

Abstract
IgG4-related disease (IgG4RD) is a novel clinical disease entity characterized by elevated serum IgG4 concentration and tumefaction or tissue infiltration by IgG4-positive plasma cells. IgG4RD may be present in a certain proportion of patients with a wide variety of diseases, including Mikulicz’s disease, autoimmune pancreatitis, hypophysitis, Riedel thyroiditis, interstitial pneumonitis, interstitial nephritis, prostatitis, lymphadenopathy, retroperitoneal fibrosis, inflammatory aortic aneurysm, and inflammatory pseudotumor. Although IgG4RD forms a distinct, clinically independent disease category and is attracting strong attention as a new clinical entity, many questions and problems still remain to be elucidated, including its pathogenesis, the establishment of diagnostic criteria, and the role of IgG4. Here we describe the concept of IgG4RD and up-to-date information on this emerging disease entity.
Keywords: IgG4-related diseases, Mikulicz’s disease, Sjögren’s syndrome, Autoimmune pancreatitis, Castleman’s disease
Introduction
In 1892, Dr. Johann von Mikulicz, also known as Jan Mikulicz-Radecki, published a paper describing a patient with symmetrical swelling of the lachrymal, parotid, and submandibular glands, with massive infiltration of these glands by mononuclear cells [1]. Following reports describing similar patients, this condition was called Mikulicz’s disease (MD). In contrast, patients with similar symptoms, but with diseases such as leukemia, malignant lymphoma, and sarcoidosis, were reported to have Mikulicz’s syndrome [2]. In 1930, Dr. Henrik Sjögren, an ophthalmologist, published a paper describing a woman with rheumatoid arthritis accompanied by keratoconjunctivitis sicca and severe swelling of the parotid glands, a condition that has been recognized as Sjögren’s syndrome (SS) [3]. In 1953, Morgan and Castleman examined 18 patients with MD and concluded that this condition is one manifestation of SS [4]. Since then, MD has attracted very little interest in western countries. In Japan, however, there have been many patients with MD, such that differences between MD and SS have been clarified [5–7]. For example, their gender distribution is quite different, in that MD occurs in both men and women, whereas SS occurs mainly in women. Second, patients with MD have relatively mild xerostomia and xerophthalmia, despite significant enlargement of their lachrymal and salivary glands. Further, MD is accompanied by more complications, such as autoimmune pancreatitis (AIP). Patients with MD show a better response to glucocorticoid therapy than patients with SS. Finally, it has become clear that MD is related to elevated serum IgG4 concentrations and infiltration of IgG4-positive cells [5–9].
Following the description of a patient with chronic pancreatitis due to an autoimmune mechanism [10], lymphoplasmacytic sclerosing pancreatitis (LPSP) was found to be a characteristic histopathological finding in patients with AIP [11]. These findings led to the concept of AIP, which has characteristics similar to those of other autoimmune diseases, such as hypergammaglobulinemia, the presence of various autoantibodies, lymphocytic infiltration into pancreatic tissue, and good responsiveness to steroids [12]. Following a report showing elevated serum IgG4 concentrations in patients with AIP [13], the pancreatic research team of the Ministry of Health, Labor and Welfare Japan (MHLW Japan) showed that AIP was related to IgG4 [14].
IgG4-positive plasma cell infiltration has also been observed in patients with other conditions, including retroperitoneal and mediastinal fibrosis [15, 16], inflammatory pseudotumor of the lung and liver [17], Küttner tumor [18], and interstitial nephritis [19], indicating that these diseases and conditions collectively constitute a new disease concept, IgG4-related disease (Fig. 1). These findings have led to the organization of two study groups by MHLW Japan to analyze the condition of IgG4-related disease. These groups consist of doctors and researchers in various fields, including rheumatology, hematology, gastroenterology, nephrology, pulmonology, ophthalmology, odontology, pathology, statistics, and basic and molecular immunology, from all over Japan. One of these groups, chaired by Professor Umehara of Kanazawa Medical University, is seeking to establish diagnostic criteria for IgG4-related multi-organ lymphoproliferative syndrome (IgG4-MOLPS), whereas the second group, chaired by Professor Okazaki of Kasai Medical University, is seeking to understand the etiology and pathogenesis of IgG4-related systemic disease.
Fig. 1.
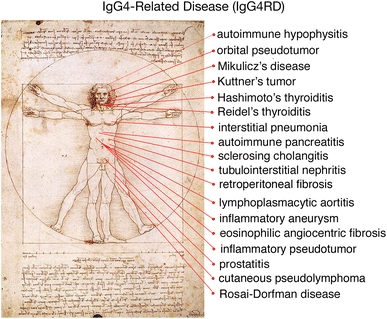
IgG4-related conditions. Many diseases have been reported to be IgG4-related
Unification of different nomenclatures for IgG4-related disease (IgG4RD)
The concept of IgG4RD arose when elevated serum IgG4 concentrations were first reported in patients with sclerosing pancreatitis [13]. Autoimmune pancreatitis (AIP) is also associated with a variety of extrapancreatic lesions, including sclerosing cholangitis, sclerosing sialadenitis, and dacryoadenitis, resulting in the concept of IgG4-related systemic disease [20], also called IgG4-related autoimmune disease [21] or IgG4-related sclerosing disease [15]. The finding of elevated serum IgG4 and IgG4-positive plasma cell infiltration in MD suggested that MD was a systemic disease, which was called systemic IgG4 plasmacytic syndrome (SIPS) [22]. Further, a comparison of patients with MD and those with typical SS resulted in the formulation of a new clinical entity, IgG4+MOLPS [23]. Although many reports from Japan and other countries have described IgG4-related conditions under different names (Table 1), these may refer to the same condition, familial multifocal fibrosclerosis (FMF). Indeed, retroperitoneal fibrosis (RPF), mediastinal fibrosis, sclerosing cholangitis, Riedel’s thyroiditis, and pseudotumor of the orbit may all be different manifestations of a single disease [24].
Table 1.
Nomenclatures of IgG-related conditions
| IgG4-related autoimmune disease | Kamisawa [21] |
| IgG4-associated multifocal systemic fibrosis | van der Vliet [76] |
| IgG4-related systemic disease | Kamisawa [20] |
| IgG4-related sclerosing disease | Kamisawa [15] |
| Hyper-IgG4 disease | Neild [59] |
| IgG4-related disease (IgG4-RD) | Zen [77] |
| Systemic IgG4 plasmacytic syndrome (SIPS) | Yamamoto [22] |
| IgG4-related multi-organ lymphoproliferative syndrome (IgG4-MOLPS) | Masaki [29] |
| IgG4-associated disease | Geyer [78] |
The name “IgG4-related sclerosing disease” is mainly based on the swelling of fibrous organs, such as the pancreas and retroperitoneum, whereas “SIPS” and “IgG4+MOLPS” are based on lymphoplasmacytic proliferation in glands and swollen lymph nodes without fibrosis. Although many patients with this condition (i.e., IgG4-related sclerosing disease, etc.) have lesions in several organs, either synchronously or metachronously, other patients show involvement of only a single organ. At this point, it is unclear whether the pathogenetic mechanism of this disease is systemic or whether it consists of manifestations in individual organs. In addition, several reports have described patients with IgG4-associated conditions concomitant with malignant tumors such as pancreatic [25, 26] and salivary [27] carcinomas, and ocular adnexal lymphoma [28]. Therefore, using the term ‘systemic’ may lead to an incorrect diagnosis of an IgG4-related condition in a patient with malignant tumors in other organs. Based on these reasons, the members of the two MHLW Japan research teams agreed, at their second meeting in Kanazawa on February 11, 2010, to use the term “IgG4-related disease (IgG4RD)”.
