for review12). These actions involve a variety of cortical and subcortical noradrenergic terminal fields. A unifying theme to this diverse array of actions is the ability of NE to facilitate the collection, processing and responding to salient information arising from an ever-changing environment (for review12). One component of this broad array of actions is the induction of an appropriate behavioral state for the detection of sensory events (i.e. waking). However, within the awake state, noradrenergic systems modulate a variety of systems/processes that facilitate rapid behavioral responding to environmental events and these actions can be both short-term and long-term (i.e. plasticity) in nature. Although many of these latter actions are state-dependent, they are independent of an arousal regulatory function.
Clinical Relevance
The wake and arousal-promoting actions of central noradrenergic neurotransmission may have clinical relevance in a number of clinical conditions associated with the dysregulation of sleep and waking and/or arousal. For example, the data reviewed above demonstrate that under normal conditions, acute activation of LC-noradrenergic signaling is incompatible with the state of sleep. Thus, inappropriate excitatory drive on this system, which can arise from upstream regions including the prefrontal cortex and amygdala, could contribute to an inability to fall or stay asleep associated with insomnia.92
Additionally, psychostimulants are efficacious in the treatment of narcolepsy, a disorder associated with excessive sedation. As reviewed elsewhere, these drugs potently increase noradrenergic signaling and a number of observations indicate that psychostimulant-induced activation of α1- and β-receptors contributes to the arousal-promoting actions of these drugs. 93 This includes the fact that amphetamine-induced arousal closely parallels drug-induced elevations in extracellular levels of brain NE and the fact that amphetamine acts in brain regions linked to NE-induced arousal.40, 94 Moreover, limited observations demonstrate that blocking actions of β-receptors prevents the EEG activating effects of intravenous amphetamine in anesthetized animals 95. Thus, the therapeutic actions of psychostimulants in the treatment of narcolepsy may well involve the arousal/wake-promoting actions of norepinephrine.
Beyond sleep disorders, certain psychiatric disorders are associated with excessive arousal, including post-traumatic stress disorder. The sedating effect of α1-receptor blockade is likely relevant to the therapeutic effects of α1-antagonists seen in the treatment of post-traumatic stress disorder.96–98 Finally, the arousal-promoting actions of noradrenergic α1- and β-receptors likely contribute to the well-documented anesthetic effects of α2-agonists, which suppress NE release and thus stimulation of post-synaptic α1- and β-receptors.99
Conclusion
The regulation of arousal is a critical aspect of normal behavior. A substantial body of work demonstrates a prominent role of the LC and other noradrenergic systems in the regulation of waking and arousal. NE-dependent waking/arousal involves additive/synergistic actions of α1- and β-receptors located within multiple subcortical regions. Combined, the available evidence indicates that under normal physiological conditions even moderate activity of the LC-NE system is incompatible with the state of sleep. Given this, excessive activity of this system may contribute to certain forms of insomnia or other conditions associated with elevated arousal levels, including stress-related disorders, such as post-traumatic stress disorder. Indeed, α1-antagonists are useful in treating hyperarousal associated with this disorder, including sleep disturbances. The wake-promoting actions of NE represent only one aspect of NE function and occur in tandem with noradrenergic modulation of an array of state-dependent physiological, cognitive and affective processes. Combined, these diverse actions of NE facilitate collecting, processing and acting upon sensory information.
Practice Points.
Locus coeruleus noradrenergic neurons display state-dependent discharge, with highest rates of norepinephrine neurotransmission associated with waking and high-vigilance states.
Norepinephrine acts at α1- and β-receptors to exert robust and sustained wake-promoting actions
α1- and β-receptors, globally within the brain, are necessary for alert waking.
Norepinephrine likely contributes to the wake-promoting effects of psychostimulants used in the treatment of narcolepsy.
Research Agenda.
To better determine the role of α1- and β-receptor subtypes (e.g. α1A, α1B, etc.) and how these different subtypes may represent new clinical targets.
To better determine how top-down regulation of LC discharge may impact sleep-wake regulation (e.g. prefrontal cortex → locus coeruleus; amygdala → locus coeruleus).
Acknowledgments
This work was supported by PHS grants MH62359, DA10681, DA00389 and the University of Wisconsin Graduate School.
