

Abstract
Neutrophil extracellular traps (NETs) comprise extracellular chromatin and granule protein complexes that immobilize and kill bacteria. NET release represents a recently discovered, novel anti-microbial strategy regulated non-exclusively by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase generation of reactive oxygen intermediates (ROIs), particularly hydrogen peroxide. This study aimed to characterize the role of ROIs in the process of NET release and to identify the dominant ROI trigger. We employed various enzymes, inhibitors and ROIs to record their effect fluorometrically on in vitro NET release by human peripheral blood neutrophils. Treatment with exogenous superoxide dismutase (SOD) supported the established link between hydrogen peroxide and NET production. However, treatment with myeloperoxidase inhibitors and direct addition of hypochlorous acid (HOCl; generated in situ from sodium hypochlorite) established that HOCl was a necessary and sufficient ROI for NET release. This was confirmed by the ability of HOCl to stimulate NET release in chronic granulomatous disease (CGD) patient neutrophils which, due to the lack of a functional NADPH oxidase, also lack the capacity for NET release in response to classical stimuli. Moreover, the exogenous addition of taurine, abundantly present within the neutrophil cytosol, abrogated NET production stimulated by phorbol myristate acetate (PMA) and HOCl, providing a novel mode of cytoprotection by taurine against oxidative stress by taurine.
Keywords: hypochlorous acid, NADPH oxidase, neutrophil, neutrophil extracellular trap, taurine
Introduction
As key effector cells of both innate and acquired immune responses, polymorphonuclear leucocytes (neutrophils) possess intracellular and extracellular killing mechanisms for elimination of pathogenic bacteria. Neutrophils are also capable of switching to a non-phlogistic phenotype during the active resolution phase of acute inflammation [1]. In addition to the classic killing mechanisms of phagocytosis and extracellular degranulation of proteases and reactive oxygen species (ROS), neutrophils are now known to extrude their decondensed nuclear chromatin complexed with granule-derived anti-microbial peptides into the extracellular space. The released structures are known as neutrophil extracellular traps (NETs) and function to both immobilize and kill microbes [2]. The release of NETs has been proposed to arise as a form of programmed cell death termed ‘NETosis’, which is distinct from apoptosis and necrosis [3,4]. Research has also demonstrated NET release from viable eosinophils [5] and viable neutrophils, where short-term stimulation releases mitochondrial NET-DNA rather than nuclear DNA and neutrophil life expectancy was unaffected [6]. NET release mechanisms demonstrate variance according to the robustness of the stimulus and the cell type investigated. Moreover, the role of NETs within human biology remains unclear, with protective functions in infectious diseases being reported [7] and also potentially pathogenic roles in autoimmune diseases proposed [8–10].
Since the first description of NETs [2], studies have attempted to elucidate the molecular signalling pathways regulating their release. While there are likely to be a multitude of converging factors regulating this process, research has focused upon the pathway involving nicotinamide adenine dinucleotide phosphate (NADPH) oxidase generation of ROS. The importance of the NADPH oxidase to NET release was first demonstrated by studies employing the oxidase inhibitor, diphenylene iodonium (DPI) which, when added extracellularly and prior to stimulation of ‘NETosis’, reduced NET release [3]. The NADPH oxidase generates superoxide which either dismutates spontaneously to hydrogen peroxide (H2O2) or is reduced more efficiently by the enzyme family of superoxide dismutases [11] (SOD; Fig. 1). The generation of H2O2 was shown to be sufficient to elicit NET release and the requirement for this signalling molecule was confirmed subsequently by studies utilizing catalase to remove H2O2 (by reduction to H2O and O2) and which was found to inhibit NET release. In contrast, the catalase inhibitor 3-aminotriazole (3-AT) increased NET release by elevating levels of available H2O2[3]. Most recently, an inhibitor of myeloperoxidase (MPO) (aminobenzoic acid hydrazide, 4-ABAH) has been reported to reduce NET release, indicating the potential requirement for this enzyme in the process [12,13]. Independently, NADPH oxidase generation of ROS has been found to be required specifically for the chromatin decondensation step that is a necessary prerequisite for NET formation [4]. The decondensation of neutrophil nuclear chromatin prior to NET extrusion into the extracellular space has also been demonstrated to require citrullination of histones by the enzyme peptidylarginine deiminase-4 [14], and also neutrophil elastase [15].