General concept of IgG4RD
After the unification of the disease name as IgG4RD, both MHLW Japan research teams have sought to determine its pathogenesis and to formulate diagnostic criteria. The two teams reached a consensus that IgG4RD can occur in various organs, including the central nervous system, salivary glands, thyroid gland, lungs, pancreas, biliary duct, liver, gastrointestinal tract, kidneys, prostate gland, retroperitoneum, and lymph nodes, but that clinical symptoms depend on the location of the lesion. IgG4RD mainly affects middle-aged to elderly men. Its clinical symptoms are relatively mild, and the condition usually comes to clinical attention due to organ swelling or damage. Many patients with IgG4RD are treated effectively by steroid therapy. Although the infiltration of IgG4-positive cells and increased serum concentrations of IgG4 are characteristic of IgG4RD, the severity of fibrosis is dependent on the individual organs involved. For example, storiform fibrosis and obliterative phlebitis are characteristic of pancreatic, biliary tract, and retroperitoneal lesions, but are very seldom found in salivary glands or lymph nodes (Fig. 2).
Fig. 2.
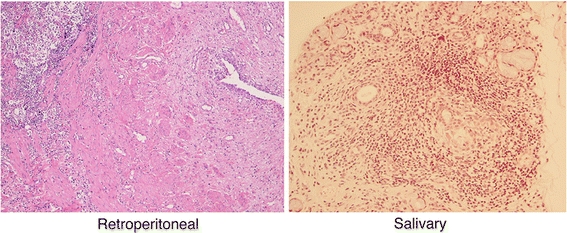
Histopathology of IgG4-related disease (IgG4RD). IgG4RD is characterized histopathologically by the infiltration of IgG4-positive plasma cells and fibrosis. However, the severity of fibrosis is dependent on the individual organs involved. For example, storiform fibrosis and obliterative phlebitis are characteristic of retroperitoneal lesions, but are very seldom observed in salivary glands (×40)
Prevalence of IgG4RD
It is difficult to ascertain the number of patients with IgG4RD because its diagnostic criteria have not yet been established, the awareness of this disease is low, and its symptoms vary. An attempt was made to estimate the number of individuals with IgG4RD throughout Japan by using as an example Ishikawa Prefecture, which has a population of 1.14 million people with little population inflow/outflow (Fig. 3). In Ishikawa Prefecture, there are two University Hospitals, Kanazawa Medical University Hospital (KMU) and Kanazawa University Hospital (KUH). Assuming that new patients with IgG4RD would visit one of these two hospitals, it was estimated that the incidence of this disease throughout Japan would be 0.28–1.08/100,000 population, with 336–1,300 patients newly diagnosed per year. Because the median age of onset of IgG4RD is 58 years and the clinical symptoms are relatively mild, with slow progression and good response to steroid therapy, life expectancy after diagnosis was estimated at 20 years. Thus, an estimated 6,700–26,000 individuals in Japan would have developed IgG4RD over the past 20 years.
Fig. 3.
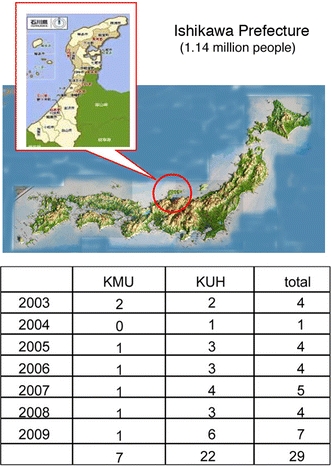
Prevalence of patients with IgG4RD. An attempt was made to estimate the number of individuals with IgG4RD throughout Japan by using as an example Ishikawa Prefecture (population 1.14 million people) with little population inflow/outflow. If all new patients with IgG4RD visit Kanazawa Medical University Hospital (KMU) or Kanazawa University Hospital (KUH), the incidence of this disease throughout Japan would be 0.28–1.08/100,000 population, with 336–1,300 patients newly diagnosed per year. If life expectancy after diagnosis is 20 years, then approximately 6,700–26,000 patients in Japan would have developed IgG4RD over the past 20 years. The numbers in the table represent the numbers of patients who visited KMU or KUH each year
Clinicopathological features of IgG4RD
Differences between IgG4-related MD and Sjögren’s syndrome
Since elevated serum IgG4 was first reported in patients with MD [6], the members of the Japanese Society of Sjögren’s Syndrome have assessed the clinical symptoms, laboratory findings, and detailed histopathology in patients with MD (characterized by symmetrical swelling of the lachrymal, submandibular, and parotid glands), nationwide, since 2004. Some patients did not show typical symptoms of MD such as swelling of the lachrymal, parotid, or submandibular glands but showed elevated serum IgG4 and other indices indicative of MD according to the criteria for the diagnosis of IgG4-related MD shown in Table 2 [8]. Sixty-four patients with MD or elevated serum IgG4 (>135 mg/dl) and characteristic histological findings were initially diagnosed with IgG4RD (formerly called IgG4+MOLPS) based on proposed guidelines for the diagnosis of IgG4RD (Table 3). A comparison of patients with IgG4RD and those with typical SS showed: (1) compared with SS patients, fewer patients with IgG4RD had symptoms of xerophthalmia, xerostomia, or arthralgia, whereas many had coexisting AIP, interstitial nephritis, allergic rhinitis, and/or bronchial asthma (Fig. 4a); (2) most patients with IgG4RD were negative for anti-SS-A and anti-SS-B antibodies, as well as for rheumatoid factor (RF) and anti-nuclear antibody (ANA) (Fig. 4b); (3) serum IgG4 and IgE concentrations were significantly higher in IgG4RD than in SS patients (Fig. 4c); and (4) steroid therapy was extremely effective in patients with IgG4RD but had limited effect in patients with SS [29].
Table 2.
Diagnostic criteria of IgG4+ Mikulicz’s disease [8] (approved by the Japanese Society for Sjögren’s Syndrome 2008)
| 1. Symmetrical swelling of at least 2 pairs of lachrymal, parotid, or submandibular glands for at least 3 months |
| AND |
| 2. Elevated serum IgG4 (>135 mg/dl) |
| OR |
| 3. Histopathological features including lymphocyte and IgG4+ plasma cell infiltration (IgG4+ plasma cells/IgG+ plasma cells >50%) with typical tissue fibrosis or sclerosis |
| Differential diagnosis is necessary to distinguish IgG4+ Mikulicz’s disease from other distinct disorders, including sarcoidosis, Castleman’s disease, Wegener’s granulomatosis, lymphoma, and cancer. The diagnostic criteria for Sjögren’s syndrome (SS) may also include some patients with IgG4+ Mikulicz’s disease; however, the clinicopathological conditions of patients with typical SS and IgG4+ Mikulicz’s disease are different |
Table 3.
Guidelines for diagnosis of IgG4RD (proposed by the Research Program for Intractable Disease Ministry of Health, Labor and Welfare Japan, G4 team)
| Clinical features highly suggestive of IgG4RD |
| 1. Symmetrical swelling of lachrymal, parotid, or submandibular glands |
| 2. Autoimmune pancreatitis |
| 3. Inflammatory pseudotumor |
| 4. Retroperitoneal fibrosis |
| 5. Suspicion of Castleman’s disease |
| Laboratory data highly suggestive of IgG4RD |
| 1. Serum IgG4 >135 mg/dl |
| 2. IgG4+ cells/IgG+ cells >40% in biopsy |
| Clinical features suggestive of IgG4RD |
| 1. Unilateral swelling of at least one lachrymal, parotid, or submandibular gland |
| 2. Orbital pseudotumor |
| 3. Sclerosing cholangitis |
| 4. Prostatitis |
| 5. Hypertrophic pachymeningitis |
| 6. Interstitial pneumonitis |
| 7. Interstitial nephritis |
| 8. Thyroiditis/hypo-function of thyroid |
| 9. Hypophysitis |
| 10. Inflammatory aneurysm |
| Laboratory data suggestive of IgG4RD |
| 1. Hypergammaglobulinemia of unknown origin |
| 2. Hypocomplementemia or existence of immune complex |
| 3. Increase of IgE or eosinophils |
| 4. Tumefactive lesions or lymph node swelling detected by gallium scan or fluoro-D-glucose positron emission tomography (FDG-PET) |
Fig. 4.