Abbreviations
- EEG
electroencephalographic
- EMG
electromyographic
- ICV
intracerebroventricular
- IP
intraperitoneal
- LC
Locus Coeruleus
- LH
lateral hypothalamus
- MSA
medial septal area
- MPOA
medial preoptic area
- MNPN
median preoptic nucleus
- NE
norepinephrine
- SI
substantia innominata
- VNAB
ventral noradrenergic bundle
- VLPOA
ventrolateral preoptic area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bushnell MC, Goldberg ME, Robinson DL. Behavioral enhancement of visual responses in monkey cerebral cortex. I. Modulation in posterior parietal cortex related to selective visual attention. J Neurophysiol. 1981;46(4):755–72. doi: 10.1152/jn.1981.46.4.755. [DOI] [PubMed] [Google Scholar]
- 2.Evarts EV. Effects of sleep and waking on spontaneous and evoked discharge of single units in visual cortex. Fed Proc. 1960;19:828–37. [PubMed] [Google Scholar]
- 3.Von Economo C. Sleep as a problem of localization. J Nerv Ment Dis. 1930;71:249–59. [Google Scholar]
- 4.Bremer F. Cerebral activity during sleep and narcosis: Contribution to the study of the mechanisms of sleep. Bull l’Acad Royale Med Belgique. 1937;4:240–75. [Google Scholar]
- 5.Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol. 1949;1:455–73. [PubMed] [Google Scholar]
- 6.Foote SL, Bloom FE, Aston-Jones G. Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63(3):844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- 7.Foote SL, Morrison JH. Extrathalamic modulation of cortical function. Annu Rev Neurosci. 1987;10:67–95. doi: 10.1146/annurev.ne.10.030187.000435. [DOI] [PubMed] [Google Scholar]
- 8.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1(8):876–86. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *9.Foote SL, Aston-Jones G, Bloom FE. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc Natl Acad Sci USA. 1980;77(5):3033–7. doi: 10.1073/pnas.77.5.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189(4196):55–8. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- 11.Vanderwolf CH, Robinson TE. Reticulo-cortical activity and behavior: A critique of the arousal theory and a new synthesis. Behav Brain Sci. 1981;4:459–514. [Google Scholar]
- 12.Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. 2003;42(1):33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 13.Abercrombie ED, Zigmond MJ. Partial injury to central noradrenergic neurons: reduction of tissue norepinephrine content is greater than reduction of extracellular norepinephrine measured by microdialysis. J Neurosci. 1989;9(11):4062–7. doi: 10.1523/JNEUROSCI.09-11-04062.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson TE, Whishaw IQ. Normalization of extracellular dopamine in striatum following recovery from a partial unilateral 6-OHDA lesion of the substantia nigra: a microdialysis study in freely moving rats. Brain Res. 1988;450(1–2):209–24. doi: 10.1016/0006-8993(88)91560-0. [DOI] [PubMed] [Google Scholar]
- 15.Castaneda E, Whishaw IQ, Robinson TE. Changes in striatal dopamine neurotransmission assessed with microdialysis following recovery from a bilateral 6-OHDA lesion: variation as a function of lesion size. J Neurosci. 1990;10(6):1847–54. doi: 10.1523/JNEUROSCI.10-06-01847.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson TE, Mocsary Z, Camp DM, Whishaw IQ. Time course of recovery of extracellular dopamine following partial damage to the nigrostriatal dopamine system. J Neurosci. 1994;14(5 Pt 1):2687–96. doi: 10.1523/JNEUROSCI.14-05-02687.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lidbrink P, Fuxe K. Effects of intracerebral injections of 6-hydroxydopamine on sleep and waking in the rat. J Pharm Pharmacol. 1973;25(1):84–7. doi: 10.1111/j.2042-7158.1973.tb09125.x. [DOI] [PubMed] [Google Scholar]
- 18.Lidbrink P. The effect of lesions of ascending noradrenaline pathways on sleep and waking in the rat. Brain Res. 1974;74(1):19–40. doi: 10.1016/0006-8993(74)90109-7. [DOI] [PubMed] [Google Scholar]