Fig. 1.

Metabolism of oxygen radicals produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.
NET biology is a relatively new area of study and with the literature growing rapidly there are various reports of apparently conflicting data concerning the mechanisms of NET release. This may be due in part to the inherent challenges associated with quantifying NET release, such that descriptive analyses form a substantial component of the reported evidence base. For example, NET release has been reported to be both NAPDH oxidase-independent [16] and NADPH oxidase-dependent [3,6,17]. The reason for the apparently discordant data may, in part, relate to different stimuli being employed; for example, although phorbol myristate acetate (PMA) and Helicobacter pylori elicit NADPH oxidase-dependent NET release, this activation occurs via different pathways, either protein kinase C (PKC)-dependent or -independent, respectively [18]. Also, when stimulated with Staphylococcus aureus, NET production has been reported to be NAPDH oxidase-independent during the first hour [16], in contrast to the NADPH oxidase-dependent NET release observed after 90 min [3]. Moreover, differences in the nature of cell stimuli has been proposed as the reason for NETs consisting of either nuclear DNA [3], mitochondrial DNA [6] or a combination of both [16]. While the majority of reports of NET release involve the eventual rupture of the neutrophil plasma membrane [3,4,15], early S. aureus-stimulated NET release (5–60 min) has been reported to occur via a process akin to exocytosis without plasma membrane rupture [16]. Furthermore, NETs comprising only mitochondrial DNA are reported to originate from cells remaining viable [6].
Here we demonstrate, for the first time, the requirement of hypochlorous acid (HOCl) for NET release and a potential regulatory role for endogenous taurine in this process. In our studies, for NET stimulation, PMA was employed to stimulate PKC in place of the endogenous activator, diacylglycerol. PMA, which is known to stimulate NADPH oxidase generation of ROS in neutrophils, has also been reported to elicit dramatic NET release [3]. Although less physiologically relevant, PMA provides direct intracellular stimulation, removing the complication of multiple simultaneous signalling pathways and responses that are likely to be evoked in neutrophils stimulated with more physiologically relevant receptor-mediated stimuli such as un-opsonized (Toll-like receptor; TLR) or opsonized (Fcγ-receptor) bacteria. In addition, this form of stimulation is consistent with many previously reported studies.
Materials and methods
Reagents
RPMI-1640 was obtained from Biosera (Ringmer, UK), Percoll from GE Healthcare (Little Chalfont, UK), SYTOX green nucleic acid stain was obtained from Invitrogen (Paisley, UK), 96-well plates were from Corning (Lowell, MA, USA), InnoZyme Myeloperoxidase Activity Kit was from Calbiochem (Nottingham, UK), micrococcal nuclease was from Worthington Biochemical Corporation (Lakewood, NJ, USA) and tryptone soy agar and broth were from Oxoid (ThermoFisher Scientific, Basingstoke, UK). All other chemicals were purchased from Sigma (Gillingham, UK).
Isolation of neutrophils
Unless specified otherwise, all ex-vivo experiments were conducted using human neutrophils from medically healthy volunteers and were isolated from venous blood by discontinuous Percoll density gradient followed by ammonium chloride lysis of red blood cells, as described previously [19]. Patients with chronic granulomatous disease (CGD) were recruited from the Department of Immunology, Birmingham Heartlands Hospital, following informed consent (West Midlands Research Ethics Committee number 10/H/1208/48).