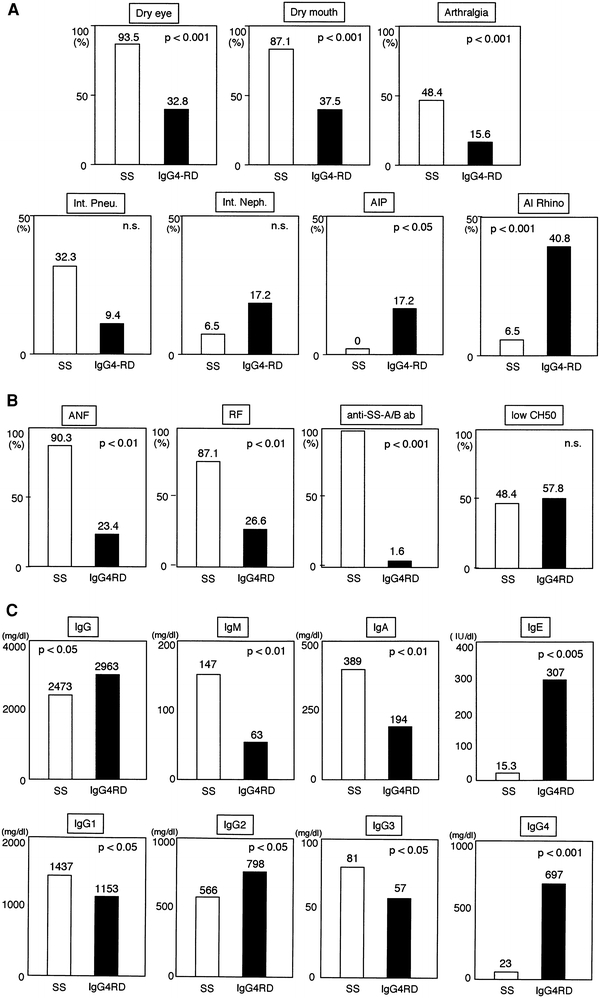
Comparison of clinical symptoms and laboratory findings in IgG4RD and typical Sjögren’s syndrome (SS) [29]. a Clinical symptoms, b immunological findings, and c subclasses of immunoglobulins and IgG observed in patients with IgG4RD (n = 61) and typical SS (n = 31). Data are expressed as percentages. P values are for comparisons of IgG4RD with typical SS. Patients with typical SS fulfilled both Japanese and European SS criteria and were positive for both anti-SSA/Ro and anti-SSB/La antibodies
The histopathological features of IgG4RD are unique, though both IgG4RD and SS show marked lymphocytic infiltration. IgG4RD is characterized by the formation of lymphoid follicles but lower levels of lymphocytic infiltration into the salivary ducts, such that their structure remains intact (Fig. 5a). Therefore, the absence of lymphoepithelial lesions in patients with IgG4RD, in contrast to SS, may explain the lower rate of dryness in the former, despite the marked swelling of lachrymal and salivary glands. The most important difference between IgG4RD and SS is that the former is characterized by marked infiltration of IgG4-positive plasma cells, with a ratio of IgG4-positive to IgG-positive cells of >40%, a finding almost never seen in patients with SS (Fig. 5b). Moreover, most patients with IgG4RD show polyclonal B-cell proliferation, with equal staining for immunoglobulin κ- and λ-chains (Fig. 5c). Thus, despite their similarities in organ involvement, IgG4-MD and SS are quite different conditions, with distinct clinical and pathological characteristics [7–9, 22, 29–31].
Fig. 5.
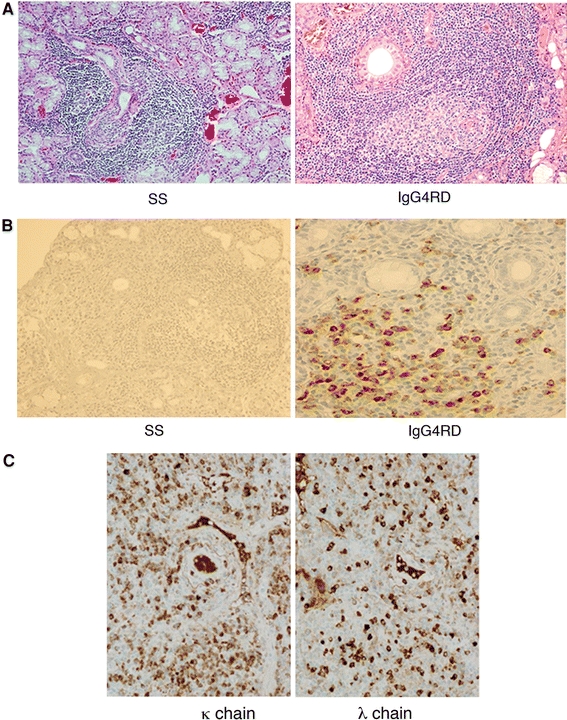
Histopathological findings of minor labial salivary gland biopsies in patients with IgG4RD and typical SS. a Massive infiltration of lymphocytes and plasma cells was observed in patients with IgG4RD and those with typical SS (×200). IgG4RD, however, was characterized by lymphoid follicle formation but ducts were intact without lymphocytic infiltration. H&E staining. b IgG4RD showed scattered IgG4+ plasma cells in the periphery of the follicles (×200), whereas typical SS showed few or no IgG4+ cells. IgG4 immunostaining. c Staining for immunoglobulin κ- and λ-chains (×200)
IgG4-related Küttner tumor
Küttner tumor, a unilateral sclerosing sialadenitis, is an IgG4RD [18]. A common feature of MD and Küttner tumor is that both manifest sialadenitis, as in IgG4RD. Histologically, Küttner tumors are very severe fibrous sclerotic lesions containing IgG4-positive plasma cells [32]. In contrast, fibrosis tends to be less severe in MD, although fibrosis in MD is frequently not examined extensively, because MD is generally diagnosed by the biopsy of minor labial salivary glands. Therefore, at present, it is difficult to set a strict boundary between MD and Küttner tumor.
IgG4-related autoimmune pancreatitis (IgG4-related AIP)
Recent studies have suggested that AIP manifests as two distinct subtypes, called types 1 and 2 (Table 4) [33]. Clinically, type 1 AIP seems to be the pancreatic manifestation of IgG4RD, characterized by: (1) mild abdominal symptoms, usually without acute attacks of pancreatitis; (2) occasional occurrence of obstructive jaundice; (3) increased serum gammaglobulin, IgG, and/or IgG4 concentrations; (4) presence of autoantibodies; (5) diffuse enlargement of the pancreas with a capsule-like low-density rim; (6) irregular narrowing of the pancreatic duct (sclerosing pancreatitis on endoscopic retrograde cholangiopancreatography [ERCP] images); (7) lymphocyte and IgG4-positive plasmacyte infiltration and fibrosis, and obliterative phlebitis; (8) occasional association with extrapancreatic lesions, such as sclerosing cholangitis similar to primary sclerosing cholangitis (PSC), sclerosing cholecystitis, sclerosing sialoadenitis, RPF, interstitial renal tubular disorders, enlarged celiac and hilar lymph nodes, chronic thyroiditis, and pseudotumor of the pancreas, liver, or lung; and (9) responsiveness to steroid therapy. Older males with IgG-related AIP often have obstructive jaundice, with both pancreatic and extrapancreatic manifestations responding to steroid therapy [12–15, 21, 33, 34].
Table 4.