- 19.Gatti PJ, Hill KJ, Da Silva AM, Norman WP, Gillis RA. Central nervous system site of action for the hypotensive effect of clonidine in the cat. J Pharmacol Exp Ther. 1988;245(1):373–80. [PubMed] [Google Scholar]
- 20.De Sarro GB, Ascioti C, Froio F, Libri V, Nistico G. Evidence that locus coeruleus is the site where clonidine and drugs acting at alpha 1- and alpha 2-adrenoceptors affect sleep and arousal mechanisms. Br J Pharmacol. 1987;90(4):675–85. doi: 10.1111/j.1476-5381.1987.tb11220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams LM, Foote SL. Effects of locally infused pharmacological agents on spontaneous and sensory-evoked activity of locus coeruleus neurons. Brain Res Bull. 1988;21(3):395–400. doi: 10.1016/0361-9230(88)90151-7. [DOI] [PubMed] [Google Scholar]
- *22.Berridge CW, Foote SL. Effects of locus coeruleus activation on electroencephalographic activity in neocortex and hippocampus. J Neurosci. 1991;11(10):3135–45. doi: 10.1523/JNEUROSCI.11-10-03135.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.Berridge CW, Page ME, Valentino RJ, Foote SL. Effects of locus coeruleus inactivation on electroencephalographic activity in neocortex and hippocampus. Neuroscience. 1993;55(2):381–93. doi: 10.1016/0306-4522(93)90507-c. [DOI] [PubMed] [Google Scholar]
- *24.Carter ME, Yizhar O, Chikahisa S, et al. Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci. 2010;13(12):1526–33. doi: 10.1038/nn.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buzsaki G, Leung LW, Vanderwolf CH. Cellular bases of hippocampal EEG in the behaving rat. Brain Res. 1983;287(2):139–71. doi: 10.1016/0165-0173(83)90037-1. [DOI] [PubMed] [Google Scholar]
- 26.Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci. 1988;8(11):4007–26. doi: 10.1523/JNEUROSCI.08-11-04007.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metherate R, Cox CL, Ashe JH. Cellular bases of neocortical activation: modulation of neural oscillations by the nucleus basalis and endogenous acetylcholine. J Neurosci. 1992;12(12):4701–11. doi: 10.1523/JNEUROSCI.12-12-04701.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaborszky L. Afferent connections of the forebrain cholinergic projection neurons, with special reference to monoaminergic and peptidergic fibers. In: Frotscher M, Misgel U, editors. Central cholinergic synaptic transmission. Basel: Birkhauser; 1989. pp. 12–32. [DOI] [PubMed] [Google Scholar]
- 29.Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol. 1975;163(4):467–505. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- 30.España RA, Berridge CW. Organization of noradrenergic efferents to arousal-related basal forebrain structures. J Comp Neurol. 2006;496(5):668–83. doi: 10.1002/cne.20946. [DOI] [PubMed] [Google Scholar]
- 31.Berridge CW, Bolen SJ, Manley MS, Foote SL. Modulation of forebrain electroencephalographic activity in halothane- anesthetized rat via actions of noradrenergic beta-receptors within the medial septal region. J Neurosci. 1996;16(21):7010–20. doi: 10.1523/JNEUROSCI.16-21-07010.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *32.Berridge CW, Foote SL. Enhancement of behavioral and electroencephalographic indices of waking following stimulation of noradrenergic beta-receptors within the medial septal region of the basal forebrain. J Neurosci. 1996;16(21):6999–7009. doi: 10.1523/JNEUROSCI.16-21-06999.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *33.Berridge CW, O’Neill J. Differential sensitivity to the wake-promoting actions of norepinephrine within the medial preoptic area and the substantia innominata. Behav Neurosci. 2001;115(1):165–74. doi: 10.1037/0735-7044.115.1.165. [DOI] [PubMed] [Google Scholar]
- *34.Berridge CW, Isaac SO, España RA. Additive wake-promoting actions of medial basal forebrain noradrenergic alpha1- and beta-receptor stimulation. Behav Neurosci. 2003;117(2):350–9. doi: 10.1037/0735-7044.117.2.350. [DOI] [PubMed] [Google Scholar]