Fluorometric quantification of NET-DNA release
Neutrophils (1 × 105) in RPMI-1640 were seeded into bovine serum albumin (BSA)-coated [1% in phosphate-buffered saline (PBS)] 96-well plates and allowed to settle for 30 min at 37°C in the presence of the inhibitors or enzymes being tested. Cells were stimulated with 25 nM PMA or 0·75 mM HOCl and incubated for 3 h at 37°C [2]. NET-DNA was quantified using a modified version of a previously published method [20–22]. Micrococcal nuclease (1 unit/ml) was added for 10 min to partially digest any released NETs and cells and debris were pelleted by centrifugation at 1800 g for 10 min. A sample of the supernatant was added to SYTOX green nucleic acid stain (1 µM) in a black 96-well plate to quantify NET-DNA fragments by fluorometry (Twinkle LB970, Berthold Technologies, Harpenden, UK; excitation 485 nm, emission 525 nm) and recorded as arbitrary fluorescent units (AFU).
Visualization of NET-DNA release
Neutrophils (1 × 105) suspended in 500 µl RPMI-1640 were seeded into BSA-coated 24-well plates and allowed to settle for 30 min at 37°C, prior to stimulation for 3 h at 37°C [2] and staining of NET-DNA using 1 µM SYTOX green. NETs and cells were observed at room temperature under a fluorescence microscope (Nikon Eclipse TE300, Kingston upon Thames, UK) using a × 20 objective and images captured by digital camera (Nikon CoolPix 450, Kingston upon Thames, UK).
Determination of MPO activity
The InnoZyme myeloperoxidase activity kit was used according to the manufacturers' instructions to examine the effect of 3-AT (1 mM) on the activity of purified human MPO (100 ng/ml).
Measurement of ROS generation
ROS generation was quantified by enhanced chemiluminescence assay [19]. Neutrophils (1 × 105) suspended in PBS supplemented with glucose (10 mM), MgCl2 (1·5 mM) and CaCl2 (1·35 mM) were seeded into a BSA-coated 96-well plate with luminol (450 µM) to detect total ROS, isoluminol (900 µM) plus horseradish peroxidase (7.5 units/ml) to detect extracellular ROS or lucigenin (50 µg/ml) to detect superoxide. Cells were allowed to settle for 30 min at 37°C prior to stimulation. ROS generation was recorded as the peak relative light units (RLU) per second recorded by microplate luminometer (Berthold LB96v) over the 2·5-h incubation period, as reported previously [19].
Determination of HOCl concentration
Sodium hypochlorite was diluted and the concentration of hypochlorite ions (OCl–) estimated by optical density at 292 nm of pH 12·0 solutions using an extinction coefficient of 350 M/cm [23]. The final pH when used in experiments was approximately the same as the pKa for HOCl (7·5), thus it was assumed that 50% existed as HOCl and 50% existed as OCl–.
Preparation of opsonized S. aureus
S. aureus (NCTC 6571) was grown aerobically at 37°C on tryptone soya agar and inoculated into tryptone soya broth. Bacteria were isolated from broth culture by centrifugation, washed three times in sterile PBS and heat-treated at 100°C for 10 min. Opsonization was performed as described previously [24] and stored as a 1·2 × 109 cells/ml suspension at −80°C.
Statistical analysis
Data were analysed using Excel 2007 (Microsoft). Each in vitro experiment was performed at least four times using independent neutrophil donors, and each experiment was performed in quadruplicate. Comparison between groups was made using two-tailed paired t-test where P-values of less than 0·05 were considered significant.
Results
H2O2 is required for NET release
It has been reported previously that NADPH oxidase-dependent generation of ROS, and specifically H2O2, is required for NET release [3]. This was demonstrated using the NADPH oxidase inhibitor DPI, which reduced NET release significantly, a finding corroborated here (Fig. 2a). NET release via this mechanism was investigated further by the addition of exogenous SOD to increase the conversion of superoxide to H2O2. SOD addition resulted in increased NET production (Fig. 2b), indicating that H2O2 mediates the release of NETs. In addition to the specific inhibitors and enzymes involved directly in the generation of ROS, the actin polymerization inhibitor cytochalasin has been shown to prevent the physical extrusion of NETs [25]. Interestingly, when this inhibitor was employed, not only was NET release reduced (Fig. 2c), but the generation of ROS as measured by enhanced chemiluminescence also decreased (Fig. 2d). This effect was more pronounced when neutrophils were stimulated by physiologically relevant particulate stimuli (bacteria) than soluble (PMA) and is therefore likely to be attributable to reduced post-phagocytic NADPH oxidase induction. Cytochalasin inhibition of NET release was also more pronounced in bacterially stimulated compared with PMA-stimulated cells. Therefore the inhibition of NET release by cytochalasin appears to have a dual mechanism, due to both reduced phagocytic induction of ROS and reduced actin cytoskeleton-mediated NET extrusion.