Subtypes of autoimmune pancreatitis (AIP) [33]
| Subtype of AIP other nomenclatures | Type 1 AIP without GEL IgG4-related, LPSP | Type 2 AIP with GEL IgG4-unrelated IDCP |
|---|---|---|
| Prevalence | Asia > USA, Europe | Europe > USA > Asia |
| Age | High age | Younger |
| Gender | Male ≫ female | Male = female (NS) |
| Symptoms | Often obstructive jaundice | Often obstructive jaundice |
| Jaundice | Rare abdominal pain | Abdominal pain like acute pancreatitis |
| Pancreas images | Swelling/diffuse | Swelling/diffuse |
| Segmental/focal | Segmental/focal | |
| Mass-forming | Mass-forming | |
| Serology | High serum IgG | Normal IgG |
| High serum IgG4 | Normal IgG4 | |
| Auto antibodies (+) | Auto antibodies (−) | |
| Other organ involvement (OOI) | Sclerosing cholangitis | Unrelated to OOI |
| Sclerosing sialadenitis | ||
| Retroperitoneal fibrosis | ||
| Other characteristics | ||
| Ulcerative colitis | Rare | Often |
| Steroid response | Responsive | Responsive |
| Relapse | High rate | Rare |
GEL, granulocyte epithelial lesion; LPSP, lymphoplasmacytic sclerosing pancreatitis; IDCP, idiopathic duct-centric chronic pancreatitis; NS, not significant
Histological examination by American and European pathologists of the resected pancreases of patients with chronic non-alcoholic pancreatitis revealed another histopathological pattern, called idiopathic duct-centric pancreatitis (IDCP) or AIP with granulocytic epithelial lesions (GELs), later called type 2 AIP [35, 36]. Type 2 AIP is characterized primarily by these GELs, often accompanied by destruction and obliteration of the pancreatic duct [36]. Patients with type 2 AIP show swelling of the pancreas, but no or very few IgG4-positive plasma cells. Type 2 AIP has different clinicopathological features than type 1 AIP. Type 2 AIP shows no elevations in serum IgG4 or IgG, no autoantibodies, and no involvement of other organs, except for inflammatory bowel disease. Inflammatory bowel disease has been observed in approximately 30% of patients with type 2 AIP. Although type 1, or IgG4-related, AIP (LPSP type) often occurs in older men and is accompanied by a variety of extrapancreatic lesions, type 2, or neutrophil-related pancreatitis (IDCP/GEL type), has no gender bias, younger age at onset (often <40 years), and is frequently associated with inflammatory bowel disease. Thus, after a worldwide debate over the diagnostic criteria for AIP, IgG4-related pancreatitis has been defined as type 1 (LPSP type) and neutrophil-related pancreatitis has been defined as type 2 (IDCP/GEL type) [34].
IgG4-related sclerosing cholangitis (IgG4-related SC)
Extrapancreatic bile duct lesions are frequently associated with AIP. For example, 73% of patients with AIP have shown wall thickening or sclerosing changes in extrapancreatic bile ducts on endoscopic ultrasonography (EUS) and intraductal ultrasonography (IDUS), though only 26% of patients with AIP demonstrated sclerosing changes by ERCP [37]. However, many individuals without AIP have shown IgG4-related SC with isolated biliary tract involvement [38, 39]. In IgG4-related SC, stenosis is usually observed in the lower part of the common bile duct. The cholangiographic appearance of stenosis in the intrahepatic or hilar hepatic bile duct is very similar to that observed in PSC [40], a progressive disease of unknown etiology that ultimately results in liver cirrhosis. IgG4-related SC is associated with older age, male predominance, obstructive jaundice, weight loss, and abdominal discomfort [40]. Although steroid therapy has shown mixed results in patients with PSC, IgG4-related SC responds dramatically to steroid therapy, as does IgG4RD [41]. The histopathological features of IgG4-related SC are similar to those of AIP and include diffuse plasmacytic infiltration, marked interstitial fibrosis with a focal storiform-like pattern, and obliterative phlebitis.
IgG4-related kidney disease (IgG4-related KD)
The kidney is a frequent target organ in IgG4RD, with tubulointerstitial nephritis (TIN) and fibrosis and abundant IgG4-positive plasma cell infiltration being diagnostically important histopathological features of this disease [42–44]. Recently, the clinicopathological features of 23 patients with IgG4-related TIN were reported to be quite uniform and similar to those observed in patients with IgG4-AIP, including high serum concentrations of IgG4 and IgE, hypocomplementemia, and TIN with infiltration of large numbers of IgG4-positive plasma cells plus fibrosis [45].
Kidney diseases in IgG4RD include conditions other than renal parenchymal lesions, such as hydronephrosis associated with RPF and tumors of the renal pelvis and urethra. However, IgG4-related TIN is considered to be representative of IgG4 renal parenchymal lesions [19]. Compared with other types of interstitial nephritis, IgG4-related TIN is often associated with extrarenal lesions, such as pancreatitis, sialadenitis, and lymphadenitis, and a high incidence of hypocomplementemia [46]. Imaging often shows heterogeneous shadows in the kidneys, such as a mass or multiple nodules (findings that are not observed in other types of interstitial nephritis). Histopathologically, the renal tubulointerstitium shows the infiltration of many lymphocytes and plasmacytes, as well as fibrosis, and IgG4 immunostaining shows a number of IgG4-positive plasma cells [47]. Although many studies have found no significant changes in the glomeruli, others have reported an association with glomerular lesions, including membranous nephropathy [46]. In the near future, the Japanese Kidney Society expects to develop diagnostic criteria for IgG4-related KD.
IgG4-related pulmonary diseases (IgG4-related PD)
IgG4-related PD has been described as inflammatory pseudotumor, interstitial pneumonitis, organizing pneumonia, and lymphomatoid granulomatosis [48]. Most (81%) patients with IgG4-related PD have been reported to be men, with a median age at diagnosis of 69 years [48], features similar to those of IgG4RD. Some patients present initially with respiratory symptoms, such as dry cough or dyspnea, whereas 75% of patients are asymptomatic and the disease is found incidentally by abnormal shadows on chest X-rays. Although IgG4-related PD is associated with a variety of radiologic abnormalities [49], diffuse lymphoplasmacytic infiltration has been observed in all lesions, with irregular fibrosis and obliterative vascular changes being more common in solid areas [48]. Hilar and pancreatic accumulation of gallium-67 has been reported as characteristic of the active stage of AIP when serum IgG4 concentrations are high [50].
Radiographically, IgG4-related PD can be divided into two types, inflammatory pseudotumors and interstitial pneumonitis. Inflammatory pseudotumors have been described as nodular or mass lesions, or infiltration, and are characterized by radiating reticular shadows surrounding the tumor. Interstitial pneumonitis presents in most patients with reticular shadows, ground-glass opacity, and interstitial fibrosis in both lower lung fields [17].
Histopathologically, inflammatory pseudotumor is a plasma cell granuloma, with infiltration mainly by plasma cells and lymphocytes, irregular fibrosis, lymphoid follicle formation, findings of interstitial pneumonitis at the periphery of the nodule, obliterating phlebitis and arteritis, and eosinophilic infiltration [17]. Interstitial pneumonitis is characterized by thickening of the alveolar septa due to infiltration by plasma cells and lymphocytes, and by diffuse fibrosis. Histopathologically, interstitial pneumonitis often shows a pattern previously classified as non-specific interstitial pneumonia (NSIP) [51]. The diagnostic criteria for IgG4-related PD are now under consideration by the Japanese Respiratory Association.
IgG4-related Hashimoto’s thyroiditis (IgG4-related HT)
Hashimoto’s thyroiditis (HT) has been considered a well-defined clinicopathological entity, characterized by the presence of goiter and serum thyroid autoantibodies. Recently, a unique subtype of HT was described, characterized by the presence of prominent fibrosis such as storiform fibrosis and swirling fibrosis, numerous IgG4-positive plasma cells, and elevated serum IgG4 [52], and called IgG4-related HT [53]. Among 23 patients with HT who underwent total thyroidectomy, 14 cases (60.8%) were IgG4-related HT, but there were no significant differences in positivity for thyroid and microsome tests between IgG4-related HT and non-IgG4 HT [54].
