

Summary
Carbon monoxide (CO) is produced during the catabolism of free haem, catalyzed by haem oxygenase (HO) enzymes, and its physiological roles include vasodilation, neurotransmission, inhibition of platelet aggregation and anti-proliferative effects on smooth muscle. In vivo preclinical studies have shown that exogenously administered quantities of CO may represent an effective treatment for conditions characterized by a dysregulated immune response. The carbon monoxide-releasing molecules (CORMs) represent a group of compounds capable of carrying and liberating controlled quantities of CO in the cellular systems. This review covers the physiological and anti-inflammatory properties of the HO/CO pathway in the central nervous system. It also discusses the effects of CORMs in preclinical models of inflammation. The accumulating data discussed herein support the possibility that CORMs may represent a novel class of drugs with disease-modifying properties in multiple sclerosis.
Keywords: carbon monoxide, CORMs, haem oxygenase, multiple sclerosis
Introduction
Carbon monoxide (CO) is a low-molecular-weight diatomic gas that is generated from the oxidation of organic materials such as wood, natural gas, coal and tobacco. Already in 1857, it was reported that CO could bind haemoglobin molecules. Indeed, CO binds about 200 times more effectively to haemoglobin than oxygen, resulting in the formation of carboxyhaemoglobin (COHb). The subsequent decrease in oxygen release to the tissues ultimately leads to death. CO has long been considered only a dangerous poisonous gas. However, Sjoestrand observed in 1949 that CO was an endogenous metabolic product and that its rate of production is elevated in pathological events, such as haemolysis [1]. More than 86% of the endogenously produced CO originates from haem degradation, while the remainder arises from other biological processes including lipid peroxidation and xenobiotic metabolism. However, it was only in the late 1960s that Tenhunen and colleagues first linked CO production to haem oxygenase (HO) activity [2].
In humans, the production rate of CO is 16·4 µmol/h, with a daily production of CO reaching more than 500 µmol [3]. However, based on normal levels of COHb of 1–2%, average physiological concentrations of CO in living tissues are in the nanomolar range.
The direct evidence for the relevance of the HO1/CO pathway to different homeostatic pathways came from the first human case of HO-1 deficiency described in Japan in 1999 [4]. This HO-1-deficient patient showed leucocytosis, anaemia, elevated serum levels of ferritin and haem, lower serum levels of bilirubin, growth retardation, thrombocytosis and hyperlipidaemia, and died at the age of 6 years.
Several in-vivo preclinical studies have shown that exogenously administered quantities of CO may represent an effective treatment for conditions characterized by a dysregulated inflammatory response, including endotoxaemia [5], collagen-induced arthritis [6,7], trinitrobenzene sulphonic acid-induced colitis [8], acute pancreatitis [9], lung injury [10], chronic rejection and arteriosclerotic lesions [11], improving clinical and immunological parameters.
In order to examine the therapeutic potential of CO, three approaches have been used: direct administration of CO by inhalation of the gas; inductors of HO-1 expression; and CO delivery via metallo-organic carbonyl compounds [12]. These molecules, known as carbon monoxide-releasing molecules (CORMs), represent a group of compounds capable of carrying and liberating controlled quantities of CO in cellular systems, thus overcoming the limitations of gaseous CO [12].
Different CORMs show different pharmacokinetics under physiological conditions, and are able to achieve safe and therapeutically effective thresholds of CO without delivering potentially toxic amounts through the respiratory system [13].
Endogenous production of CO
The majority of CO in the mammalian organism is produced by enzymatic haem metabolism, which occurs in the reticulo-endothelial system of the spleen and liver [14] and is catalyzed by HO (Fig. 1). Three isoforms of HO have been characterized, among which HO-1 represents the inducible isoform. Increased cellular stress levels are known to up-regulate de-novo transcription of HO-1 [15]. Conversely, both HO-2 and HO-3 are expressed constitutively in many mammalian cells.
Fig. 1.
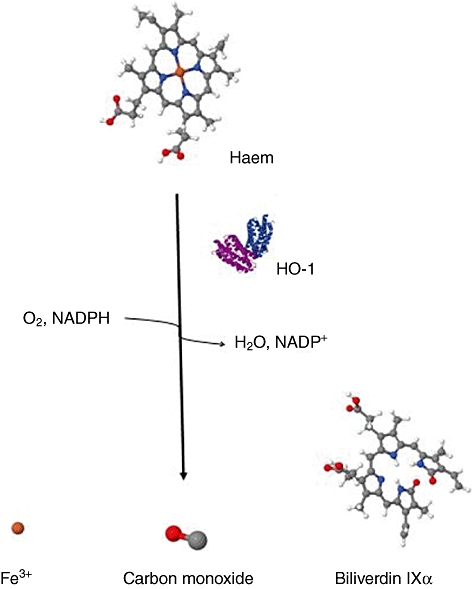
Haem metabolism pathway. In the presence of haem oxygenase enzymes, the porphyrin ring of haem is oxidized producing equimolar amounts of carbon monoxide (CO), ferrous iron and biliverdin. This pathway also requires nicotinamide adenine dinucleotide phosphate (NADPH) and O2.
Consensus binding sites for oxidative stress-responsive transcription factors, including nuclear factor-κB (NF-κB), activator protein-1 (AP-1), AP-2, Sp1, upstream stimulatory factor, c-myc/max and interleukin (IL)-6 response elements have been found in the promoter region of the human ho-1 gene [16–18], thus suggesting a potential role for these factors in modulating HO-1 expression.
HO-1, also known as heat shock protein 32 (HSP32), can be induced by several factors including haem and haem derivatives, heat shock, heavy metals, NO, oxidized lipids, hyperoxia, lipopolysaccharides, phorbol ester, sodium arsenite [19], radiation, ultraviolet, hydrogen peroxide, hypoxia, endotoxin, platelet-derived growth factor (PDGF), transforming growth factor (TGF)-β, electrophiles, okadaic acid, methylglyoxal [20], curcumin [21], oxidative stress [22], cytokines [IL-1, IL-6, IL-10, tumour necrosis factor (TNF)-α, interferon (IFN)-γ], shear stress [23], intensive light, angiotensin II, glucose deprivation and by exogenous CO [24,25]. HO-1 induction seems to be mediated by different signalling pathways, including cyclic adenosine-5′-monophosphate (cAMP)-dependent mechanisms [26], protein kinase C (PKC), Ca2-calmodulin-dependent protein kinase and the phosphoinositol pathway [27]. Mitogen-activated protein kinases (ERK and P38) and tyrosine phosphorylation have been also involved in HO-1 induction in some tissues [28–30].
