

Abstract
T cell receptor transgenic (TCR-Tg) mice specific for the arthritogenic 5/4E8 epitope in the G1 domain of cartilage proteoglycan were generated and back-crossed into arthritis-prone BALB/c background. Although more than 90% of CD4+ T cells of all TCR-Tg lines were 5/4E8-specific, one (TCR-TgA) was highly sensitive to G1-induced or spontaneous arthritis, while another (TCR-TgB) was less susceptible. Here we studied whether fine differences in TCR signalling controlled the onset and severity of arthritis. Mice from the two TCR-Tg lines were immunized side by side with purified recombinant human G1 (rhG1) domain for G1 domain of cartilage proteoglycan (PG)-induced arthritis (GIA). TCR-TgA mice developed severe and early-onset arthritis, whereas TCR-TgB mice developed weaker arthritis with delayed onset, although TCR-TgB CD4+ T cells expressed approximately twice more TCR-Vβ4 chain protein. The more severe arthritis in TCR-TgA mice was associated with higher amounts of anti-G1 domain-specific antibodies, larger numbers of B cells and activated T helper cells. Importantly, TCR-TgB CD4+ T cells were more sensitive to in vitro activation-induced apoptosis, correlating with their higher TCR and CD3 expression and with the increased TCR signal strength. These findings indicate that TCR signal strength determines the clinical outcome of arthritis induction: ‘optimal’ TCR signal strength leads to strong T cell activation and severe arthritis in TCR-TgA mice, whereas ‘supra-optimal’ TCR signal leads to enhanced elimination of self-reactive T cells, resulting in attenuated disease.
Keywords: rheumatoid arthritis, TcR signalling, transgenic mice
Introduction
Rheumatoid arthritis (RA), a systemic autoimmune disease affecting 1% of the human population, progresses from severe inflammation to deformities and loss of function of peripheral joints. Several animal models have been developed to mimic one or more characteristics of this human disease. Cartilage proteoglycan (PG)-induced arthritis (PGIA) in BALB/c mice [1] is a T cell-dependent and B cell/antibody-mediated autoimmune disease. Several lines of evidence indicate the role of T cell involvement in the pathogenesis of PGIA [2–4]. Activation and proliferation of T helper type 1 (Th1)/Th17-polarized CD4+ T cells during the initiation/development of the disease have been described [5–7]. Systemic depletion of CD4+ T cells prevents the development of PGIA [8], and both T and B cells are required for the adoptive transfer of the disease [6,9].
The cartilage PG (aggrecan) molecule consists of a large core protein (>200 kDa) to which hundreds of glycosaminoglycan side chains are attached [3]. The G1 globular domain of cartilage PG contains several dominant/arthritogenic epitopes, whereas a few cryptic or subdominant epitopes are located in the other regions of the PG molecule [10,11]. In a recent study, we replaced the full-length PG molecule (the PGIA model) with a recombinant human G1 (rhG1) domain to immunize BALB/c mice, resulting in G1 domain-induced arthritis (the GIA model) [12]. The clinical phenotype, histopathological abnormalities and laboratory test results in the GIA model were very similar to those described in ‘parental’ PGIA [1,2,4,12,13]. Because the dominant and possibly most arthritogenic ‘5/4E8’ T cell epitope (70ATEGRVRVNSAYQDK84; the core sequence is underlined) is located in the G1 domain [3] and a 5/4E8 epitope-specific T cell hybridoma has been shown to induce arthritis [14], we generated T cell receptor (TCR) transgenic (TCR-Tg) mice [15,16], in which more than 90% of the CD4+ T cells expressed the Vα1·1 and Vβ4 chains recognizing the 5/4E8 epitope within the G1 domain of human PG.
The first 5/4E8 epitope (PG)-specific TCR-Tg line (henceforth TCR-TgA) had been used for a number of immunological studies and adoptive transfer experiments [15,16], whereas the second transgenic line (henceforth TCR-TgB) had not yet been characterized. As both TgA and TgB strains express the same epitope (5/4E8)-specific TCR, we expected that these mice would develop arthritis similarly upon rhG1 immunization. Contrary to this, TCR-TgB mice exhibited delayed onset and less severe arthritis than TCR-TgA mice in response to either PG or rhG1 immunization, and they failed to develop spontaneous arthritis at an advanced age, whereas it is a characteristic phenotype of TCR-TgA mice [17]. In the present study, we investigated the possible underlying mechanisms of these profound differences. Our results confirm that the TCR signal in TCR-TgB mice was significantly stronger than in TCR-TgA mice, which led to extensive activation-induced cell death (AICD) of CD4+ T cells attenuating the arthritic phenotype. We conclude that TCR signal strength controls the onset and severity of arthritis by regulating AICD of T cells.