Fig. 2.
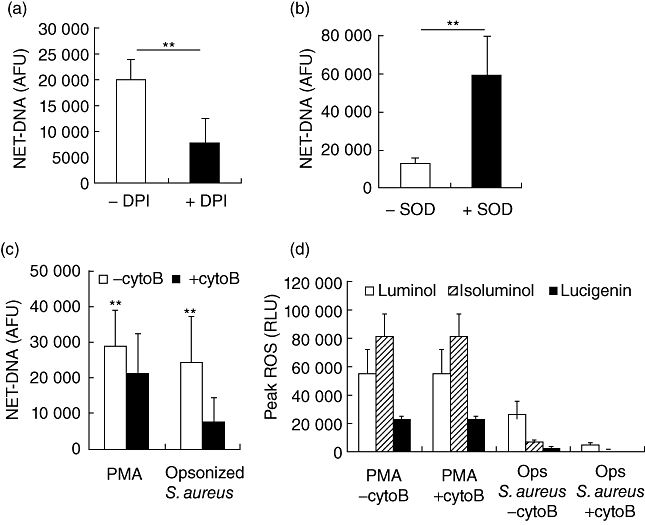
Fluorometric quantification of neutrophil extracellular trap (NET)-DNA from phorbol myristate acetate (PMA) (25 nM)-stimulated neutrophils after 3 h incubation in the absence or presence of (a) diphenylene iodonium (DPI) 25 µM and (b) superoxide dismutase (SOD) 95 units/ml. Data represent six independent experiments ± standard deviation (s.d.). Fluorometric quantification of NET-DNA (c) and reactive oxygen species (ROS) generation (d) from PMA (25 nM) or opsonized Staphylococcus aureus (300 per neutrophil)-stimulated neutrophils after 3 h incubation in the absence or presence of cytochalasin B (cytoB; 10 µg/ml). Data represent at least five independent experiments ± s.d. Luminol measures total ROS, isoluminol measures extracellular ROS and lucigenin measures extracellular superoxide. **P ≤ 0·01 by two-tailed paired t-test.
H2O2 metabolites regulate NET release
H2O2 is metabolized enzymatically via several pathways within the cell (Fig. 1), and enzyme supplementation and inhibition studies have been employed to demonstrate the dependency of NET release upon H2O2. Catalase performs an intracellular anti-oxidant role, removing H2O2 to form water and oxygen. Inhibition of catalase by 3-AT has been reported to increase NET release by allowing accumulation of H2O2[3]. However, under our experimental conditions we found 3-AT treatment had no significant effect upon NET release (Fig. 3a; P = 0·55 by two-tailed t-test). Interestingly, total ROS detection in PMA stimulated neutrophils when treated with 3-AT decreased unexpectedly (Fig. 3b). The specific inhibition of catalase by 3-AT would be expected to increase H2O2 concentrations and subsequent luminol detection of ROS. We therefore hypothesized that the 3-AT inhibitor was not specific to catalase and, consistent with previous reports, may also inhibit MPO [26,27]. To confirm this, a MPO activity assay was performed which revealed that 3-AT reduced the activity of purified human MPO by 22% (Fig. 3c). This inhibition was not observed when 3-AT was only present prior to washing and therefore indicated a reversible inhibition.
Fig. 3.

