

Abstract
The Hyp mouse is a commonly used model for the study of the phosphate wasting disease X-linked hypophosphataemia. The defect in this mouse line is a deletion that includes exons 16 to 22 of Phex, although the exact extent of this X chromosome deletion remains unknown. This complicates genotyping which increases costs, time and difficulty of working with this important model. We aimed to determine the molecular breakpoints of this deletion in order develop a robust assay for its detection. We designed short mapping PCRs around the Phex locus to refine the putative breakpoint locations, then used gap PCR to amplify a product containing the breakpoint junction. DNA sequencing showed the deleted region was approximately 297 kb, significantly larger than previous reports, but did not contain any genes other than Phex. DNA sequence analysis revealed that this deletion may be the result of microhomology-mediated end joining. Finally, we designed a multiplex PCR assay for genotyping Hyp colonies and validated it using a panel of Hyp colony mice. This study provides confirmation of the Hyp phenotype as a single gene defect, a potential mechanism for its formation and an improved method for genotyping that will make working with this strain significantly easier.
Keywords: X-linked hypophosphataemia, Hyp mouse, deletion
Introduction
X-linked hypophosphataemia (XLH) is the most common inherited form of hypophosphataemic rickets, a dominant disorder that affects phosphate homeostasis. XLH is characterised by abnormal bone mineralisation, abnormal renal vitamin D metabolism, osteomalacia, and hyposphosphataemia. XLH is caused exclusively by mutations in PHEX [1], a 22 exon gene that encodes a protein called phosphate regulating endopeptidase homolog, X-linked. While the specific function of this protein is unknown, it is thought to exert its effects on phosphate balance via increased fibroblast growth factor 23 signalling [2]. The Hyp mouse strain arose from a spontaneous mutation and its phenotypic similarity to human XLH patients has led to wide use as a model of the human disease [3]. Several attempts to map the Hyp defect have been made. Initial studies showed that the Hyp phenotype mapped to the distal end of the long arm of chromosome X [3]. Later studies showed that the Phex mRNA lacked exons 16 to 22, due to a deletion that had one breakpoint in intron 15 and another in the region to the 3’ of the gene [4][5]. The most comprehensive mapping of the deletion was performed by Sabbagh et al. who used PCR to refine the location of the breakpoint in intron 15 and determined the other was approximately 10 kb downstream of Phex, consistent with a deletion of between 53 and 58 kb [6]. Despite this, they were unable to amplify a product containing the deletion breakpoints and so the precise extent of the deletion remains to be determined.
As the exact boundaries of the Hyp deletion are unknown, genotyping it is problematic. The deletion is generally genotyped using either PCR or Southern hybridisation with primers or a probe in the known deleted region, respectively [5][7]. While these methods can be used to determine male genotypes by the absence of amplification or hybridisation, this may lead to misclassification due to methodological errors and, in the case of Southern hybridisation, is slow and technically demanding. This is also unsuitable for discriminating between heterozygous and wild-type (WT) females due to the presence of at least one normal X chromosome. These factors complicate breeding and increase both the number of mice used and the associated costs.
In this study, we aimed to determine the molecular extent of the Hyp deletion to provide an improved method for genotyping this strain. We designed PCR primers to amplify short products around the proposed breakpoints of the deletion which allowed us to refine the breakpoints sufficiently so that we were able to amplify a product containing the breakpoint junction. DNA sequencing showed the Hyp deletion was approximately 297 kb, significantly larger than previously thought, but does not affect any genes other than Phex. Molecular characterisation of the deletion enabled us to design a rapid multiplex PCR assay for Hyp genotyping that will simplify work with this important mouse model of XLH.
Methods and Materials
Animals
Hyp (stock number 000528) and C57BL/6J (stock number 000664) mouse strains were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). All studies were performed with IACUC approval.
DNA Extraction
DNA was extracted from C57BL/6J and Hyp mouse tail clips using a DNeasy Blood and Tissue kit according to manufacturer’s instructions (QIAGEN, Hilden, Germany).
Mapping PCRs
The mouse genome sequence from exon 15 of Phex to exon 6 of Sat1 on chromosome X was retrieved from the University of California Santa Cruz mouse genome browser (http://genome.ucsc.edu; April 2011, assembly NCBI37/mm9) and this sequence was filtered for putative repeat DNA sequences using RepeatMasker (www.repeatmasker.org) and CENSOR (www.girinst.org/censor/) [8]. PCR primer pairs were designed in repeat-free regions to amplify products of between 114 and 337 bp. PCRs were performed using Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s specifications, at an annealing temperature of 60°C. Products were analysed by electrophoresis through a 2% agarose gel and stained with ethidium bromide. All mapping PCRs were performed in duplicate, one with Hyp male DNA and one with wild-type (WT) C57BL/6J DNA. Sequences for all primers used in this study are provided in Supplementary Table 1.
Gap PCR
PCR primer pairs were designed in regions closely flanking putative breakpoints, shown to be intact in the Hyp mouse by mapping PCR. PCRs were performed using Platinum Taq High Fidelity DNA Polymerase (Invitrogen), according to manufacturer’s specifications, at an annealing temperature of 62°C.