- 35.Kumar V, Datta S, Chhina GS, Gandhi N, Singh B. Sleep-awake responses elicited from medial preoptic area on application of norepinephrine and phenoxybenzamine in free moving rats. Brain Res. 1984;322(2):322–5. doi: 10.1016/0006-8993(84)90125-2. [DOI] [PubMed] [Google Scholar]
- 36.Sood S, Dhawan JK, Ramesh V, John J, Gopinath G, Kumar VM. Role of medial preoptic area beta adrenoceptors in the regulation of sleep-wakefulness. Pharmacol Biochem Behav. 1997;57(1–2):1–5. doi: 10.1016/s0091-3057(96)00384-x. [DOI] [PubMed] [Google Scholar]
- 37.Berridge CW, Stellick RL, Schmeichel BE. Wake-promoting actions of medial basal forebrain beta2 receptor stimulation. Behav Neurosci. 2005;119(3):743–51. doi: 10.1037/0735-7044.119.3.743. [DOI] [PubMed] [Google Scholar]
- 38.Conway PG, Tejani-Butt S, Brunswick DJ. Interaction of beta adrenergic agonists and antagonists with brain beta adrenergic receptors in vivo. J Pharmacol Exp Ther. 1987;241(3):755–62. [PubMed] [Google Scholar]
- 39.Buzsaki G, Gage FH. The cholinergic nucleus basalis: a key structure in neocortical arousal. EXS. 1989;57:159–71. doi: 10.1007/978-3-0348-9138-7_16. [DOI] [PubMed] [Google Scholar]
- 40.Berridge CW, O’Neil J, Wifler K. Amphetamine acts within the medial basal forebrain to initiate and maintain alert waking. Neuroscience. 1999;93(3):885–96. doi: 10.1016/s0306-4522(99)00271-7. [DOI] [PubMed] [Google Scholar]
- 41.Cape EG, Jones BE. Differential modulation of high-frequency gamma-electroencephalogram activity and sleep-wake state by noradrenaline and serotonin microinjections into the region of cholinergic basalis neurons. J Neurosci. 1998;18(7):2653–66. doi: 10.1523/JNEUROSCI.18-07-02653.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vetrivelan R, Mallick HN, Kumar VM. Unmasking of alpha1 adrenoceptor induced hypnogenic response from medial preoptic area. Physiol Behav. 2005;84(4):641–50. doi: 10.1016/j.physbeh.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 43.Vetrivelan R, Mallick HN, Kumar VM. Tonic activity of alpha1 adrenergic receptors of the medial preoptic area contributes towards increased sleep in rats. Neuroscience. 2006;139(3):1141–51. doi: 10.1016/j.neuroscience.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 44.Szymusiak R, Gvilia I, McGinty D. Hypothalamic control of sleep. Sleep Med. 2007;8(4):291–301. doi: 10.1016/j.sleep.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 45.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437(7063):1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 46.Osaka T, Matsumura H. Noradrenaline inhibits preoptic sleep-active neurons through alpha 2-receptors in the rat. Neurosci Res. 1995;21(4):323–30. doi: 10.1016/0168-0102(94)00871-c. [DOI] [PubMed] [Google Scholar]
- 47.Manns ID, Lee MG, Modirrousta M, Hou YP, Jones BE. Alpha 2 adrenergic receptors on GABAergic, putative sleep-promoting basal forebrain neurons. Eur J Neurosci. 2003;18(3):723–7. doi: 10.1046/j.1460-9568.2003.02788.x. [DOI] [PubMed] [Google Scholar]
- 48.Modirrousta M, Mainville L, Jones BE. Gabaergic neurons with alpha2-adrenergic receptors in basal forebrain and preoptic area express c-Fos during sleep. Neuroscience. 2004;129(3):803–10. doi: 10.1016/j.neuroscience.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 49.Colom LV, Nassif-Caudarella S, Dickson CT, Smythe JW, Bland BH. In vivo intrahippocampal microinfusion of carbachol and bicuculline induces theta-like oscillations in the septally deafferented hippocampus. Hippocampus. 1991;1(4):381–90. doi: 10.1002/hipo.450010406. [DOI] [PubMed] [Google Scholar]
- 50.Stone EA, McEwen BS, Herrera AS, Carr KD. Regulation of alpha and beta components of noradrenergic cyclic AMP response in cortical slices. Eur J Pharmacol. 1987;141(3):347–56. doi: 10.1016/0014-2999(87)90551-6. [DOI] [PubMed] [Google Scholar]