(a) Fluorometric quantification of neutrophil extracellular trap (NET)-DNA from phorbol myristate acetate (PMA) 25 nM-stimulated neutrophils after 3 h incubation in the absence or presence of catalase inhibitor, 3-aminotriazole (3-AT) 1 mM. Data represent six independent experiments ± standard deviation (s.d.) (b) Peak PMA 25 nM stimulated total reactive oxygen species (ROS) production measured by luminol. Data represent three independent experiments ± standard deviation (s.d.). *P ≤ 0·05 by two-tailed paired t-test. (c) Effect of 3-AT 1 mM on the activity of purified myeloperoxidase (MPO) 100 ng/ml compared to the specific MPO inhibitor, aminobenzoic acid hydrazide (4-ABAH) 1 mM (positive control). Experiment performed in triplicate ± s.d. Fluorometric quantification of NET-DNA from PMA 25 nM-stimulated neutrophils after 3 h incubation in the absence or presence of (d) glutathione precursor, NAC 10 mM or (e) 4-ABAH 0·1 mM. Data represent six independent experiments ± s.d. **P ≤ 0·01 by two-tailed paired t-test.
Another enzyme present within neutrophils which functions to metabolize H2O2 is glutathione peroxidase (Fig. 1). As reported previously in nitric oxide donor-stimulated neutrophils [12], addition of the cell permeable precursor for glutathione (N-acetyl-cysteine; NAC) reduced PMA-stimulated NET release (Fig. 3d). This data further supported the requirement for H2O2 for NET release. MPO also metabolizes H2O2, in this case to form HOCl. The requirement for this enzyme in the process of NET release was confirmed using the specific inhibitor 4-ABAH which reduced NET production significantly, as reported previously [12,13].
HOCl regulates NET release
Data confirm that, under our experimental conditions, inhibition of MPO by 4-ABAH inhibits the formation of NETs. Therefore the product of MPO, HOCl was supplemented directly to neutrophils and was observed to elicit NET release (Fig. 4a,b). This effect was found to be specific to the product of MPO as another chlorine-based acid (hydrochloric acid) evoked no detectable NET release (Fig. 4c). To confirm the physiological relevance of our hypothesis, we then exposed neutrophils obtained from patients with CGD to HOCl to ascertain whether NET release could be evoked, despite the absence of a functional NADPH oxidase system (confirmed by chemiluminescent assay, data not shown). Neutrophils from CGD patients did not release NETs when stimulated with PMA, but were able to release NETs upon exposure to exogenous HOCl (Fig. 4d).
Fig. 4.
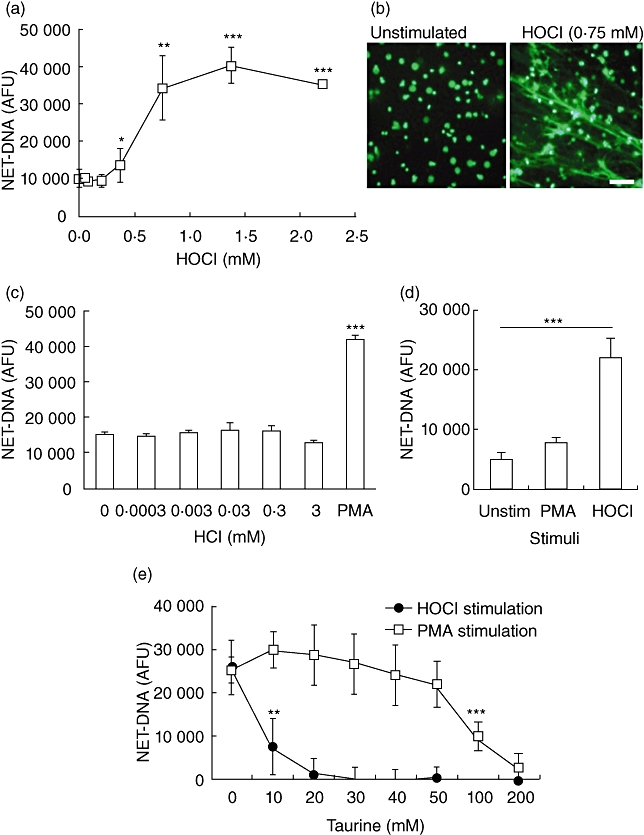
(a) Fluorometric quantification of neutrophil extracellular trap (NET)-DNA from hypochlorous acid (HOCl)-stimulated neutrophils after 3 h incubation. Data represent six independent experiments ± standard deviation (s.d.). (b) Images of SYTOX-stained NET-DNA released after 3 h in the absence or presence of HOCl 0·75 mM. Bar represents 50 µm. (c) Fluorometric quantification of NET-DNA from hydrochloric acid (HCl) stimulated neutrophils after 3 h incubation. Experiment performed in quadruplicate ± s.d.; phorbol myristate acetate (PMA) 25 nM stimulation included as positive control. (d) Fluorometric quantification of NET-DNA released from chronic granulomatous disease (CGD) neutrophils after 3 h incubation with HOCl 0·75 mM. Data representative of three independent experiments ± s.d. *P ≤ 0·05, **P ≤ 0·01, ***P ≤ 0·001 by two-tailed paired t-test in comparison to unstimulated cells. (e) Fluorometric quantification of NET-DNA from PMA 25 nM or HOCl 0·75 mM-stimulated neutrophils after 3 h incubation in the absence or presence of taurine. Data represent four independent experiments ± s.d. **P ≤ 0·01, ***P ≤ 0·01 by two-tailed paired t-test in comparison to 0 mM taurine.