Riedel’s thyroiditis was first described in 1896 in two patients with hard goiter and tracheal compressive symptoms. One-third of patients with Riedel’s thyroiditis have multifocal fibrosclerosis, including sclerosing cholangitis, salivary gland fibrosis, RPF, or fibrotic orbital pseudotumor. Therefore, despite the lack of immunohistochemical staining for IgG4, certain proportions of Riedel’s thyroiditis were considered a type of IgG4RD. Although one patient with IgG4RD showed involvement of the lachrymal gland and pulmonary and biliary tracts as well as Riedel’s thyroiditis [32], it is still unclear whether Riedel’s thyroiditis is a type of IgG4RD.
IgG4-related lymphadenopathy and Castleman’s disease
Concomitant lymphadenopathy is common in patients with IgG4RD, and there have been several reports dealing with the morphological and immunohistological findings of lymph node lesions [55–57]. Although IgG4-related lymphadenopathy is occasionally characterized by systemic lymphadenopathy, polyclonal hyperimmunoglobulinemia, especially elevated IgG and IgE concentrations, and positivity for various autoantibodies, patients with IgG4RD with generalized lymphadenopathy should only be evaluated for lymphoma, sarcoidosis, multicentric Castleman’s disease, and other malignancies.
IgG4-related lymphadenopathy can be characterized into five histological subtypes: Castleman’s disease-like morphology (type I), reactive follicular hyperplasia (type II), interfollicular plasmacytosis and immunoblastosis (type III), progressive transformation of germinal center-like (type IV), and inflammatory pseudotumor-like morphology (type V) [57]. In addition, IgG4-related lymphadenopathy can be classified into two types based on the infiltrative patterns of IgG4-positive cells: interfollicular plasmacytosis (types I, II, III, and V) and intragerminal center plasmacytosis (type IV). Patients with systemic IgG4-related lymphadenopathy were significantly older (68.8 vs. 43.3 years) and had significantly lower C-reactive protein (0.29 vs. 8.71 mg/dl) and interleukin (IL)-6 (8.45 vs. 34.82 pg/ml) concentrations than patients with multicentric Castleman’s disease [56].
IgG4-related retroperitoneal fibrosis (IgG4-related RPF)
RPF is a chronic inflammatory condition with marked fibrosis in retroperitoneal tissue. In patients with advanced RPF a retroperitoneal mass covers the abdominal aorta and compresses the ureters, leading to urinary obstruction. Its etiology is unknown, but it has many causes, including infection, radiation, drugs, malignant tumor, and trauma. Three patients with RPF and elevated serum IgG4 have been described [58], and the histology of all 12 patients with RPF was reported to be similar to that seen in AIP, including fibrosis, intense inflammatory cell infiltration with plasma cells, venulitis, and obliterative arteritis [59]. Of 17 patients with RPF, 10 had both elevated serum IgG4 and histopathological features typical of IgG4RD, suggesting that RPF could be categorized as IgG4-related [60]. However, in RPF, fibrosis gradually progresses during chronic inflammation, with lymphocyte infiltration predominant during the early stages and a fibroinflammatory process occurring later. Therefore, determining the stage of illness seems important for diagnosis and prediction of response to steroid treatment [61].
IgG4-related aortitis
There have been several recent reports of inflammatory aneurysms in the abdominal or thoracic aorta [62–64]. For example, 40% of inflammatory abdominal aortic aneurysms (AAAs) were IgG4RD, with elevated IgG4 in serum and abundant infiltration of IgG4+ plasma cells and obliterative phlebitis [62]. These findings suggested that inflammatory AAAs can be classified into 2 groups: IgG4-related and IgG4-unrelated [62]. Although IgG4RD shows good response to steroid therapy, treatment with the anti-CD20 monoclonal antibody, rituximab, may result not only in clinical improvement, but in the tapering or discontinuation of steroids or other drugs [65].
Pathogenesis and pathophysiology of IgG4RD
At present, the pathogenetic mechanism and underlying immunological abnormalities in IgG4RD remain unclear. The elevated serum IgG4 concentration and tissue infiltration of IgG4-positive plasma cells are characteristic features of IgG4RD. Because IgG4 antibodies are dynamic molecules that can exchange Fab arms by swapping a heavy chain and attached light chain, IgG4 can form bi-specific antibodies, as well as functioning as a monovalent molecule [66, 67]. These properties may protect against type I allergy by inhibiting IgE functions, and may prevent type II and III allergy by blocking the Fc-mediated effector functions of IgG1 and inhibiting the formation of large immune complexes. The predominant expression of IgG4 under conditions of chronic antigen exposure is compatible with the clinical features of IgG4RD, including its slow progression and relatively weak immune response.
Some autoantibodies, including those to pancreatic trypsin inhibitor (PSTI), lactoferrin (LF), and carbonic anhydrase (CA), have been detected in patients with IgG4RD, especially in those with IgG4-related AIP [34]. Although IgG4 from the patients was able to bind the normal epithelia of the pancreatic ducts, gallbladder, and salivary gland ducts [68], IgG4-type autoantibodies have not been detected in patients with IgG4RD.
Aberrant immunological findings have been observed in patients with IgG4RD. For example, the Th2-dominant immune response and the production of Th2-type cytokines, such as IL-4, IL-5, IL-10, and IL-13, are increased [69–71]. Furthermore, the numbers of regulatory T cells (Treg) expressing CD4+CD25+Foxp3 are significantly higher in the affected tissues and peripheral blood of patients with IgG4RD than the numbers in patients with autoimmune and nonautoimmune diseases [72–74]. Overexpression of the regulatory cytokines IL-10 and transforming growth factor β (TGF-β) has also been reported in patients with IgG4RD [74, 75]. IL-10 and TGF-β have potent activities in directing B cells to produce IgG4 and induce fibroplasia, respectively. IL-4, IL-5, and IL-13 are important for class switching to IgE production and eosinophil migration. Therefore, abnormalities in the production of these cytokines may be involved in the pathogenesis of IgG4RD.
Perspectives on IgG4RD
Although IgG4RD is a novel clinical entity, it is not a rare disease. Despite the effectiveness of steroid therapy, for IgG4RD, the condition has often been misdiagnosed as a malignant tumor, lymphoma, Sjögren’s syndrome, or other diseases. To date, the clinical diagnostic criteria for IgG4RD have not been established. Because IgG4RD may occur in a variety of organs throughout the body, comprehensive discussions with the cooperation of many clinicians from various specialized fields is needed to establish uniform diagnostic criteria. At present, the diagnostic criteria for IgG4-MD (Table 2) [8] and those for IgG4-AIP type 1 (Table 5) [14] have been established.
Table 5.
Clinical diagnostic criteria of autoimmune pancreatitis; revised proposal in Japan (2006) [79]
| 1. Diffuse or segmental narrowing of the main pancreatic duct with irregular wall and diffuse or localized enlargement of the pancreas on imaging modalities, such as abdominal ultrasound (US), computed tomography (CT), and magnetic resonance imaging (MRI) |
| 2. High-serum F-globulin, IgG, or IgG4, or the presence of autoantibodies, such as antinuclear antibodies and rheumatoid factor |
| 3. Marked interlobular fibrosis and prominent infiltration of lymphocytes and plasma cells into the periductal area, with occasional lymphoid follicles in the pancreas |
| For diagnosis, criterion 1 must be present, together with criteria 2 and/or 3 |
| However, it is necessary to exclude malignant diseases such as pancreatic and biliary cancers |
Consensus has been reached on two diagnostic criteria for IgG4RD: (1) serum IgG4 concentration >135 mg/dl, and (2) >40% of IgG-positive plasma cells being IgG4-positive. The MHLW Japan team has proposed guidelines for the diagnosis of IgG4RD; these are shown in Table 3. The formulation of organ-specific (i.e., kidney and pulmonary) diagnostic criteria for IgG4RD requires cooperation with the relevant societies. Although IgG4RD responds well to steroid therapy, recurrence and relapse occur following the early reduction or withdrawal of prednisone. Therefore, it is necessary to develop treatment guidelines to establish initial doses of steroids, tapering procedures, and maintenance doses. The MHLW Japan team is currently pursuing a “Prospective study for creating IgG4-related disease treatment guidelines”, and unified clinical guidelines are expected in the near future.