Physiological roles for CO
CO stimulates soluble guanylate cyclase (sGC) activity in vitro by binding to the ferrous haem moiety of the enzyme [31], probably by forming a hexaco-ordinate complex. Given the relatively low affinity of CO for the haem-iron of sGC, CO signalling might become relevant under oxidative stress or pathophysiological conditions where HO-1 is significantly induced. The mobilization of CO for signalling may be regulated by the availability of the substrate haem for enzymatic degradation, and the availability of active HO enzymes, a process which, in turn, may be orchestrated by the transcriptional activation of the ho-1 gene by stress. Indeed, ultraviolet A (UVA) irradiation or hydrogen peroxide treatment of human skin fibroblasts leads to an immediate release of haem from microsomal haem proteins [32].
Several physiological roles for CO have been reported which directly involve the modulation of cGMP levels, including vasodilation, neurotransmission, inhibition of platelet aggregation and anti-proliferative effects on smooth muscle [31–33]. Overexpression of HO-1 in pigs inhibits phenylephrine-induced vasoconstriction in isolated aortic rings. Furthermore, the effects of HO-1 expression were subject to inhibition by ZnPPIX. The induction of guanylate cyclase in cultured olfactory neurones by olfactory stimulants can be inhibited by metalloporphyrin inhibitors of HO such as ZnPPIX [34]. Body hyperthermia or elevation of cGMP levels in the heart correlate with the transcriptional induction of HO-1. cGMP increase following hypoxia is associated with HO-1 elevation, and it can be inhibited by SnPPIX and the CO scavenger haemoglobin [32]. Both exogenously applied CO or hypoxia-induced CO exert anti-proliferative effects on vascular smooth muscle cells (VSMC), which is associated with elevation of cGMP, and inhibition of transcription factor E2F, a regulator of cell cycle control [33]. Transfection of VSMC with an adenovirus vector bearing the ho-1 gene (AdHO-1) stimulated cGMP production, and inhibited cell proliferation in vitro by G1/G0 arrest [34].
Anti-inflammatory properties of CO
At the concentration range of 100–500 parts per million (ppm), CO differentially and selectively inhibited the expression of lipopolysaccharide (LPS)-induced proinflammatory cytokines TNF-α, interleukin-1β and macrophage inflammatory protein-1β and increased the LPS-induced expression of the anti-inflammatory cytokine IL-10 from macrophages [35] and the secretion of IL-2 from activated T cells [41]. The anti-inflammatory effect of CO is mediated by p38 kinase, but independent of the cGMP pathway [35]. In turn, IL-10 up-regulates HO-1 expression with consequent increase in CO production [36]. In this manner, CO self-amplifies its anti-inflammatory effect.
The anti-inflammatory effect of endogenous CO is related closely to the expression of HO-1. Indeed, HO-1 null mice show enlarged spleen and lymph nodes, high white blood cell counts and high splenic and lymph node CD4 : CD8 T cell ratios with numerous activated CD4 T cells, thus indicating a chronic inflammatory condition [37].
One important mechanism by which the HO-1/CO system might mediate its anti-inflammatory effect is the down-regulation of adhesion molecules, i.e. selectin, integrin and the immunoglobulin superfamily [38]. Induction of HO-1 has indeed found to down-regulate the H2O2-induced P-selectin expression, with consequent decreased leucocyte binding [39].
The anti-inflammatory and cytoprotective effects elicited by CO are mediated mainly by the activation of the mitogen-activated protein kinase (MAPK) and the nuclear factor (NF)-κB pathway. It has been reported that exposing macrophages to 250 ppm of CO leads to reduced production of LPS-induced granulocyte macrophage colony-stimulating factor (GM-CSF) [40]. This effect was mediated by a decreased phosphorylation of Ik-Bα and consequently by the inhibition of the transcription factor NF-κB. Also, in activated CD4 T cells, CO inhibited extracellular-regulated kinase (ERK) phosphorylation and suppressed cell proliferation [41].
These anti-inflammatory effects of CO were confirmed in vivo in a model of endotoxaemia where mice were injected with LPS with or without CO pretreatment. CO dose-dependently inhibited LPS-induced serum TNF-α levels and increased IL-10 production. These effects seemed to be mediated by MAPK kinase 3 (MKK3), as CO failed to down-regulate serum TNF-α levels or up-regulate IL-10 levels in LPS-injected MKK3−/− mice.
The above-mentioned data suggest that the anti-inflammatory effect exerted by CO involves the MKK3/p38 MAPK pathway.
CORMs
Although the use of small amounts of CO gas in medicine might be feasible [42], gaseous compounds are generally difficult to deliver to organisms in an accurate and safe manner. CORMs represent a good alternative to CO gas both from a pharmaceutical perspective and in terms of specificity of action. The preferential reactivity of CO towards metals allows to bind CO covalently to transition metal carbonyls in order to obtain agents aimed at delivering controlled amounts of CO to tissues and organs under appropriate conditions [13]. In fact, CO liberated from CORMs can be delivered precisely at given concentrations through all possible routes of administration, unlike CO gas, which can be delivered effectively only by inhalation. Also, it is evident that HbCO concentrations do not increase to life-threatening levels when CORMs are used at doses that are pharmacologically effective [43,44].
The first in class compound is the dimethylsulphoxide (DMSO)-soluble metal carbonyl complex tricarbonyldichlororuthenium(II) dimer {[Ru(CO)3Cl2]2}, known as CORM-2. This compound, when in contact with reduced deoxymyoglobin or deoxyhaemoglobin rapidly transfers ca. one CO equivalent to the protein haem forming carboxymyoglobin (COMb) or carboxyhaemoglobin (COHb), respectively. CORM-3 {[Ru(CO)3Cl(glycinate]} and CORM-A1 [Na2(H3BCO2)] are instead water-soluble CO-releasing agents [46], with different chemical structures and properties. In the same reaction with deoxymyoglobin, CORM-3 has a short half-life (<1 min), while CORM-A1, a boron-containing carboxylic acid, releases CO in a pH-dependent manner which under physiological conditions attains a half-life of 21 min [13]. The pharmacokinetic differences of these two compounds explain why CORM-3 elicits a prompt and rapid vasodilatory effect, whereas CORM-A1 promotes mild vasorelaxation and hypotension [45]. (Fig. 2)
Fig. 2.