Taurine inhibits NET formation
Taurine is found abundantly within the cytoplasm of neutrophils (at ∼50 mM [28]) and is known to neutralize HOCl by forming taurine chloramine and essentially removing H2O2 and HOCl to promote cell survival [29]. Indeed, taurine chloramine activates Nrf2 and a battery of downstream cytoprotective anti-oxidant enzymes (including haem-oxygenase-1; glutathione-transferase; peroxiredoxin; thioredoxin), thus promoting cell survival [29]. Therefore the role of taurine was examined by its addition prior to stimulation of NET release using both PMA (to stimulate endogenous HOCl generation) and also following direct addition of HOCl (0·75 mM). Taurine treatment reduced NET release significantly in response to PMA at 100 mM and in response to HOCl at only 10 mM (Fig. 4e). This difference is likely to be due to both taurine and HOCl being added exogenously, and therefore the HOCl was likely to have been neutralized prior to entering the cell, unlike PMA which stimulates HOCl generation by direct intracellular activation of PKC.
HOCl stimulates NET release in 1 h
Direct neutrophil exposure to 0·75 mM HOCl resulted in the release of NET structures between 30 and 70 min (Fig. 5a), whereas stimulation with PBS did not result in release of nuclear DNA (Fig. 5b) and treatment with 1% Triton X-100 killed neutrophils almost instantly to release non-NET DNA (Fig. 5c).
Fig. 5.

Time–course fluorescence photomicrographs (SYTOX staining) of neutrophils stimulated with (a) 0·75 mM hypochlorous acid (HOCl); (b) 1% Triton X-100 to induce cell lysis; (c) phosphate-buffered saline (PBS) negative control. PBS does not affect neutrophil viability to release nuclear DNA. Triton lyses cells immediately (0 min) to release DNA without neutrophil extracellular trap (NET) structure formation. HOCl stimulates NET-DNA release between 30 and 70 min post-stimulation. Bar represents 100 µm.
Discussion
The recent discovery of NET release [2] led to a plethora of studies describing their potential physiological role as a vitally important anti-microbial strategy in humans. However, their apparent complete dependence upon ROS activity suggests that the physiological heterogeneity surrounding ROS generation probably also pertains to NET release. Thus neutrophil hyperactivity and hyper-reactivity [19] with respect to ROS release may lead to disproportionate, physiologically discordant and/or displaced NET release with potentially pathogenic sequelae, such as autoimmune disease [8–10]. Several publications describing the process of NET release contain apparently conflicting reports as to whether NET release is dependent upon the generation of ROS by NADPH oxidase activation [3,6,16,17]. While it has been reported that DPI merely delays PMA-stimulated NET release such that it is not detectable until 5 h after stimulation [4], the majority of reported studies [3,6,17,18] demonstrate that DPI inhibits NET release during at least the initial 3 h of stimulation (which is the phase examined in our reported studies).