DNA Sequencing
PCR products were purified by centrifugation at 750× g through a Montage MultiScreen PCR96 microtitre plate (Millipore, Billerica, MA, USA), then resuspended in nuclease free water. Purified PCR products were sequenced using BigDye Terminator v. 3.1 chemistry (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Sequencing data was analysed using SeqScape v. 2.6 software (Applied Biosystems).
Multiplex genotyping PCR Assay
Hyp genotyping PCRs contained 1× Hot Start Supermix (Biopioneer Inc, San Diego, CA, USA) with primers WT-F (62.5 nM), WT-R (62.5 nM), Hyp-F (500 nM) and Hyp-R (500 nM), according to manufacturer’s specifications, at an annealing temperature of 60°C.
Results and Discussion
The location of the Hyp deletion on the X chromosome makes mapping it by PCR significantly easier than an autosomal deletion where the normal chromosome would mask the deletion. We exploited this property by designing short PCRs, which enabled mapping of the deletion by the presence or absence of a product from male Hyp DNA. We used the findings of the most extensive previous attempt to characterise the deletion as a starting point [6]. In that study, Sabbagh et al. refined breakpoint locations to between ~10 to 13 kb into intron 15 and between ~ 8 to 10 kb from the 3’ end of Phex. We were able to reproduce their results (not shown) and subsequently designed further primers in proximity to these to refine the deletion boundaries. Using this approach, we found the intron 15 breakpoint was between PCRs 5 and 6 (Figure 1A and B), which corresponds to a region of approximately 2.0 kb between nucleotides 153,645,729 and 153,647,703. We were unable to amplify any further products (primer pairs A, B, C and D) in the vicinity of the primer pair used by Sabbagh et al. (designated F6R6) to delineate the boundary of the deletion. Gap PCR, with multiple combinations of primers designed to amplify across the deletion junction, also failed to amplify any Hyp specific products.
Figure 1.

(A) Mapping PCRs of Hyp deletion locus. Numbers (1 to 5) and letters (A to N) indicate PCRs in the telomeric (intron 15) and centromeric (intergenic) breakpoint regions respectively. Absence of a product for PCR 6 indicates a breakpoint between PCRs 5 and 6. Lack of a product for H denotes a breakpoint between H and I, which was narrowed to between K and L in a subsequent round of mapping PCR. (B) Summary of Hyp deletion mapping PCRs showing the extent of the Hyp deletion (grey box). Boxed regions are enlarged in the lower panels. Circles represent PCRs used in this study and squares denote results of Sabbagh et al for comparison (not to scale). Filled and unfilled shapes represent presence or absence of PCR products from male Hyp mouse DNA, respectively.
We reviewed the primer sequences and putative products described in Sabbagh et al. and found that the primer pair F6R6 (filled square in Figure1B) amplifies a 620 bp fragment of a LINE element, which occurs 202 further times within the mouse genome with 98% or higher sequence identity. This indicates a false positive and that the Hyp deletion is likely more extensive than predicted in that study. We designed further PCR primer pairs (E, F, G, H, I and J) extending approximately 300 kb into the intergenic region. These showed a breakpoint between PCRs H and I (between nucleotides 153,336,911 and 153,376,320) Further mapping PCRs resolved the breakpoint to between K and L (between nucleotides 153,347,029 and 153,348,980), a region of approximately 2.3 kb. PCR primers were designed between PCRs 5 and 6 and between K and L to amplify across the breakpoint junction. A single product of 518 bp was amplified from Hyp DNA, but not from WT DNA, and was subsequently sequenced (Figure 2A).
Figure 2.

Gap PCR and DNA sequence of a product containing the Hyp deletion junction. (A) PCR with primers flanking the putative deletion breakpoints amplified a 518 bp Hyp specific product that was not observed with wild-type (WT) DNA. The left lane contains 2 log DNA marker. (B) DNA sequence of the PCR product from (A) revealed the location of the Hyp deletion junction. Arrows show the X chromosome nucleotide position of the unique sequence flanking the junction. Bold sequence shows AT microsatellite sequence common to both ends of the deletion and underlined sequence represents the proposed junction.
DNA sequencing showed unique sequence ending at nt 153,348,569 and restarting at nt 153,646,285 with 48 base pairs of AT microsatellite sequence common to both breakpoints in between (Figure 2B). This represents a deletion of 297,752 bp, significantly greater than the 53 to 58 kb previously predicted. Despite the greater extent of the deletion, it does not include any genes other than Phex, as the next closest gene (Ptchd1) is located approximately 1.8 Mb from the end of the deletion. This confirms the Hyp mouse phenotype is due solely to a Phex defect and is not a contiguous gene phenotype.