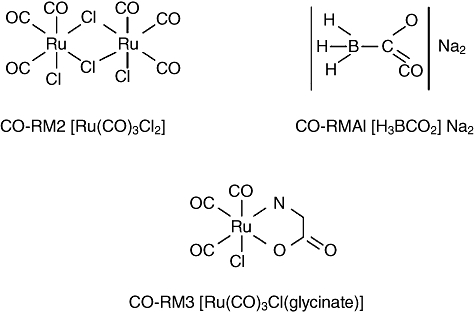
Chemical structure of carbon monoxide (CO)-releasing molecules (CORMs). The first in class CORM compound is the lipid-soluble metal carbonyl complex tricarbonyl dichlororuthenium(II) dimer ([Ru(CO)3Cl2]2), known as CORM2. CORM3 and {[Ru(CO)3Clglycinate]} and CORMA1 [Na2(H3bCO2] are instead examples of water-soluble CO-releasing agents with different chemical structures and pharmacokinetics.
The effects of these CORMs include cardioprotection against both ischaemia and myocardial infarction [46]; reduction of cardiac graft rejection and positive inotropic effects on the heart [46,47]; attenuation of the acute inflammatory response and amelioration of neuroinflammatory responses in microglia [48–50]; reduction of histamine release from guinea pig mast cells and human neutrophils [51]; anti-hypertensive effects and inhibition of platelet aggregation [52]; vasodilatation and anti-apoptotic effects in the cerebral circulation [53,54]; reduction of hepatic leucocyte sequestration and systemic inflammatory response during severe burn injury [55]; mitigation of photocarcinogenesis in the skin [56]; and improved kidney function following cold ischaemia occurring during organ preservation and protection against cisplatin-induced nephrotoxicity [57,58].
The HO1 and CO axis in inflammatory and degenerative events of the CNS: friends or foes?
HO-2 is expressed widely in the central nervous system (CNS) where it is present in mitral cells in the olfactory bulb, pyramidal cells in the cortex and hippocampus [34], as well as in granule cells of the dentate gyrus, in the thalamus and hypothalamus, cerebellum and caudal brainstem. It is thought that HO-2 may protect these neurones from oxidative stress by reducing lipid peroxidation through the catabolism of free haem. Conversely, HO-1 expression has been found in both neuronal and non-neuronal cells of the forebrain, diencephalon, cerebellum and brainstem regions. The relevance of HO-1 expression, however, becomes more evident in certain pathological conditions, such as Alzheimer's disease (AD), Parkinson's disease and multiple sclerosis (MS). Both neuronal and non-neuronal CNS cells rapidly up-regulate HO-1 in response to stress [59]. Upon cerebral ischaemia, transgenic mice that overexpress HO-1 in neurones exhibit diminished tissue staining for lipid peroxidation end-products, enhanced expression of the anti-apoptotic factor bcl-2 and reduced infarct volumes in comparison with wild-type mice [60]. Glial HO-1 expression has also been shown in traumatic brain injury animal models to contribute to neuroprotection. Also, cerebellar granule cells collected from HO-1 transgenic mice appear to be relatively resistant to glutamate and H2O2-related oxidative stress [61].
Increased HO-1 expression has been reported in AD brains in association with neurofibrillary tangles [62,63], senile plaques and glial fibrillary acidic protein-positive astrocytes [64]. This HO-1 up-regulation could be induced by the increased free haem levels associated with neuronal death and may represent a compensatory mechanism to convert haem into the anti-oxidant molecules, biliverdin and bilirubin [64]. Furthermore, substantia nigral dopaminergic neurones of Parkinson's disease patients exhibit up-regulated HO-1 in response to oxidative stress [65].
Oligodendrocytes are extremely sensitive to oxidative stress [66] mediated by reactive oxygen species (ROS) that arise from extensive elaborations of membranes [66] because of the relatively low levels of anti-oxidant defences, such as superoxide dismutase and catalase [67]. Conversely, experimental evidences show that ROS are a key feature of inflammatory demyelinating disease. Indeed, ROS are produced massively both in experimental autoimmune encephalomyelitis (EAE) and MS [68] and damage to lipid membranes by ROS has been observed in EAE and MS [69]. A recent study by Dallas and co-workers has also gained insight into the possible mode of action by which CO may exert its protective effects against oxidant-induced apoptosis in inflammatory and degenerative events of the CNS [70]. An early step in this process is the loss of intracellular K(+) via K(+) channels, and evidence indicates that K(v)2·1 is of particular importance in this regard, being inserted rapidly into the plasma membrane in response to apoptotic stimuli. The authors demonstrate that CO reversibly inhibits K(v)2·1. Channel inhibition by CO involves ROS and protein kinase G activity. Overexpression of K(v)2·1 in HEK293 cells increases their vulnerability to oxidant-induced apoptosis, and this is reversed by CO. In hippocampal neurones, CO selectively inhibits K(v)2·1, reverses the dramatic oxidant-induced increase in K(+) current density and provides marked protection against oxidant-induced apoptosis. Hence, these results provide a novel mechanism to account for the neuroprotective effects of CO against oxidative apoptosis, which has potential for therapeutic exploitation to provide neuronal protection in situations of oxidative stress.
However, other experimental evidences suggest that the up-regulated expression of HO1 in inflammatory and degenerative pathologies of the CNS may represent a pathogenetic event (Fig. 3).
Fig. 3.
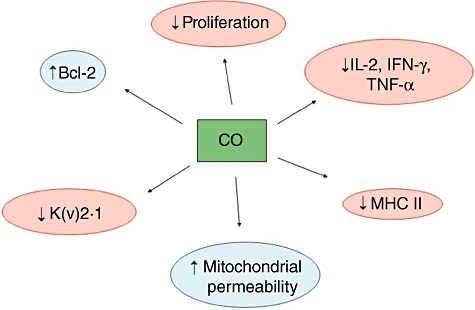
Potential mechanisms of action of carbon monoxide (CO) in neuroinflammation and neurodegenerative disorders. The beneficial effects of CO seems to occur at multiple cellular and molecular levels, including redox control, modulation of proliferation and of the immune response.