Following agreement with the findings of other investigators using the oxidase inhibitor DPI under our experimental conditions, we attempted to identify the specific ROS necessary for NET release; in particular, whether H2O2 or other reactive intermediates downstream of H2O2 were responsible. Initially, we applied exogenous SOD for novel evidence in support of the hypothesis of H2O2-mediated NET release. Although SOD is believed to gain intracellular access relatively slowly [30], lucigenin chemiluminescence, which specifically detects superoxide (the substrate for SOD), was decreased in the presence of exogenous SOD (data not shown). These data indicate that the catalyzed dismutation of superoxide was enhanced, and whether or not this arose intra- or extracellularly, the H2O2 generated is membrane-permeable and triggered NET release. Additionally, H2O2 was able to elicit NET release in the absence of any other stimuli, as reported previously [14,25] (data not shown).
Having confirmed and reinforced the link between H2O2 and NET release we subsequently examined the contribution of metabolites of H2O2 in the process of NET release. Various enzymatic pathways exist within the neutrophil to provide strict regulation of the neutrophils oxidative status by either removing H2O2, to prevent cytotoxicity to neighbouring host cells, or by converting it to further reactive oxidants such as HOCl in order to enhance microbicidal processes. One such H2O2 eliminator is glutathione peroxidase, promotion of which (by addition of its reduced glutathione substrate precursor, NAC) reduced NET release. We then analysed the effects of catalase inhibition using 3-AT, reported previously to increase NET release [3]. However, under our experimental conditions no effect was detected, which our subsequent experiments demonstrated to be due to a lack of catalase specificity of this inhibitor, which we found also reduced MPO activity (Fig. 3c). Specific inhibition of MPO demonstrated that the MPO product HOCl may be responsible for the regulation of NET release. In confirmation of this thesis, HOCl was able to stimulate NET release directly in the absence of any other stimuli (Fig. 4a). This finding was verified by demonstrating the ability of HOCl to stimulate NET release in CGD neutrophils lacking a functional NADPH oxidase to generate superoxide and downstream H2O2 and HOCl. While concentrations of HOCl reported extracellularly at sites of inflammation are of the order of 100 µM [31], higher levels of the exogenous species would be required to evoke NET release compared with locally generated endogenous HOCl. Therefore we employed physiologically relevant concentrations in our ex-vivo studies (Fig. 4a). A time–course study demonstrated NET-DNA release between 30 and 70 min (Fig. 5a), and as expected the process was more rapid than mechanisms involving receptor-ligand binding or phagocytosis.
Finally, given the in-vivo abundance of taurine within neutrophils and its cytoprotective role in removing HOCl and thus upstream H2O2 by forming taurine chloramines, we investigated the effects of adding taurine exogenously to stimulated neutrophils. The taurine effectively prevented both PMA and HOCl-induced NET release, confirming previous studies performed prior to the advent of NET biology, that taurine is capable of rescuing neutrophils from programmed cell death. As discussed previously, whether NET release is followed immediately by cell death or whether cells remain viable after NET release appears to depend upon the conditions of stimulation, and both outcomes are documented. Although the reports of NETs released from viable cells were not performed using the same stimuli as reported here, the fate of the cells after NET release was not the focus of these studies, and it is recognized that cells may remain viable for a significant period following NET extrusion.
The data reported in the current paper demonstrate a pivotal role for HOCl in NET release by peripheral blood neutrophils, identifying for the first time the trigger-point downstream of H2O2. Further studies of this pathway may provide opportunities for therapeutic developments in patients with CGD or in sepsis where NET production may enhance the resolution of infection or, conversely, may contribute to autoimmune and/or autoinflammatory disease mechanisms.
Acknowledgments
This work was funded by the University of Birmingham.
Disclosure
The authors declare that they have no competing interests.