Acknowledgments
This work was supported by the Research Program for Intractable Diseases, Health and Labor Sciences Research Grants from the Ministry of Health, Labor and Welfare, Japan. We sincerely thank the many contributing researchers and collaborators who participated in the MHLW Japan G4 team.
Conflict of interest None.
Glossary
- IgG4RD
IgG4-related disease
- MD
Mikulicz’s disease
- SS
Sjögren’s syndrome
- MHLW Japan
Ministry of Health, Labor and Welfare Japan
- LPSP
Lymphoplasmacytic sclerosing pancreatitis
- AIP
Autoimmune pancreatitis
- FMF
Familial multifocal fibrosclerosis
- ANA
Anti-nuclear antibody
Appendix
The authors thank the many patients who participated in this registry. In addition to the listed authors, other professional collaborators in the Research Program for Intractable Disease supported by the Ministry of Health, Labor and Welfare (MHLW) Japan G4 team, include: Keita Fujikawa (Isahaya Hospital); Hideaki Hamano, Keiji Kubo, and Hiroshi Yamamoto (Shinshu University); Mitsuyosi Hirokawa (Kuma Hospital); Kunihiko Itoh (Shizuoka Prefectural University); Terumi Kamisawa (Tokyo Metropolitan Research Institute); Daisuke Kawabata (Kyoto University); Morio Matsumoto (Nishi Gunma Hospital); Seijiro Minamoto (Osaka Respiratory and Allergy Center); Kayoko Murayama (Gunma Cancer Institute); Susumu Nishiyama (Kurashiki Hospital); Yoko Ogawa (Keio University); Tomoki Origuchi (Nagasaki University); Norihide Oyama (Niigata University); Yasuharu Sato (Okayama University); Masao Seto (Aichi Cancer Center); Susumu Sugai (Kudou Hospital); Norifumi Tsukamoto (Gunma University); Masayuki Takahira (Kanazawa University); Hiroki Takahashi (Sapporo Medical University); Hiroto Tsuboi (Tsukuba University); Yuko Waseda (Kanazawa University); and Kazuko Kitagawa, Takayuki Nojima, Hitoshi Yokoyama, Hisao Tonami, Toshihiro Fukushima, Masao Tanaka, Yoshimasa Fujita, Toshioki Sawaki, Takafumi Kawanami, Miyuki Miki, Haruka Iwao, Akio Nakajima, and Takuji Nakamura (Kanazawa Medical University).
Footnotes
H. Umehara and K. Okazaki contributed equally to this work.
Names of the collaborators of The Research Program for Intractable Disease by Ministry of Health, Labor and Welfare (MHLW) Japan G4 team are listed in Appendix.
References
- 1.Mikulicz J. Uber Eine Eigenartige Symmetrishe Erkrankung Der Tranen Und Mundspeicheldrusen. Stuttgart: BeitrzChirFesrschrf Theodor Billroth; 1982. p. 610–30 (in German).
- 2.Schaffer JM, Tilley FW. Further investigations of the relation between the chemical constitution and the germicidal activity of alcohols and phenols. J Bacteriol. 1927;14(4):259–273. doi: 10.1128/jb.14.4.259-273.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sjogren H. Keratoconjunctivitis sicca and chronic polyarthritis. Acta Med Scand. 1948;30:484–488. [PubMed] [Google Scholar]
- 4.Morgan WS, Castleman B. A clinicopathologic study of Mikulicz’s disease. Am J Pathol. 1953;29:471–503. [PMC free article] [PubMed] [Google Scholar]
- 5.Tsubota K, Fujita H, Tadano K, Onoda N, Tsuzaka K, Takeuchi T. Abnormal expression and function of Fas ligand of lacrimal glands and peripheral blood in Sjögren’s syndrome patients with enlarged exocrine glands. Clin Exp Immunol. 2002;129:177–182. doi: 10.1046/j.1365-2249.2002.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto M, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, Takahashi H, et al. Elevated IgG4 concentrations in serum of patients with Mikulicz’s disease. Scand J Rheumatol. 2004;33:432–433. doi: 10.1080/03009740410006439. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, et al. A new conceptualization for Mikulicz’s disease as an IgG4-related plasmacytic disease. Mod Rheumatol. 2006;16(6):335–340. doi: 10.1007/s10165-006-0518-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masaki Y, Sugai S, Umehara H. IgG4-related diseases including Mikulicz’s disease and sclerosing pancreatitis: diagnostic insights. J Rheum. 2010;37:1380–1385. doi: 10.3899/jrheum.091153. [DOI] [PubMed] [Google Scholar]
- 9.Masaki Y, Iwao H, Nakajima A, Miki M, Sugai S, Umehara H. IgG4-related disease (IgG4+MOLPS)—diagnostic criteria and diagnostic problems. Curr Immunol Rev. 2011;7:172–177. [Google Scholar]
- 10.Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis. 1961;6:688–698. doi: 10.1007/BF02232341. [DOI] [PubMed] [Google Scholar]
- 11.Kawaguchi K, Koike M, Tsuruta K, Okamoto A, Tabata I, Fujita N. Lymphoplasmacytic sclerosing pancreatitis with cholangitis: a variant of primary sclerosing cholangitis extensively involving pancreas. Hum Pathol. 1991;22(4):387–395. doi: 10.1016/0046-8177(91)90087-6. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561–1568. doi: 10.1007/BF02285209. [DOI] [PubMed] [Google Scholar]
- 13.Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–738. doi: 10.1056/NEJM200103083441005. [DOI] [PubMed] [Google Scholar]
- 14.Okazaki K, Kawa S, Kamisawa T, Itoh T, Inui K, Irie H, et al. Japanese clinical guideline for autoimmune pancreatitis. Pancreas. 2009;38:849–866. doi: 10.1097/MPA.0b013e3181b9ee1c. [DOI] [PubMed] [Google Scholar]
- 15.Kamisawa T, Nakajima H, Egawa N, et al. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology. 2006;6:132–137. doi: 10.1159/000090033. [DOI] [PubMed] [Google Scholar]
- 16.Zen Y, Sawazaki A, Miyayama S, et al. A case of retroperitoneal and mediastinal fibrosis exhibiting elevated levels of IgG4 in the absence of sclerosing pancreatitis (autoimmune pancreatitis) Hum Pathol. 2006;37:239–243. doi: 10.1016/j.humpath.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Zen Y, Kitagawa S, Minato H, Kuramaya H, Katayanagi K, Masuda S, et al. IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol. 2005;36:710–717. doi: 10.1016/j.humpath.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 18.Kitagawa S, Zen Y, Harada K, Sasaki M, Sato Y, Minato H, et al. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Küttner’s tumor) Am J Surg Pathol. 2005;29:783–791. doi: 10.1097/01.pas.0000164031.59940.fc. [DOI] [PubMed] [Google Scholar]
- 19.Saeki T, Nishi S, Ito T, Yamazaki H, Miyamura S, Emura I, et al. Renal lesions in IgG4-related systemic disease. Intern Med. 2007;46(17):1365–1371. doi: 10.2169/internalmedicine.46.0183. [DOI] [PubMed] [Google Scholar]
- 20.Kamisawa T, Funata N, Hayashi Y. Lymphoplasmacytic sclerosing pancreatitis is a pancreatic lesion of IgG4-related systemic disease. Am J Surg Pathol. 2004;28(8):1114. doi: 10.1097/01.pas.0000126634.43301.45. [DOI] [PubMed] [Google Scholar]
- 21.Kamisawa T, Funata N, Hayashi Y, Eishi Y, Koike M, Tsuruta K, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982–984. doi: 10.1007/s00535-003-1175-y. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Takahashi H, Hasebe K, Suzuki C, Naishiro Y, Hayashi T, et al. The analysis of interleukin-6 in patients with systemic IgG4-related plasmacytic syndrome–expansion of SIPS to the territory of Castleman’s disease. Rheumatology (Oxford) 2009;48:860–862. doi: 10.1093/rheumatology/kep098. [DOI] [PubMed] [Google Scholar]