Mehindate et al. [71] measured HO-1 mRNA levels and mitochondrial uptake of [(55)Fe]Cl(3)-derived iron in rat astroglial cultures exposed to IL-1β or TNF-α alone or in combination with the HO-1 inhibitors, tin mesoporphyrin (SnMP) or dexamethasone (DEX), or IFN-β. HO-1 expression in astrocytes was evaluated by immunohistochemical staining of spinal cord tissue derived from MS and control subjects. IL-1β or TNF-α promoted sequestration of non-transferrin-derived (55)Fe by astroglial mitochondria. HO-1 inhibitors, mitochondrial permeability transition pore (MTP) blockers and anti-oxidants significantly attenuated cytokine-related mitochondrial iron sequestration in these cells. IFN-β decreased HO-1 expression and mitochondrial iron sequestration in IL-1β- and TNF-α-challenged astroglia. The percentage of astrocytes co-expressing HO-1 in affected spinal cord from MS patients (57·3% ± 12·8%) was significantly greater (P < 0·05) than in normal spinal cord derived from control subjects (15·4% ± 8·4%). These data indicate that HO-1 is over-expressed in MS spinal cord astroglia and may promote mitochondrial iron deposition in MS plaques and that, in MS, IFN-β may attenuate glial HO-1 gene induction and aberrant mitochondrial iron deposition accruing from exposure to proinflammatory cytokines.
Stahnke et al. [71] have analysed autopsy and biopsy brain samples of patients with MS and acute disseminated leucoencephalomyelitis (ADEM) and spinal cord lesions of mouse EAE, which was induced actively by immunization with myelin oligodendrocyte glycoprotein (MOG35-55) peptide, for the presence of HO-1. HO-1 was observed in glial cells during different stages: (i) during acute phases of mainly inflammatory diseases (EAE and ADEM) expression of HO-1 was prominent in microglia/macrophages and astrocytes and up-regulation correlated with inflammation; and (ii) in early MS lesions HO-1 was expressed in oligodendrocytes. Furthermore, in glial cell cultures, they have shown that up-regulation of HO-1 in oligodendrocytes was paralleled by severe morphological damage. Oligodendrocytes underwent apoptotic cell death at a concentration of hydrogen peroxide (50–200 µM), which did not affect astrocytes or microglia. Using oligodendroglial OLN-93 cells, they demonstrated that oxidative stress led to mitochondrial impairment and the disorganization of the microtubule network. Zinc protoporphyrin, an inhibitor of HO-1, augmented the cytotoxic consequences of hydrogen peroxide in OLN-93 cells. Based on these data the authors suggest that stress-induced HO-1 initially plays a protective role, while its chronic up-regulation might contribute to oligodendroglial cell death rather than providing protection [71].
Can the HO1 and CO axis represent potential therapeutic targets in MS?
MS is an autoimmune-mediated disease that affects the CNS and is characterized by inflammatory lesions, demyelination and axonal loss. Common symptoms of MS include weakness, fatigue, sensory impairment and poor cognition [72]. Currently, first-line therapy for different clinical forms of MS includes type I interferons (IFN-β) and glatiramer acetate (Copaxone), natalizumab, cyclophosphamide, mitoxantrone and, more recently, fingolimod. These drugs have different effects on the function and proliferation of macrophage and T cells, influence their migration to the CNS and seem to shift the cytokine secretory profile towards a type 2, type 3 profile [73–75]. However, their effectiveness is still limited and the relapse rates and the number of new lesions are reduced by only 30–40%. In addition, most of these drugs have several side effects and are costly, and prolonged treatment with IFN-β and natalizumab induce production of neutralizing antibodies that limit their therapeutic efficacy. EAE, the animal model for MS, can be induced in susceptible rodent strains by active immunization of myelin antigens and has been used largely as an in-vivo tool to dissect pathogenetic mechanisms of the human disease counterpart as well as to provide proof of concept studies for the in-vivo efficacy of test compounds that may be worthy of development for the clinical setting. Interestingly, all current therapies approved for MS ameliorate the EAE disease course [76]. Thus, as new therapeutic interventions for MS are strongly required, the EAE models offer a practical tool for evaluating the effectiveness of novel putative pharmacological approaches.
Neuroinflammation shares several similarities with inflammation in peripheral tissues and, indeed, increased levels of HO-1, linked probably to a physiological anti-inflammatory and cytoprotective response have been observed in the CNS in MS [71,77] and EAE [78]. In agreement with this hypothesis, Liu et al. [78] have shown high expression of HO-1 in lesions of EAE, and demonstrated that haemin, an inducer of HO-1, significantly inhibited EAE. In contrast, SnMP treatment, which inhibits HO-1, markedly exacerbated the clinical course of EAE. These data suggest that endogenous HO-1 plays an important protective role in EAE, and that targeted induction of HO-1 overexpression may represent a novel treatment option for MS.
Chora et al. [79] have provided clear-cut evidence that HO-1 orchestrates the pathological outcome of EAE. Induction of EAE in Hmox1(−/−) mice leads to increased CNS demyelination, paralysis and mortality compared with wild-type mice. Induction of HO-1 by cobalt protoporphyrin IX (CoPPIX) administration reversed paralysis and disease relapse. The protective effect of HO-1 was associated with inhibition of major histocompatibility complex (MHC) class II expression by antigen-presenting cells (APCs) and inhibition of CD4+ and CD8+ T cell accumulation, proliferation and effector function within the CNS. Exogenous CO administration mimicked these effects, suggesting that CO represents the key molecule that mediates the protective action of HO-1. In addition, the HO-1 system seemed to stabilize the blood–spinal cord barrier, thus limiting the infiltration of leucocytes [80].
Along the same lines, we have demonstrated recently that the water-soluble CORM-A1 prevents clinical and histological signs of EAE in mice with proteolipid protein (PLP)-induced EAE [81]. The early prophylactic treatment with CORM-A1 significantly reduced both the cumulative score and the duration of the disease compared to vehicle-treated controls. Moreover, the incidence of the disease in the mice treated with CORM-A1 was lower than in the vehicle-treated mice. These protective effects were maintained for a significant period of time after interruption of the treatment. Interestingly, the level of protection exerted by the prophylactic treatment with CORM-A1 was superior to that reached by the reference compound dexamethasone. A trend of reduction in the cumulative scores, disease duration and cumulative incidence was also observed in mice treated with CORM-A1 under the late prophylactic regimen. Significant reduction in the infiltration of polymorphonucleated cells in the spinal cord was also observed in the mice treated prophylactically with CORM-A1. Interestingly, the levels of COHb reached at a dose of 2 mg/kg used in this EAE study never exceeded 3% COHb levels, which are relatively safe and only slightly above basal values, indicating that CORM-A1 can exert its therapeutic activity within a range of COHb levels far lower than those considered to be toxic to humans.
That HO-1 and CO may play a significant role in reducing neuroinflammation is demonstrated further by a paper from Pamplona et al. [82], showing a marked suppressive effect of HO-1 and CO in the pathogenesis of murine cerebral malaria, which is a condition of the CNS that shares some immunoinflammatory pathways similar to EAE.