- 51.Marston HM, West HL, Wilkinson LS, Everitt BJ, Robbins TW. Effects of excitotoxic lesions of the septum and vertical limb nucleus of the diagonal band of Broca on conditional visual discrimination: relationship between performance and choline acetyltransferase activity in the cingulate cortex. J Neurosci. 1994;14(4):2009–19. doi: 10.1523/JNEUROSCI.14-04-02009.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saper CB. Organization of cerebral cortical afferent systems in the rat. II. Magnocellular basal nucleus. J Comp Neurol. 1984;222(3):313–42. doi: 10.1002/cne.902220302. [DOI] [PubMed] [Google Scholar]
- 53.Stewart DJ, MacFabe DF, Leung LW. Topographical projection of cholinergic neurons in the basal forebrain to the cingulate cortex in the rat. Brain Res. 1985;358(1–2):404–7. doi: 10.1016/0006-8993(85)90994-1. [DOI] [PubMed] [Google Scholar]
- 54.Meibach RC, Siegel A. Efferent connections of the septal area in the rat: an analysis utilizing retrograde and anterograde transport methods. Brain Res. 1977;119:1–20. doi: 10.1016/0006-8993(77)90088-9. [DOI] [PubMed] [Google Scholar]
- 55.Swanson LW, Cowan WM. The connections of the septal region in the rat. JComp Neur. 1979;186:621–56. doi: 10.1002/cne.901860408. [DOI] [PubMed] [Google Scholar]
- 56.Ino T, Itoh K, Kamiya H, Shigemoto R, Akiguchi I, Mizuno N. Direct projections of non-pyramidal neurons of Ammon’s horn to the supramammillary region in the cat. Brain Res. 1988;460(1):173–7. doi: 10.1016/0006-8993(88)91219-x. [DOI] [PubMed] [Google Scholar]
- 57.Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172(1):49–84. doi: 10.1002/cne.901720104. [DOI] [PubMed] [Google Scholar]
- 58.Berridge CW, Wifler K. Contrasting effects of noradrenergic beta-receptor blockade within the medial septal area on forebrain electroencephalographic and behavioral activity state in anesthetized and unanesthetized rat. Neuroscience. 2000;97(3):543–52. doi: 10.1016/s0306-4522(00)00047-6. [DOI] [PubMed] [Google Scholar]
- 59.Danysz W, Dyr W, Plaznik A, Kostowski W. The effect of microinjections of clonidine into the locus coeruleus on cortical EEG in rats. Pol J Pharmacol Pharm. 1989;41(1):45–50. [PubMed] [Google Scholar]
- 60.De Sarro GB, Bagetta G, Ascioti C, Libri V, Nistico G. Microinfusion of clonidine and yohimbine into locus coeruleus alters EEG power spectrum: effects of aging and reversal by phosphatidylserine. BrJ Pharmacol. 1988;95(4):1278–86. doi: 10.1111/j.1476-5381.1988.tb11765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loughlin SE, Foote SL, Bloom FE. Efferent projections of nucleus locus coeruleus: topographic organization of cells of origin demonstrated by three-dimensional reconstruction. Neuroscience. 1986;18(2):291–306. doi: 10.1016/0306-4522(86)90155-7. [DOI] [PubMed] [Google Scholar]
- 62.Fort P, Khateb A, Pegna A, Muhlethaler M, Jones BE. Noradrenergic modulation of cholinergic nucleus basalis neurons demonstrated by in vitro pharmacological and immunohistochemical evidence in the guinea-pig brain. Eur J Neurosci. 1995;7(7):1502–11. doi: 10.1111/j.1460-9568.1995.tb01145.x. [DOI] [PubMed] [Google Scholar]
- 63.Sarter M, Bruno JP. Abnormal regulation of corticopetal cholinergic neurons and impaired information processing in neuropsychiatric disorders. Trends Neurosci. 1999;22(2):67–74. doi: 10.1016/s0166-2236(98)01289-2. [DOI] [PubMed] [Google Scholar]
- 64.Berntson GG, Shafi R, Knox D, Sarter M. Blockade of epinephrine priming of the cerebral auditory evoked response by cortical cholinergic deafferentation. Neuroscience. 2003;116(1):179–86. doi: 10.1016/s0306-4522(02)00702-9. [DOI] [PubMed] [Google Scholar]
- 65.Stellar EM. The physiology of motivation. Psychol Rev. 1954;154:5–22. doi: 10.1037/h0060347. [DOI] [PubMed] [Google Scholar]
- 66.Olds J. Hypothalamic substrates of reward. Physiol Rev. 1962;42(554):604. doi: 10.1152/physrev.1962.42.4.554. [DOI] [PubMed] [Google Scholar]