References
- 1.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolving lipid mediators. Nat Rev Immunol. 2008;8:349–61. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–41. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Remijsen Q, Vanden Berghe T, Wirawan E, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yousefi S, Gold JA, Andina N, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14:949–53. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- 6.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–44. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114:2619–22. doi: 10.1182/blood-2009-05-221606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kessenbrock K, Krumbholz M, Schönermarck U, et al. Netting neutophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–5. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutteridge JMC, Halliwell B. Antioxidants in nutrition, health, and disease. New York: Oxford University Press; 1994. [Google Scholar]
- 12.Patel S, Kumar S, Jyoti A, et al. Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide. 2010;22:226–34. doi: 10.1016/j.niox.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Metzler KD, Fuchs TA, Nauseef WM, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2010;117:953–9. doi: 10.1182/blood-2010-06-290171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–62. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–91. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilsczek FH, Salina D, Poon KK, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–25. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 17.Juneau RA, Pang B, Weimer KE, Armbruster CE, Swords WE. Nontypeable Haemophilus influenzae initiates formation of neutrophil extracellular traps. Infect Immun. 2011;79:431–8. doi: 10.1128/IAI.00660-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hakkim A, Fuchs TA, Martinez NE, et al. Activation of the Raf–MEK–ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75–7. doi: 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 19.Matthews JB, Wright HJ, Roberts A, Cooper PR, Chapple IL. Hyperactivity and reactivity of peripheral blood neutrophils in chronic periodontitis. Clin Exp Immunol. 2007;147:255–64. doi: 10.1111/j.1365-2249.2006.03276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jovanović B, Anastasova L, Rowe EW, Zhang Y, Clapp AR, Palić D. Effects of nanosized titanium dioxide on innate immune system of fathead minnow (Pimephales promelas Rafinesque, 1820) Ecotoxicol Environ Saf. 2010;74:675–83. doi: 10.1016/j.ecoenv.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 21.Chuammitri P, Ostojić J, Andreasen CB, Redmond SB, Lamont SJ, Palić D. Chicken heterophil extracellular traps (HETs): novel defense mechanism of chicken heterophils. Vet Immunol Immunopathol. 2009;129:126–31. doi: 10.1016/j.vetimm.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Gabriel C, McMaster WR, Girard D, Descoteaux A. Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J Immunol. 2010;185:4319–27. doi: 10.4049/jimmunol.1000893. [DOI] [PubMed] [Google Scholar]
- 23.Morris JC. The acid ionization constant of HOCl from 5 to 35°. J Phys Chem. 1966;70:3798–805. [Google Scholar]
- 24.Bergstrom K, Åsman B. Luminol enhanced Fc-receptor dependent chemiluminescence from peripheral PMNL cells. A methodological study. Scand J Clin Lab Invest. 1993;53:171–7. doi: 10.3109/00365519309088404. [DOI] [PubMed] [Google Scholar]
- 25.Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun. 2009;1:194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nauseef WM, Metcalf JA, Root RK. Role of myeloperoxidase in the respiratory burst of human neutrophils. Blood. 1983;61:483–92. [PubMed] [Google Scholar]
- 27.Myzak MC, Carr AC. Myeloperoxidase-dependent caspase-3 activation and apoptosis in HL-60 cells: protection by the antioxidants ascorbate and (dihydro)lipoic acid. Redox Rep. 2002;7:47–53. doi: 10.1179/135100002125000181. [DOI] [PubMed] [Google Scholar]
- 28.Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta. 1991;1073:91–7. doi: 10.1016/0304-4165(91)90187-l. [DOI] [PubMed] [Google Scholar]
- 29.Sun Jang J, Piao SU, Cha Y-N, Kim C. Taurine chloramines activates Nrf2, increases HO-1 expression and protects cells from death caused by hydrogen peroxide. J Clin Biochem Nutr. 2009;45:37–43. doi: 10.3164/jcbn.08-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emerit I, Garban F, Vassy J, Levy A, Filipe P, Freitas J. Superoxide-mediated clastogenesis and anticlastogenic effects of exogenous superoxide dismutase. Proc Natl Acad Sci USA. 1996;93:12799–804. doi: 10.1073/pnas.93.23.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression. Involvement of myelo-peroxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1309–14. doi: 10.1161/01.ATV.0000131784.50633.4f. [DOI] [PubMed] [Google Scholar]