It is, however, important to mention that in apparent contradiction with all these data which point to a beneficial effect of endogenous or exogenous up-regulation of the HO1/CO axis in EAE and MS, Chakrabarty et al. have demonstrated that the administration of the putative inhibitor [tin-protoporphyrin IX (Sn-PP IX)] of HO-1 to SJL mice during active disease attenuated clinical scores, weight loss and some signs of pathology in comparison to vehicle treatment. Glutathione levels were greater in treated EAE mice than in those receiving vehicle, indicating lower oxidative stress in the former group. These data suggest that inhibition of HO-1 attenuated disease and suppressed free radical production. In the SJL murine model of EAE, extravasated blood is present in the CNS, and iron released by HO-1 from this haem source may not be sequestered adequately by ferritin, allowing for iron-mediated tissue damage [83]. Thus, HO-1 may act to amplify the disease process in this model. The reasons for these diverging results, that may lend support to the previously discussed concept of the HO1 system as pathogenetic effector in EAE and MS, are difficult to understand and require confirmation from independent studies and in different rodent models of EAE. None the less, these data advocate caution regarding the possibility that during EAE and MS development endogenous HO1 may orchestrate immunoinflammatory events in a dicothomic and possibly concentration-dependent fashion, and that excessive up-regulation of the HO1 system in this setting may become proinflammatory. Because this dicothomic action has not so far been reported with the end product of HO1, CO, this would favour targeting the development of CO, rather than HO1 for the treatment of MS.
Although the precise mechanistic mode of action by which HO1 and CO may exert their beneficial effects in EAE remains to be demonstrated, Chora et al. have provided convincing in-vitro and in-vivo evidence that the beneficial effect of HO-1 induction may occur at the level of antigen presentation, in particular through induction of HO-1 expression in dendritic cells (DC). In their study, induction of HO-1 leads to specific inhibition of MHC class II in APCs, including dendritic cells (DC), microglia and CNS-infiltrating macrophages. Induction of HO-1 expression in microglia suppressed signal transducer and activator of transcription 1 (STAT1) phosphorylation as well as MHC class II transactivator (CIITA) expression, two critical events for MHC class II expression in APCs. Mice lacking the CIITA show tissue-specific impairment of MHC class II expression, as well as in the reactivation of myelin-reactive T helper (Th) cells in the CNS [84]. This effect is likely to contribute to the overall protective effect of HO-1 induction, as MHC class II expression in microglia is thought to be involved in EAE pathogenesis and progression (reviewed in [85]).
Another possibility could be that HO-1 promotes the accumulation of regulatory T cells and/or up-regulates their activity within the CNS and/or in the periphery. This would be consistent with widespread evidence that regulatory T cells can control the pathogenesis of EAE [86] as well as with the hypothesis that HO-1 expression may control regulatory T cell function. This point, however, remains to be clarified, as despite its ability to suppress ongoing EAE, induction of HO-1 failed to modulate the number of CNS-infiltrating regulatory T cells [79].
Chora et al. [79] have also shown that upon induction of HO-1 in APCs, the effector function of myelin-reactive Th cells in the CNS is modulated in a manner that suppresses their pathogenicity with consequent suppression of neuroinflammatory, i.e. IFN-γ, but not neuroprotective, i.e. IL-10 and TNF-α cytokine expression by CNS-infiltrating Th cells [79]. It also remains to be studied whether HO-1 prevents EAE progression not only by immunomodulation but also by its cytoprotective properties on oligodendrocytes or neurones in the CNS [87]. Such an effect would be consistent with the observed arrest of EAE progression [88], as well as with previous observation that cytoprotection afforded by HO-1 can prevent the rejection of transplanted organs [89].
Conclusions
The use of controlled amounts of CO in preclinical experimental models of disease has produced some interesting data indicating that its therapeutic delivery to mammals could alleviate multiple pathological conditions, and in particular inflammatory disorders.
To date, there is clear evidence that the HO system plays an important role in neuroinflammatory disorders such as MS and its animal counterpart, and that controlled CO delivery through CORMs may represent a useful therapeutic tool for the treatment of these diseases. Indeed, CORM-A1 treatment exerts significant immunomodulatory effects in the PLP-induced EAE model in mice. The prolonged prophylactic treatment with CORM-A1 improved the clinical and histological signs of relapsing–remitting EAE. This was evident from the reduced cumulative score, shorter duration and lower incidence of the disease and reduced inflammatory infiltration of the spinal cords compared to vehicle-treated animals.
The results generated in the last few years indicate that CORMs exert effective anti-inflammatory effects in multiple pathological conditions characterized by an altered immune response. Even though caution must be exercised when translating pathogenic concepts and pharmacological data from rodent EAE to human MS, the multiple converging and independent in-vitro and in-vivo data that have been generated and that we have discussed herein highlight the immunopharmacological properties of the HO1, and, in particular, of the CO system in CNS neuroinflammation, and lend strong support to the possibility that CORMs may represent a novel class of drugs with disease-modifying properties in MS and, as such, are worthy of being developed further for their application in this clinical setting.
Disclosure
Professor Nicoletti's laboratory has received fees for services payment from Alfama. Gianni Garotta is a consultant and Member of the Scientific Advisory Board of Alfama. Carlos Romao is Member of the Board of Directors of Alfama.