- 67.España RA, Baldo BA, Kelley AE, Berridge CW. Wake-promoting and sleep-suppressing actions of hypocretin (orexin): Basal forebrain sites of action. Neuroscience. 2001;106(4):699–715. doi: 10.1016/s0306-4522(01)00319-0. [DOI] [PubMed] [Google Scholar]
- 68.Sutcliffe JG, de Lecea L. The hypocretins: setting the arousal threshold. Nat Rev Neurosci. 2002;3(5):339–49. doi: 10.1038/nrn808. [DOI] [PubMed] [Google Scholar]
- 69.Baldo BA, Daniel RA, Berridge CW, Kelley AE. Overlapping distributions of orexin/hypocretin- and dopamine-beta-hydroxylase immunoreactive fibers in rat brain regions mediating arousal, motivation, and stress. J Comp Neurol. 2003;464(2):220–37. doi: 10.1002/cne.10783. [DOI] [PubMed] [Google Scholar]
- 70.Yoshida K, McCormack S, España RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. 2005;494(5):845–61. doi: 10.1002/cne.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Berridge CW, Schmeichel BE. Norepinephrine Acts Within the Lateral Hypothalamus to Promote Waking: Relevance to Psychostimulant-Induced Arousal. Soc Neurosci Abst. 2007:37. [Google Scholar]
- 72.Yamanaka A, Muraki Y, Tsujino N, Goto K, Sakurai T. Regulation of orexin neurons by the monoaminergic and cholinergic systems. Biochem Biophys Res Commun. 2003;303(1):120–9. doi: 10.1016/s0006-291x(03)00299-7. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, van Den Pol AN. Direct and indirect inhibition by catecholamines of hypocretin/orexin neurons. J Neurosci. 2005;25(1):173–83. doi: 10.1523/JNEUROSCI.4015-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bayer L, Eggermann E, Serafin M, et al. Opposite effects of noradrenaline and acetylcholine upon hypocretin/orexin versus melanin concentrating hormone neurons in rat hypothalamic slices. Neuroscience. 2005;130(4):807–11. doi: 10.1016/j.neuroscience.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 75.Mukhametov LM, Rizzolatti G, Tradardi V. Spontaneous activity of neurones of nucleus reticularis thalami in freely moving cats. J Physiol. 1970;210(3):651–67. doi: 10.1113/jphysiol.1970.sp009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- 77.Isaac SO, Berridge CW. Wake-promoting actions of dopamine D1 and D2 receptor stimulation. J Pharmacol Exp Ther. 2003;307(1):386–94. doi: 10.1124/jpet.103.053918. [DOI] [PubMed] [Google Scholar]
- 78.Thakkar MM. Histamine in the regulation of wakefulness. Sleep Med Rev. 2011;15(1):65–74. doi: 10.1016/j.smrv.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stevens DR, Birnstiel S, Gerber U, McCarley RW, Greene RW. Nicotinic depolarizations of rat medial pontine reticular formation neurons studied in vitro. Neuroscience. 1993;57(2):419–24. doi: 10.1016/0306-4522(93)90073-o. [DOI] [PubMed] [Google Scholar]
- 80.Stevens DR, McCarley RW, Greene RW. The mechanism of noradrenergic alpha 1 excitatory modulation of pontine reticular formation neurons. J Neurosci. 1994;14(11 Pt 1):6481–7. doi: 10.1523/JNEUROSCI.14-11-06481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aston-Jones G, Akaoka H, Charlety P, Chouvet G. Serotonin selectively attenuates glutamate-evoked activation of noradrenergic locus coeruleus neurons. J Neurosci. 1991;11(3):760–9. doi: 10.1523/JNEUROSCI.11-03-00760.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tononi G, Pompeiano M, Pompeiano O. Modulation of desynchronized sleep through microinjection of beta- adrenergic agonists and antagonists in the dorsal pontine tegmentum of the cat. Pflugers Arch. 1989;415(2):142–9. doi: 10.1007/BF00370584. [DOI] [PubMed] [Google Scholar]
- 83.Berridge CW, España RA. Synergistic sedative effects of noradrenergic alpha(1)- and beta- receptor blockade on forebrain electroencephalographic and behavioral indices. Neuroscience. 2000;99(3):495–505. doi: 10.1016/s0306-4522(00)00215-3. [DOI] [PubMed] [Google Scholar]