- 23.Masaki Y, Umehara H. IgG4-related disease–the diagnostic confusion and how to avoid it. Jpn J Clin Immunol. 2009;32:478–483. doi: 10.2177/jsci.32.478. [DOI] [PubMed] [Google Scholar]
- 24.Comings D, Skubi K, Eyes J, Motulsky A. Familial multifocal fibrosclerosis. Findings suggesting that retroperitoneal fibrosis, mediastinal fibrosis, sclerosing cholangitis, Riedel’s thyroiditis, and pseudotumor of the orbit may be different manifestations of a single disease. Ann Intern Med. 1967;66:884–892. doi: 10.7326/0003-4819-66-5-884. [DOI] [PubMed] [Google Scholar]
- 25.Kamisawa T, Chen PY, Tu Y, Nakajima H, Egawa N, Tsuruta K, et al. Pancreatic cancer with a high serum IgG4 concentration. World J Gastroenterol. 2006;12(38):6225–6228. doi: 10.3748/wjg.v12.i38.6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Witkiewicz AK, Kennedy EP, Kennyon L, Yeo CJ, Hruban RH. Synchronous autoimmune pancreatitis and infiltrating pancreatic ductal adenocarcinoma: case report and review of the literature. Hum Pathol. 2008;39(10):1548–1551. doi: 10.1016/j.humpath.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 27.Gill J, Angelo N, Yeong ML, McIvor N. Salivary duct carcinoma arising in IgG4-related autoimmune disease of the parotid gland. Hum Pathol. 2009;40(6):881–886. doi: 10.1016/j.humpath.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Cheuk W, Yuen H, Chan A, Shih L, Kuo T, Ma M, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:1159–1167. doi: 10.1097/PAS.0b013e31816148ad. [DOI] [PubMed] [Google Scholar]
- 29.Masaki Y, Dong L, Kurose N, Kitagawa K, Morikawa Y, Yamamoto M, et al. Proposal for a new clinical entity, IgG4-positive multi-organ lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis. 2009;63:1310–1315. doi: 10.1136/ard.2008.089169. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto M, Takahashi H, Ohara M, Suzuki C, Naishiro Y, Yamamoto H, et al. A case of Mikulicz’s disease (IgG4-related plasmacytic disease) complicated by autoimmune hypophysitis. Scand J Rheumatol. 2006;35:410–411. doi: 10.1080/03009740600758110. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto M, Takahashi H, Sugai S, Imai K. Clinical and pathological characteristics of Mikulicz’s disease (IgG4-related plasmacytic exocrinopathy) Autoimmun Rev. 2005;4(4):195–200. doi: 10.1016/j.autrev.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Dahlgren M, Khosroshahi A, Nielsen GP, Deshpande V, Stone JH. Riedel’s thyroiditis and multifocal fibrosclerosis are part of the IgG4-related systemic disease spectrum. Arthritis Care Res (Hoboken) 2010;62(9):1312–1318. doi: 10.1002/acr.20215. [DOI] [PubMed] [Google Scholar]
- 33.Okazaki K, Uchida K, Miyoshi H, Ikeura T, Takaoka M, Nishio A. Recent concepts of autoimmune pancreatitis and IgG4-related disease. Clin Rev Allergy Immunol 2011. doi:10.1007/s12016-010-8214-2. [DOI] [PubMed]
- 34.Okazaki K, Uchida K, Koyabu M, Miyoshi H, Takaoka M. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J Gastroenterol. 2011;46(3):277–288. doi: 10.1007/s00535-011-0386-x. [DOI] [PubMed] [Google Scholar]
- 35.Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol. 2003;27(8):1119–1127. doi: 10.1097/00000478-200308000-00009. [DOI] [PubMed] [Google Scholar]
- 36.Zamboni G, Luttges J, Capelli P, Frulloni L, Cavallini G, Pederzoli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004;445(6):552–563. doi: 10.1007/s00428-004-1140-z. [DOI] [PubMed] [Google Scholar]
- 37.Hamano H, Arakura N, Muraki T, Ozaki Y, Kiyosawa K, Kawa S. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol. 2006;41(12):1197–1205. doi: 10.1007/s00535-006-1908-9. [DOI] [PubMed] [Google Scholar]
- 38.Hamano H, Kawa S, Uehara T, Ochi Y, Takayama M, Komatsu K, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc. 2005;62(1):152–157. doi: 10.1016/S0016-5107(05)00561-4. [DOI] [PubMed] [Google Scholar]
- 39.Zen Y, Harada K, Sasaki M, Sato Y, Tsuneyama K, Haratake J, et al. IgG4-related sclerosing cholangitis with and without hepatic inflammatory pseudotumor, and sclerosing pancreatitis-associated sclerosing cholangitis: do they belong to a spectrum of sclerosing pancreatitis? Am J Surg Pathol. 2004;28(9):1193–1203. doi: 10.1097/01.pas.0000136449.37936.6c. [DOI] [PubMed] [Google Scholar]
- 40.Kamisawa T, Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol. 2008;14(25):3948–3955. doi: 10.3748/wjg.14.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghazale A, Chari ST, Zhang L, Smyrk TC, Takahashi N, Levy MJ, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134(3):706–715. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 42.Uchiyama-Tanaka Y, Mori Y, Kimura T, Sonomura K, Umemura S, Kishimoto N, et al. Acute tubulointerstitial nephritis associated with autoimmune-related pancreatitis. Am J Kidney Dis. 2004;43(3):e18–e25. doi: 10.1053/j.ajkd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Takeda S, Haratake J, Kasai T, Takaeda C, Takazakura E. IgG4-associated idiopathic tubulointerstitial nephritis complicating autoimmune pancreatitis. Nephrol Dial Transplant. 2004;19(2):474–476. doi: 10.1093/ndt/gfg477. [DOI] [PubMed] [Google Scholar]
- 44.Saeki T, Saito A, Yamazaki H, Emura I, Imai N, Ueno M, et al. Tubulointerstitial nephritis associated with IgG4-related systemic disease. Clin Exp Nephrol. 2007;11(2):168–173. doi: 10.1007/s10157-007-0464-9. [DOI] [PubMed] [Google Scholar]
- 45.Saeki T, Nishi S, Imai N, Ito T, Yamazaki H, Kawano M, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78(10):1016–1023. doi: 10.1038/ki.2010.271. [DOI] [PubMed] [Google Scholar]
- 46.Saeki T, Imai N, Ito T, Yamazaki H, Hishi S. Membranous nephropathy associated with IgG4-related systemic disease and without autoimmune pancreatitis. Clin Nephrol. 2009;2:173–178. doi: 10.5414/cnp71173. [DOI] [PubMed] [Google Scholar]
- 47.Saeki T, Nishi S, Ito T, Yamazaki H, Miyamura S, Emura I, et al. Renal lesion in IgG4-related systemic disease. Intern Med. 2007;46:1365–72. [DOI] [PubMed]
- 48.Zen Y, Inoue D, Kitao A, Onodera M, Abo H, Miyayama S, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33(12):1886–1893. doi: 10.1097/PAS.0b013e3181bd535b. [DOI] [PubMed] [Google Scholar]
- 49.Inoue D, Zen Y, Abo H, Gabata T, Demachi H, Kobayashi T, et al. Immunoglobulin G4-related lung disease: CT findings with pathologic correlations. Radiology. 2009;251(1):260–270. doi: 10.1148/radiol.2511080965. [DOI] [PubMed] [Google Scholar]