References
- 1.Sjoestrand T. Endogenous formation of carbon monoxide in man. Nature. 1949;164:580. doi: 10.1038/164580a0. [DOI] [PubMed] [Google Scholar]
- 2.Landaw SA, Callahan EW, Schmid R. Catabolism of heme in vivo: comparison of the simultaneous production of bilirubin and carbon monoxide. J Clin Invest. 1970;49:914–25. doi: 10.1172/JCI106311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coburn RF, Williams WJ, Forster RE. Effect of erythrocyte destruction on carbon monoxide production in man. J Clin Invest. 1965;43:1098–103. doi: 10.1172/JCI104994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yachie A, Niida Y, Wada T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103:129–35. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizuguchi S, Stephen J, Bihari R, et al. CORM-3-derived CO modulates polymorphonuclear leukocyte migration across the vascular endothelium by reducing levels of cell surface-bound elastase. Am J Physiol Heart Circ Physiol. 2009;297:H920–9. doi: 10.1152/ajpheart.00305.2009. [DOI] [PubMed] [Google Scholar]
- 6.Ferrándiz ML, Maicas N, Garcia-Arnandis I, et al. Treatment with a CO-releasing molecule (CORM-3) reduces joint inflammation and erosion in murine collagen-induced arthritis. Ann Rheum Dis. 2008;67:1211–17. doi: 10.1136/ard.2007.082412. [DOI] [PubMed] [Google Scholar]
- 7.Takagi T, Naito Y, Inoue M, et al. Inhalation of carbon monoxide ameliorates collagen-induced arthritis in mice and regulates the articular expression of IL-1beta and MCP-1. Inflammation. 2009;32:83–8. doi: 10.1007/s10753-009-9106-6. [DOI] [PubMed] [Google Scholar]
- 8.Takagi T, Naito Y, Mizushima K, et al. Inhalation of carbon monoxide ameliorates TNBS-induced colitis in mice through the inhibition of TNF-α expression. Dig Dis Sci. 2010;55:2797–804. doi: 10.1007/s10620-009-1112-x. [DOI] [PubMed] [Google Scholar]
- 9.Chen P, Sun B, Chen H, et al. Effects of carbon monoxide releasing molecule-liberated CO on severe acute pancreatitis in rats. Cytokine. 2010;49:15–23. doi: 10.1016/j.cyto.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 10.Hoetzel A, Schmidt R, Vallbracht S, et al. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med. 2009;37:1708–15. doi: 10.1097/CCM.0b013e31819efa31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otterbein LE, Zuckerbraun BS, Haga M, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. 2003;9:183–90. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 12.Foresti R, Bani-Hani MG, Motterlini R. Use of carbon monoxide as a therapeutic agent: promises and challenges. Intens Care Med. 2008;34:649–58. doi: 10.1007/s00134-008-1011-1. [DOI] [PubMed] [Google Scholar]
- 13.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728–43. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- 14.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms and clinical applications. FASEB J. 1988;2:2557–68. [PubMed] [Google Scholar]
- 15.Tyrrell R. Redox regulation and oxidant activation of heme oxygenase-1. Free Radic Res. 1999;31:335–40. doi: 10.1080/10715769900300901. [DOI] [PubMed] [Google Scholar]
- 16.Lavrovsky Y, Schwartzman ML, Levere RD, Kappas A, Abraham NG. Identification of binding sites for transcription factors NF-B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc Natl Acad Sci USA. 1994;91:5987–91. doi: 10.1073/pnas.91.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato M, Ishizawa S, Yoshida T, Shibahara S. Interaction of upstream stimulatory factor with the human heme oxygenase gene promoter. Eur J Biochem. 1990;188:231–7. doi: 10.1111/j.1432-1033.1990.tb15394.x. [DOI] [PubMed] [Google Scholar]
- 18.Tyrrell RM, Applegate LA, Tromvoukis Y. The proximal promoter region of the human heme oxygenase gene contains elements involved in stimulation of transcriptional activity by a variety of agents including oxidants. Carcinogenesis. 1993;14:761–5. doi: 10.1093/carcin/14.4.761. [DOI] [PubMed] [Google Scholar]
- 19.Sardana MK, Drummond GS, Sassa S, Kappas A. The potent heme oxygenase inducing action of arsenic and parasiticidal arsenicals. Pharmacology. 1981;23:247–53. doi: 10.1159/000137557. [DOI] [PubMed] [Google Scholar]
- 20.Wu L. The pro-oxidant role of methylglyoxal in mesenteric artery smooth muscle cells. Can J Physiol Pharmacol. 2005;83:63–8. doi: 10.1139/y04-112. [DOI] [PubMed] [Google Scholar]
- 21.Motterlini R, Foresti R, Bassi R, Green CJ. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic Biol Med. 2000;28:1303–12. doi: 10.1016/s0891-5849(00)00294-x. [DOI] [PubMed] [Google Scholar]
- 22.Nath KA, Grande JP, Haggard JJ, et al. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol. 2001;158:893–903. doi: 10.1016/S0002-9440(10)64037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner CT, Durante W, Christodoulides N, Hellums JD, Schafer AI. Hemodynamic forces induce the expression of heme oxygenase in cultured vascular smooth muscle cells. J Clin Invest. 1997;100:589–96. doi: 10.1172/JCI119569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ndisang JF, Wang R. Age-related alterations in soluble guanylyl cyclase and cGMP pathway in spontaneously hypertensive rats. J Hypertens. 2003;21:1117–24. doi: 10.1097/00004872-200306000-00011. [DOI] [PubMed] [Google Scholar]
- 25.Carraway MS, Ghio AJ, Suliman HB, Carter JD, Whorton AR, Piantadosi CA. Carbon monoxide promotes hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2002;282:L693–L702. doi: 10.1152/ajplung.00211.2001. [DOI] [PubMed] [Google Scholar]
- 26.Durante W, Christodoulides N, Cheng K, Peyton KJ, Sunahara RK, Schafer AI. cAMP induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle. Am J Physiol. 1997;273:H317–23. doi: 10.1152/ajpheart.1997.273.1.H317. [DOI] [PubMed] [Google Scholar]
- 27.Terry CM, Clikeman JA, Hoidal JR, Callahan KS. TNF-alpha and IL-1alpha induce heme oxygenase-1 via protein kinase C, Ca2+ and phospholipase A2 in endothelial cells. Am J Physiol. 1999;276:H1493–H1501. doi: 10.1152/ajpheart.1999.276.5.H1493. [DOI] [PubMed] [Google Scholar]
- 28.Elbirt KK, Whitmarsh AJ, Davis RJ, Bonkovsky HL. Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J Biol Chem. 1998;273:8922–31. doi: 10.1074/jbc.273.15.8922. [DOI] [PubMed] [Google Scholar]
- 29.Alam J, Wicks C, Stewart D, et al. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem. 2000;275:27694–702. doi: 10.1074/jbc.M004729200. [DOI] [PubMed] [Google Scholar]
- 30.Chen K, Maines MD. Nitric oxide induces heme oxygenase-1 via mitogen activated protein kinases ERK and p38. Cell Mol Biol. 2000;46:609–17. [PubMed] [Google Scholar]