Examination of breakpoints in genomic rearrangements can yield interesting insight into the mechanisms by which they form. While the most common explanations for simple genomic deletions are non-homologous end joining or unequal homologous recombination (reviewed in [9]), the DNA sequence at the Hyp breakpoint junction does not suggest either of these mechanisms. The most parsimonious explanation is microhomology-mediated end joining (MMEJ), a more recently described alternative mechanism of DNA double strand break (DSB) repair (reviewed in [10]). MMEJ is a multistep DSB repair process that begins with resection to expose microhomologous regions at both breakpoints, which allows annealing of single stranded DNA. After error-prone DNA polymerases fill in gaps the processed, annealed ends are ligated and the DSB is repaired, albeit with loss of the interstitial sequence. The defining feature of MMEJ is short tracts of homology between the ends of the deletion, such as the AT microsatellites we observed. MMEJ junctions also usually have inserted nucleotides, introduced by the low fidelity polymerases involved in repair. Here we found a short AAATAA sequence (underlined in Fig 2B) in the otherwise perfect AT microsatellite repeat. This is the likely site of ligation, indicating the true break points are after nucleotide 153,348,586 in intron 15 and before 153,646,258 in the region downstream of the gene.
Having characterised the Hyp deletion, we were able to design a multiplex PCR assay for rapid genotyping. In this assay, a mutant specific primer pair amplifies a product of 346 bp, while a second primer pair located in the deleted region amplifies a product of 234 bp for the WT allele. We used this assay to genotype a series of 11 mice from our Hyp colony and were able to assign genotypes to all mice in this group, validating the assay (Figure 3). This provides a robust, rapid, direct detection of all genotypes, in contrast to existing methods in which a genotype is inferred by absence of amplification or hybridisation [5][7].
Figure 3.
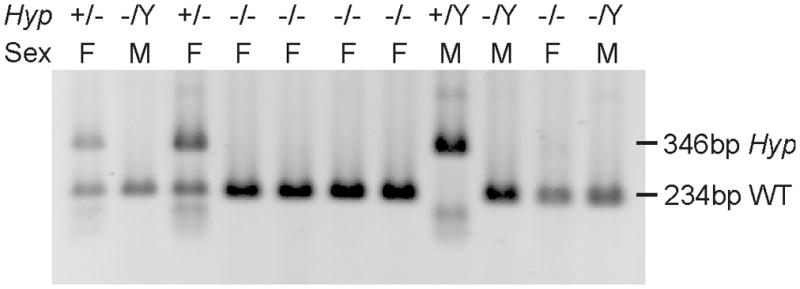
Genotyping PCRs of Hyp mouse colony DNA. Bands at 346 bp and 234 bp represent the Hyp (+) and wild type (−) alleles, respectively. All mice are the offspring of female (F) Hyp +/− (inferred) and male (M) -/Y parents.
The Hyp mouse is a widely used model of human XLH, however the exact extent of the deletion causing this defect was not defined. Here we exploited the X-linked nature of the mutation to clearly map and delineate the Hyp deletion and provide some insight into its mechanism of formation. We also report an improved assay for its direct detection, which will simplify breeding and colony management for this important mouse model of XLH.
Supplementary Material
Highlights.
The deletion in the Hyp mouse model of X-linked hypophosphataemia was fully characterised.
The Hyp deletion was more extensive than previously described but only affects Phex.
A genotyping method for direct detection of the Hyp deletion was established.
Acknowledgments
This work was supported in part by grant AR47908 from the National Institutes of Health, USA.
Abbreviations
- XLH
X-linked hypophosphataemia
- WT
wild-type
- MMEJ
microhomology-mediated end joining
- DSB
double strand break
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Genay O. W. Pilarowski, Email: pilarogo@whitman.edu.
Wei Wang, Email: wwang@sanfordburnham.org.
Jose Luis Millan, Email: millan@sanfordburnham.org.
References
- 1.The HYP Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 2.Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth F R. 2005;16:221–32. doi: 10.1016/j.cytogfr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Eicher EM, Southard JL, Scriver CR, Glorieux FH. Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc Natl Acad Sci U S A. 1976;73:4667–4671. doi: 10.1073/pnas.73.12.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck L, Soumounou Y, Martel J, Krishnamurthy G, Gauthier C, Goodyer CG, et al. Pex/PEX tissue distribution and evidence for a deletion in the 3’ region of the Pex gene in X-linked hypophosphatemic mice. J Clin Invest. 1997;99:1200–1209. doi: 10.1172/JCI119276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strom TM, Francis F, Lorenz B, Boddrich A, Econs MJ, Lehrach H, et al. Pex gene deletions in Gy and Hyp mice provide mouse models for X-linked hypophosphatemia. Hum Mol Genet. 1997;6:165–171. doi: 10.1093/hmg/6.2.165. [DOI] [PubMed] [Google Scholar]
- 6.Sabbagh Y, Gauthier C, Tenenhouse HS. The X chromosome deletion in Hyp mice extends into the intergenic region but does not include the Sat gene downstream from Phex. Cytogenet Genome Res. 2002;99:344–349. doi: 10.1159/000071613. [DOI] [PubMed] [Google Scholar]
- 7.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol-Endoc M. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 8.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 10.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.