- 50.Saegusa H, Momose M, Kawa S, Hamano H, Ochi Y, Takayama M, et al. Hilar and pancreatic gallium-67 accumulation is characteristic feature of autoimmune pancreatitis. Pancreas. 2003;27(1):20–25. doi: 10.1097/00006676-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 51.Takato K, Yasui M, Ichikawa Y, Fujimura M, Nakao S, Zen Y, et al. Nonspecific interstitial pneumonia with abundant IgG4-positive cells infiltration, which was thought as pulmonary involvement of IgG4-related autoimmune disease. Intern Med. 2008;47:291–294. doi: 10.2169/internalmedicine.47.0411. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Bai Y, Liu Z, Ozaki T, Taniguchi E, Mori I, et al. Immunohistochemistry of IgG4 can help subclassify Hashimoto’s autoimmune thyroiditis. Pathol Int. 2009;59(9):636–641. doi: 10.1111/j.1440-1827.2009.02419.x. [DOI] [PubMed] [Google Scholar]
- 53.Kakudo K, Li Y, Hirokawa M, Ozaki T. Diagnosis of Hashimoto’s thyroiditis and IgG4-related sclerosing disease. Pathol Int. 2011;61(4):175–183. doi: 10.1111/j.1440-1827.2011.02661.x. [DOI] [PubMed] [Google Scholar]
- 54.Kojima M, Hirokawa M, Kuma H, Nishihara E, Masawa N, Nakamura N, et al. Distribution of IgG4- and/or IgG-positive plasma cells in Hashimoto’s thyroiditis: an immunohistochemical study. Pathobiology. 2010;77(5):267–272. doi: 10.1159/000319873. [DOI] [PubMed] [Google Scholar]
- 55.Cheuk W, Yuen HK, Chu SY, Chiu EK, Lam LK, Chan JK. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32(5):671–681. doi: 10.1097/PAS.0b013e318157c068. [DOI] [PubMed] [Google Scholar]
- 56.Sato Y, Kojima M, Takata K, Morito T, Asaoku H, Takeuchi T, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol. 2009;22(4):589–599. doi: 10.1038/modpathol.2009.17. [DOI] [PubMed] [Google Scholar]
- 57.Sato Y, Notohara K, Kojima M, Takata K, Masaki Y, Yoshino T. IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int. 2010;60(4):247–258. doi: 10.1111/j.1440-1827.2010.02524.x. [DOI] [PubMed] [Google Scholar]
- 58.Hamano H, Kawa S, Ochi Y, Unno H, Shiba N, Wajiki M, et al. Hydronephrosis associated with retroperitoneal fibrosis and sclerosing pancreatitis. Lancet. 2002;359(9315):1403–1404. doi: 10.1016/S0140-6736(02)08359-9. [DOI] [PubMed] [Google Scholar]
- 59.Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med. 2006;4:23. doi: 10.1186/1741-7015-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zen Y, Onodera M, Inoue D, Kitao A, Matsui O, Nohara T, et al. Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol. 2009;33(12):1833–1839. doi: 10.1097/PAS.0b013e3181b72882. [DOI] [PubMed] [Google Scholar]
- 61.Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23(1):57–66. doi: 10.1097/BOR.0b013e3283418057. [DOI] [PubMed] [Google Scholar]
- 62.Kasashima S, Zen Y, Kawashima A, Konishi K, Sasaki H, Endo M, et al. Inflammatory abdominal aortic aneurysm: close relationship to IgG4-related periaortitis. Am J Surg Pathol. 2008;32(2):197–204. doi: 10.1097/PAS.0b013e3181342f0d. [DOI] [PubMed] [Google Scholar]
- 63.Stone JH, Khosroshahi A, Hilgenberg A, Spooner A, Isselbacher EM, Stone JR. IgG4-related systemic disease and lymphoplasmacytic aortitis. Arthritis Rheum. 2009;60(10):3139–3145. doi: 10.1002/art.24798. [DOI] [PubMed] [Google Scholar]
- 64.Ishida F, Kitano K, Kobayashi H, Saito H, Kiyosawa K. Elevated IgG4 levels in a case with multicentric Castleman’s disease. Br J Haematol. 1997;99(4):981–982. [PubMed] [Google Scholar]
- 65.Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62(6):1755–1762. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 66.Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002;105(1):9–19. doi: 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van der Nuet Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317(5844):1554–1557. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 68.Aoki S, Nakazawa T, Ohara H, Sano H, Nakao H, Joh T, et al. Immunohistochemical study of autoimmune pancreatitis using anti-IgG4 antibody and patients’ sera. Histopathology. 2005;47(2):147–158. doi: 10.1111/j.1365-2559.2005.02204.x. [DOI] [PubMed] [Google Scholar]
- 69.Miyake K, Moriyama M, Aizawa K, Nagano S, Inoue Y, Sadanaga A, et al. Peripheral CD4+ T cells showing a Th2 phenotype in a patient with Mikulicz’s disease associated with lymphadenopathy and pleural effusion. Mod Rheumatol. 2008;18(1):86–90. doi: 10.1007/s10165-007-0010-3. [DOI] [PubMed] [Google Scholar]
- 70.Akitake R, Watanabe T, Zaima C, Uza N, Ida H, Tada S, et al. Possible involvement of T helper type 2 responses to Toll-like receptor ligands in IgG4-related sclerosing disease. Gut. 2010;59(4):542–545. doi: 10.1136/gut.2009.200972. [DOI] [PubMed] [Google Scholar]
- 71.Suzuki K, Tamaru J, Okuyama A, Kameda H, Amano K, Nagasawa H, et al. IgG4-positive multi-organ lymphoproliferative syndrome manifesting as chronic symmetrical sclerosing dacryo-sialadenitis with subsequent secondary portal hypertension and remarkable IgG4-linked IL-4 elevation. Rheumatology (Oxford) 2010;49(9):1789–1791. doi: 10.1093/rheumatology/keq113. [DOI] [PubMed] [Google Scholar]
- 72.Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobin G4-related sclerosing pancreatitis and cholangitis. Hepatology. 2007;45(6):1538–1546. doi: 10.1002/hep.21697. [DOI] [PubMed] [Google Scholar]
- 73.Miyoshi H, Uchida K, Taniguchi T, Yazumi S, Matsushita M, Takaoka M, et al. Circulating naive and CD4+CD25 high regulatory T cells in patients with autoimmune pancreatitis. Pancreas. 2008;36(2):133–140. doi: 10.1097/MPA.0b013e3181577553. [DOI] [PubMed] [Google Scholar]
- 74.Zen Y, Nakanuma Y. Pathogenesis of IgG4-related disease. Curr Opin Rheumatol. 2011;23(1):114–118. doi: 10.1097/BOR.0b013e3283412f4a. [DOI] [PubMed] [Google Scholar]
- 75.Nakashima H, Miyake K, Moriyama M, Tanaka A, Watanabe M, Abe Y, et al. An amplification of IL-10 and TGF-beta in patients with IgG4-related tubulointerstitial nephritis. Clin Nephrol. 2010;73(5):385–391. doi: 10.5414/cnp73385. [DOI] [PubMed] [Google Scholar]
- 76.Vliet HJ, Perenboom RM. Multiple pseudotumors in IgG4-associated multifocal systemic fibrosis. Ann Intern Med. 2004;141(11):896–897. doi: 10.7326/0003-4819-141-11-200412070-00033. [DOI] [PubMed] [Google Scholar]
- 77.Zen Y, Fujii T, Sato Y, Masuda S, Nakanuma Y. Pathological classification of hepatic inflammatory pseudotumor with respect to IgG4-related disease. Mod Pathol. 2007;20(8):884–894. doi: 10.1038/modpathol.3800836. [DOI] [PubMed] [Google Scholar]
- 78.Geyer JT, Ferry JA, Harris NL, Stone JH, Zukerberg LR, Lauwers GY, et al. Chronic sclerosing sialadenitis (Kuttner tumor) is an IgG4-associated disease. Am J Surg Pathol. 2010;34(2):202–210. doi: 10.1097/PAS.0b013e3181c811ad. [DOI] [PubMed] [Google Scholar]
- 79.Okazaki K, Kawa S, Kamisawa T, Naruse S, Tanaka S, Nishimori I, et al. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol. 2006;41(7):626–631. doi: 10.1007/s00535-006-1868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]