Materials and methods
Chemicals
All chemicals, unless indicated otherwise, were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA) or Fisher Scientific (Chicago, IL, USA). Phosphate-buffered saline (PBS; pH 7·4) was used for washing and short-term storage of cells until use. Cell surface labelling with monoclonal antibodies (mAbs) was performed in flow cytometry staining wash buffer (PBS containing 0·1% NaN3 and 0·1% bovine serum albumin). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum under standard tissue culture conditions.
Animals
We used two lines (Tg ‘A’ and Tg ‘B’) of the TCR-Tg mice, both expressing the TCR Vα1·1 and Vβ4 chains specific for the major dominant arthritogenic ‘5/4E8’ epitope (ATEGRVRVNSAYQDK) of the G1 domain of human cartilage PG. The two transgenic (TCR-TgA and TCR-TgB) lines were generated from different pronuclear injections using the same construct. Transgene-positive founders were back-crossed 12 times into the BALB/c (Charles River Laboratory, Kingston Colony, NY, USA) background. All animal procedures were conducted according to the protocol approved by the Institutional Animal Care and Use Committee at Rush University Medical Center (Chicago, IL, USA).
Antigens, immunization, clinical assessment of arthritis and sample collection
Recombinant human G1-domain (rhG1) was purified as described [12]. Wild-type (WT) BALB/c mice (Charles River) and age-matched 3-month-old female transgenic (TCR-TgA and TCR-TgB) mice were immunized intraperitoneally with 20 µg rhG1 in an emulsion of 2 mg dimethyldioctadecylammonium bromide adjuvant (in 100 µl of PBS) on days 0, 21 and 42 [12]. The mice were examined three to four times a week after the second immunization for the clinical assessment of arthritis. The onset time and incidence of arthritis were recorded, and disease severity was scored visually based on the degree of swelling and redness of each paw, ranging from 0 to 4, yielding a maximum severity score of 16 per mouse [18].
From each transgenic line and WT BALB/c mice, four animals were killed at different time-points: before immunization (naive mice), 10 days after the first immunization, 4 days before the second and third immunizations and 5 days after the second and third immunizations. These six time-points were determined in preliminary experiments. Blood samples, joint-draining lymph nodes [brachial, axillary, inguinal and popliteal lymph nodes (LNs)] and spleens were collected for flow cytometry analysis and cell culture. Serum samples were collected for measurements of antibody and cytokine concentrations.
Measurement of antigen (rhG1)-specific antibodies by enzyme-linked immunosorbent assay (ELISA) and serum cytokines by cytokine bead array (CBA)
Serum anti-rhG1 IgG1 and IgG2a antibodies were measured with ELISA as described previously [12]. Serum interleukin (IL)-1β, IL-4, IL-6, tumour necrosis factor (TNF)-α, IL-17A, IL-12p70 and interferon (IFN)-γ were measured using the CBA mouse/rat soluble protein flex set assay (BD Biosciences, San Jose, CA, USA), according to the manufacturer's instructions. The samples were measured using a BD fluorescence activated cell sorter (FACS)Canto II flow cytometer equipped with a High Throughput Sampler (HTS) module (BD Biosciences); 500 events (measured twice) per analysis were recorded. Data were analysed using FCAPArray software (Soft Flow Hungary Ltd, Pécs, Hungary).
mAbs
The following mAbs and reagents were purchased from BD Biosciences: fluorescein isothiocyanate (FITC)-conjugated anti- mouse TCR Vβ4 (clone KT4), allophycocyanin (APC)-Cy7-conjugated anti-mouse CD3 (clone 145-2C11), peridinin chlorophyll (PerCP)-cyanin-5·5 (Cy5·5)-conjugated anti-mouse CD4 (clone RM4-5), phycoerythrin (PE)-conjugated anti-mouse CD8 (clone 53–6·7), PE-Cy7-conjugated anti-mouse B220 (clone RA3-6B2), APC-conjugated anti-mouse CD25 (clone 3C7), PE-conjugated anti-mouse CD28 (clone 37·51), AlexaFluor 488-conjugated anti-mouse CD44 (clone IM7), PE-conjugated anti-mouse CD62L (clone MEl-14), PE-conjugated anti-mouse CTLA-4 (clone UC10-4F10-11), PE-conjugated anti-mouse PD-1 (clone J43), FITC-conjugated anti-mouse annexin V and 7-amino actinomycin (7-AAD, similar to propidium iodide). Biotinylated anti-mouse inducible T cell co-stimulator (ICOS) (clone 7E.17G9) were purchased from eBioscience.