- 84.Buzsaki G, Kennedy B, Solt BV, Ziegler M. Noradrenergic control of thalamic oscillation: the role of α-2 receptors. Eur J Neurosci. 1991;3:222–9. doi: 10.1111/j.1460-9568.1991.tb00083.x. [DOI] [PubMed] [Google Scholar]
- 85.Kumar VM, Sharma R, Wadhwa S, Manchanda SK. Sleep-inducing function of noradrenergic fibers in the medial preoptic area. Brain Res Bull. 1993;32(2):153–8. doi: 10.1016/0361-9230(93)90069-n. [DOI] [PubMed] [Google Scholar]
- 86.Kumar VM, Vetrivelan R, Mallick HN. Noradrenergic afferents and receptors in the medial preoptic area: Neuroanatomical and neurochemical links between the regulation of sleep and body temperature. Neurochem Int. 2007;50(6):783–90. doi: 10.1016/j.neuint.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 87.Berridge CW, Dunn AJ. DSP-4-induced depletion of brain norepinephrine produces opposite effects on exploratory behavior 3 and 14 days after treatment. Psychopharmacology. 1990;100(4):504–8. doi: 10.1007/BF02244003. [DOI] [PubMed] [Google Scholar]
- 88.Diaz J, Ellison G, Masouka D. Stages of recovery from central norepinephrine lesions in enriched and impoverished environments: a behavioral and biochemical study. Exp Brain Res. 1978;31(1):117–30. doi: 10.1007/BF00235809. [DOI] [PubMed] [Google Scholar]
- 89.Mogilnicka E. Increase in beta- and alpha 1-adrenoceptor binding sites in the rat brain and in the alpha 1-adrenoceptor functional sensitivity after the DSP-4-induced noradrenergic denervation. Pharmacol Biochem Behav. 1986;25(4):743–6. doi: 10.1016/0091-3057(86)90380-1. [DOI] [PubMed] [Google Scholar]
- 90.Kumar VM, Vetrivelan R, Mallick HN. Alpha-1 adrenergic receptors in the medial preoptic area are involved in the induction of sleep. Neurochem Res. 2006;31(8):1095–102. doi: 10.1007/s11064-006-9109-8. [DOI] [PubMed] [Google Scholar]
- 91.Vetrivelan R, Mallick HN, Kumar VM. Sleep induction and temperature lowering by medial preoptic alpha(1) adrenergic receptors. Physiol Behav. 2006;87(4):707–13. doi: 10.1016/j.physbeh.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 92.Reyes BA, Carvalho AF, Vakharia K, Van Bockstaele EJ. Amygdalar peptidergic circuits regulating noradrenergic locus coeruleus neurons: linking limbic and arousal centers. Exp Neurol. 2011;230(1):96–105. doi: 10.1016/j.expneurol.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berridge CW. Neural substrates of psychostimulant-induced arousal. Neuropsychopharmacology. 2006;31(11):2332–40. doi: 10.1038/sj.npp.1301159. [DOI] [PubMed] [Google Scholar]
- 94.Berridge CW, Stalnaker TA. Relationship between low-dose amphetamine-induced arousal and extracellular norepinephrine and dopamine levels within prefrontal cortex. Synapse. 2002;46(3):140–9. doi: 10.1002/syn.10131. [DOI] [PubMed] [Google Scholar]
- *95.Berridge CW, Morris MF. Amphetamine-induced activation of forebrain EEG is prevented by noradrenergic beta-receptor blockade in the halothane-anesthetized rat. Psychopharmacology. 2000;148(3):307–13. doi: 10.1007/s002130050055. [DOI] [PubMed] [Google Scholar]
- 96.Raskind MA, Peskind ER, Hoff DJ, et al. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry. 2007;61(8):928–34. doi: 10.1016/j.biopsych.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 97.Taylor F, Raskind MA. The alpha1-adrenergic antagonist prazosin improves sleep and nightmares in civilian trauma posttraumatic stress disorder. J Clin Psychopharmacol. 2002;22(1):82–5. doi: 10.1097/00004714-200202000-00013. [DOI] [PubMed] [Google Scholar]
- 98.Byers MG, Allison KM, Wendel CS, Lee JK. Prazosin versus quetiapine for nighttime posttraumatic stress disordersymptoms in veterans: an assessment of long -term comparative effectiveness and safety. J Clin Psychopharmacol. 2010;30(3):225–9. doi: 10.1097/JCP.0b013e3181dac52f. [DOI] [PubMed] [Google Scholar]
- 99.Nelson LE, Lu J, Guo T, Saper CB, Franks NP, Maze M. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98(2):428–36. doi: 10.1097/00000542-200302000-00024. [DOI] [PubMed] [Google Scholar]