- 31.Furchgott RF, Jothianandan D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels. 1991;28:52–61. doi: 10.1159/000158843. [DOI] [PubMed] [Google Scholar]
- 32.Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci USA. 1995;92:1475–9. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem. 1997;272:32804–9. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 34.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259:381–4. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 35.Otterbein LE, Bach FH, Alam J, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–8. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 36.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–6. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 37.Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci USA. 1997;94:10925–30. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagener FA, Volk H, Willis D, et al. Different faces of the heme–heme oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–71. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi S, Takamiya R, Yamaguchi T, et al. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: role of bilirubin generated by the enzyme. Circ Res. 1999;85:663–71. doi: 10.1161/01.res.85.8.663. [DOI] [PubMed] [Google Scholar]
- 40.Sarady JK, Otterbein SL, Liu F, Otterbein LE, Choi AMK. Carbon monoxide modulates endotoxin-induced production of granulocyte macrophage colony-stimulating factor in macrophages. Am J Respir Cell Mol Biol. 2002;27:739–45. doi: 10.1165/rcmb.4816. [DOI] [PubMed] [Google Scholar]
- 41.Pae HO, Oh GS, Choi BM, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol. 2004;172:4744–51. doi: 10.4049/jimmunol.172.8.4744. [DOI] [PubMed] [Google Scholar]
- 42.Ryter SW, Otterbein LE. Carbon monoxide in biology and medicine. BioEssays. 2004;26:270–80. doi: 10.1002/bies.20005. [DOI] [PubMed] [Google Scholar]
- 43.Vera T, Henegar JR, Drummond HA, Rimoldi JM, Stec DE. Protective effect of carbon monoxide-releasing compounds in ischemia-induced acute renal failure. J Am Soc Nephrol. 2005;16:950–8. doi: 10.1681/ASN.2004090736. [DOI] [PubMed] [Google Scholar]
- 44.Ryan MJ, Jernigan NL, Drummond HA, et al. Renal vascular responses to CORM-A1 in the mouse. Pharmacol Res. 2006;54:24–9. doi: 10.1016/j.phrs.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Foresti R, Hammad J, Clark JE, et al. Vasoactive properties of CORM-3, a novel water-soluble carbon monoxide-releasing molecule. Br J Pharmacol. 2004;142:453–60. doi: 10.1038/sj.bjp.0705825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark JE, Naughton P, Shurey S, et al. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–e8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- 47.Musameh MD, Fuller BJ, Mann BE, Green CJ, Motterlini R. Positive inotropic effects of carbon monoxide-releasing molecules (CO-RMs) in the isolated perfused rat heart. Br J Pharmacol. 2006;149:1104–12. doi: 10.1038/sj.bjp.0706939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–10. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bani-Hani MG, Greenstein D, Mann BE, Green CJ, Motterlini R. Modulation of thrombin-induced neuroinflammation in BV-2 microglia by a carbon monoxide-releasing molecule (CORM-3) J Pharmacol Exp Ther. 2006;318:1315–22. doi: 10.1124/jpet.106.104729. [DOI] [PubMed] [Google Scholar]
- 50.Freitas A, Alves-Filho JC, Secco DD, et al. Heme oxygenase/carbon monoxide–biliverdin pathway down regulates neutrophil rolling, adhesion and migration in acute inflammation. Br J Pharmacol. 2006;149:345–54. doi: 10.1038/sj.bjp.0706882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vannacci A, Giannini L, Fabrizi F, et al. Effects of the carbon monoxide releasing molecule CORM-3 in a coincubation model of rat mast cells with human neutrophils. Inflamm Res. 2007;56:S13–S14. doi: 10.1007/s00011-006-0506-x. [DOI] [PubMed] [Google Scholar]
- 52.Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Motterlini R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc Res. 2006;71:393–401. doi: 10.1016/j.cardiores.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 53.Parfenova H, Basuroy S, Bhattacharya S, et al. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: contributions of HO-1 and HO-2 to cytoprotection. Am J Physiol Cell Physiol. 2005;290:C1399–C1410. doi: 10.1152/ajpcell.00386.2005. [DOI] [PubMed] [Google Scholar]
- 54.Li MH, Cha YN, Surh YJ. Carbon monoxide protects PC12 cells from peroxynitrite-induced apoptotic death by preventing the depolarization of mitochondrial transmembrane potential. Biochem Biophys Res Commun. 2006;342:984–90. doi: 10.1016/j.bbrc.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 55.Sun BW, Chen ZY, Chen X, Liu C. Attenuation of leukocytes sequestration by carbon monoxide-releasing molecules: liberated carbon monoxide in the liver of thermally injured mice. J Burn Care Res. 2007;28:173–81. doi: 10.1097/BCR.0b013E31802CA491. [DOI] [PubMed] [Google Scholar]
- 56.Allanson M, Reeve VE. Carbon monoxide signalling reduces photocarcinogenesis in the hairless mouse. Cancer Immunol Immunother. 2007;56:1807–15. doi: 10.1007/s00262-007-0324-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandouka A, Fuller BJ, Mann BE, Green CJ, Foresti R, Motterlini R. Treatment with carbon monoxide releasing molecules (CO-RMs) during cold storage improves renal function at reperfusion. Kidney Int. 2006;69:239–47. doi: 10.1038/sj.ki.5000016. [DOI] [PubMed] [Google Scholar]
- 58.Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R. Protection against cisplatin-induced nephrotoxicity by a carbon monoxide-releasing molecule. Am J Physiol Renal Physiol. 2006;290:F789–F794. doi: 10.1152/ajprenal.00363.2005. [DOI] [PubMed] [Google Scholar]
- 59.Dwyer BE, Nishimura RN, Lu SY. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Brain Res Mol Brain Res. 1995;30:37–47. doi: 10.1016/0169-328x(94)00273-h. [DOI] [PubMed] [Google Scholar]
- 60.Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem. 1999;72:1187–203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- 61.Fukuda K, Richmon JD, Sato M, Sharp FR, Panter SS, Noble LJ. Induction of heme oxygenase-1 (HO-1) in glia after traumatic brain injury. Brain Res. 1996;736:68–75. doi: 10.1016/0006-8993(96)00680-4. [DOI] [PubMed] [Google Scholar]
- 62.Premkumar DR, Smith MA, Richey PL, et al. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer's disease. J Neurochem. 1995;65:1399–402. doi: 10.1046/j.1471-4159.1995.65031399.x. [DOI] [PubMed] [Google Scholar]
- 63.Takahashi M, Dore S, Ferris CD, et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer's disease. Neuron. 2000;28:461–73. doi: 10.1016/s0896-6273(00)00125-2. [DOI] [PubMed] [Google Scholar]
- 64.Schipper HM, Chertkow H, Mehindate K, Frankel D, Melmed C, Bergman H. Evaluation of heme oxygenase-1 as a systemic biological marker of sporadic AD. Neurology. 2000;54:1297–304. doi: 10.1212/wnl.54.6.1297. [DOI] [PubMed] [Google Scholar]
- 65.Yoo MS, Chun HS, Son JJ, et al. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: correlation with the known pathology in Parkinson's disease. Brain Res Mol Brain Res. 2003;110:76–84. doi: 10.1016/s0169-328x(02)00586-7. [DOI] [PubMed] [Google Scholar]
- 66.Smith KJ, Kapoor R, Felts PA. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinteaux E, Perraut M, Tholey G. Distribution of mitochondrial manganese superoxide dismutase among rat glial cells in culture. Glia. 1998;22:408–14. [PubMed] [Google Scholar]
- 68.Ruuls SR, Bauer J, Sontrop K, Huitinga I, ‘t Hart BA, Dijkstra CD. Reactive oxygen species are involved in the pathogenesis of experimental allergic encephalomyelitis in Lewis rats. J Neuroimmunol. 1995;56:207–17. doi: 10.1016/0165-5728(94)00154-g. [DOI] [PubMed] [Google Scholar]
- 69.Toshniwal PK, Zarling EJ. Evidence for increased lipid peroxidation in multiple sclerosis. Neurochem Res. 1992;17:205–7. doi: 10.1007/BF00966801. [DOI] [PubMed] [Google Scholar]
- 70.Dallas ML, Boyle JP, Milligan CJ, et al. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J. 2011;25:1519–30. doi: 10.1096/fj.10-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stahnke T, Stadelmann C, Netzler A, Brück W, Richter-Landsberg C. Differential upregulation of heme oxygenase-1 (HSP32) in glial cells after oxidative stress and in demyelinating disorders. J Mol Neurosci. 2007;32:25–37. doi: 10.1007/s12031-007-0005-8. [DOI] [PubMed] [Google Scholar]
- 72.Fugger L, Friese MA, Bell JI. From genes to function: the next challenge to understanding multiple sclerosis. Nat Rev Immunol. 2009;9:408–17. doi: 10.1038/nri2554. [DOI] [PubMed] [Google Scholar]
- 73.Nicoletti F, Di Marco R, Patti F, et al. Short-term treatment of relapsing remitting multiple sclerosis patients with interferon (IFN)-beta1B transiently increases the blood levels of interleukin (IL)-6, IL-10 and IFN-gamma without significantly modifying those of IL-1beta, IL-2, IL-4 and tumour necrosis factor-alpha. Cytokine. 2000;12:682–7. doi: 10.1006/cyto.1999.0616. [DOI] [PubMed] [Google Scholar]
- 74.Nicoletti F, Di Marco R, Patti F, et al. Blood levels of transforming growth factor-beta 1 (TGF-beta1) are elevated in both relapsing remitting and chronic progressive multiple sclerosis (MS) patients and are further augmented by treatment with interferon-beta 1b (IFN-beta1b) Clin Exp Immunol. 1998;113:96–9. doi: 10.1046/j.1365-2249.1998.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nicoletti F, Patti F, DiMarco R, et al. Circulating serum levels of IL-1ra in patients with relapsing remitting multiple sclerosis are normal during remission phases but significantly increased either during exacerbations or in response to IFN-beta treatment. Cytokine. 1996;8:395–400. doi: 10.1006/cyto.1996.0054. [DOI] [PubMed] [Google Scholar]
- 76.Emerson MR, Gallagher RJ, Marquis JG, LeVine SM. Enhancing the ability of experimental autoimmune encephalomyelitis to serve as a more rigorous model of multiple sclerosis through refinement of the experimental design. Comp Med. 2009;59:112–28. [PMC free article] [PubMed] [Google Scholar]
- 77.Schipper HM. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med. 2004;37:1995–2011. doi: 10.1016/j.freeradbiomed.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y, Zhu B, Luo L, Li P, Paty DW, Cynader MS. Heme oxygenase-1 plays an important protective role in experimental autoimmune encephalomyelitis. Neuroreport. 2001;12:1841–5. doi: 10.1097/00001756-200107030-00016. [DOI] [PubMed] [Google Scholar]
- 79.Chora AA, Fontoura P, Cunha A, et al. Heme oxygenase-1 and carbon monoxide suppress autoimmune neuroinflammation. J Clin Invest. 2007;117:438–47. doi: 10.1172/JCI28844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin Y, Vreman HJ, Wong RJ, Tjoa T, Yamauchi T, Noble-Haeusslein LJ. Heme oxygenase-1 stabilizes the blood-spinal cord barrier and limits oxidative stress and white matter damage in the acutely injured murine spinal cord. J Cereb Blood Flow Metab. 2007;27:1010–21. doi: 10.1038/sj.jcbfm.9600412. [DOI] [PubMed] [Google Scholar]
- 81.Fagone P, Mangano K, Quattrocchi C, et al. Prevention of clinical and histological signs of proteolipid protein (PLP)-induced experimental allergic encephalomyelitis (EAE) in mice by the water-soluble carbon monoxide-releasing molecule (CORM)-A1. Clin Exp Immunol. 2011;163:368–74. doi: 10.1111/j.1365-2249.2010.04303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pamplona A, Ferreira A, Balla J, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. 2007;13:703–10. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 83.Chakrabarty A, Emerson MR, LeVine SM. Heme oxygenase-1 in SJL mice with experimental allergic encephalomyelitis. Mult Scler. 2003;9:372–81. doi: 10.1191/1352458503ms928oa. [DOI] [PubMed] [Google Scholar]
- 84.Stüve O, Youssef S, Slavin AJ, et al. The role of the MHC class II transactivator in class II expression and antigen presentation by astrocytes and in susceptibility to central nervous system autoimmune disease. J Immunol. 2002;169:6720–32. doi: 10.4049/jimmunol.169.12.6720. [DOI] [PubMed] [Google Scholar]
- 85.Becher B, Bechmann I, Greter M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J Mol Med. 2006;84:532–43. doi: 10.1007/s00109-006-0065-1. [DOI] [PubMed] [Google Scholar]
- 86.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–16. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 87.Bishop A, Yet SF, Lee ME, Perrella MA, Demple B. A key role for heme oxygenase-1 in nitric oxide resistance in murine motor and glia. Biochem Biophys Res Commun. 2004;325:3–9. doi: 10.1016/j.bbrc.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 88.Hövelmeyer N, Hao Z, Kranidioti K, et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5875–84. doi: 10.4049/jimmunol.175.9.5875. [DOI] [PubMed] [Google Scholar]
- 89.Soares MP, Lin Y, Anrather J, et al. Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat Med. 1998;4:1073–7. doi: 10.1038/2063. [DOI] [PubMed] [Google Scholar]