Flow cytometry
Cell surface markers of peripheral blood, LN and spleen leukocytes were analysed by multi-colour flow cytometry, as described [7]. Data acquisition and analysis was performed using a FACS Canto II flow cytometer with an HTS module and FACS DIVA software (BD Biosciences). Initial gating was performed on lymphoid cells based on the forward-/side-scatter (FSC/SSC) parameters. The following cell populations were defined based on the cell surface markers: B220+: total B cells; CD3+: total T cells; CD3+/CD4+: CD4+ T cells; CD3+/CD8+: CD8+ T cells; CD3+/CD4+/CD25high: activated T cells; CD3+/CD4+/CD44high: activated (memory) T cells.
Cell separation, culture condition and in vitro antigen stimulation
T cells were purified from the spleens of TCR-Tg mice using an EasySep magnetic T cell enrichment kit (Stem Cell Technologies, Vancouver, BC, Canada). The purified T cells (8 × 105) were seeded onto irradiated A20 (BALB/c B cell lymphoma) antigen-presenting cells (ATCC, Rockville, MD, USA) that can present the 5/4E8 peptide [19]. A20 cells (1 × 105 cells/well) were plated in 48-well plates, precultured with or without the synthetic 5/4E8 peptide (5 µg/ml) for 12 h and then washed with serum-free DMEM. For apoptosis studies, purified T cells from spleen were co-cultured with these pretreated and washed A20 cells in 600 µl of DMEM containing 10% fetal bovine serum for 3 days. For signalling studies, 3 × 105 purified T cells from spleen were spun onto a layer of pretreated A20 cells by short centrifugation (900 g, 3 min) and harvested after 1 h of co-culture.
Saturation binding test
CD4+ T cells were separated from the spleens of TCR-TgA and TCR-TgB mice by magnetic enrichment. Exactly the same number of cells from each line was stained with increasing concentrations of fluorescently labelled anti-TCR Vβ4, anti-CD3 or anti-CD4 mAbs, and binding was analysed by flow cytometry. The mean fluorescence intensity (MFI) values were corrected by subtracting the MFI values of isotype controls, and the binding curves were fitted to the titration data points using GraphPad Prism 4·0 (GraphPad, San Diego, CA, USA).
Detection of TCR Vα1·1 and Vβ4 chain genomic copy numbers by real-time quantitative polymerase chain reaction (qPCR)
Tail genomic DNA was isolated from homozygous TCR-TgA and TCR-TgB animals by proteinase K digestion followed by phenol–chloroform extraction. Extracted genomic DNA samples were ethanol-precipitated and resuspended in Tris-ethylenediamine tretraacetic Acid (EDTA) buffer. The DNA samples were fragmented by restriction endonuclease digestion with enzymes cutting outside the PCR-investigated regions. Digested DNA was purified using a Qiaquick kit (Qiagen, Carlsbad, CA, USA), and DNA concentration was determined photometrically; qPCR was performed in triplicate with 10, 5, 2·5 and 1·25 ng of input DNA using SsoFast™ Probes Supermix (Bio-Rad). PrimeTime™ qPCR primers and dual-labelled probes (IDT, Coralville, IA, USA) were applied to determine the copy numbers of TCR alpha and beta chains (sequences of PCR primers and dual-labelled probes are available upon request); qPCR was performed using an iQ5 instrument (Bio-Rad, Hercules, CA, USA). For normalization, we used the promoter region of the TATA-box binding protein (TBP) gene as a single-copy control.
Apoptosis detection by annexin V/7-AAD staining
Annexin V/7-AAD staining was used to distinguish between early and late apoptotic T cells. Labelling was performed according to the manufacturer's (BD Biosciences) instructions. The cells were analysed by flow cytometry immediately. Annexin V-/7-AAD- cells were considered non-apoptotic. Annexin V+/7-AAD- cells were considered early apoptotic, and annexin V+/7-AAD+ cells were considered late apoptotic [7].
Determination of phosphorylation of TCR signalling proteins
Phosphorylation of zeta-chain-associated protein kinase 70 (ZAP-70), extracellular regulated kinase (ERK)1/2 and p38 was detected using the phospho-flow technique according to the manufacturer's instructions using phosphorylated protein-specific mAbs (BD Biosciences) [20]. After in vitro TCR stimulation, cells were labelled with anti-CD4-PerCP-Cy5·5 and phospho-specific antibodies: PE-conjugated anti-mouse pZAP-70 (clone 17A/P-ZAP-70) recognizing pY319 of ZAP-70, PE-conjugated anti-mouse pERK1/2 (clone 20A) recognizing pT203/pY205 in ERK1 and pT183/pY185 in ERK2, and PE-conjugated anti-mouse p38 (clone 36/p38 (pT180/pY182) recognizing pT180/pY182 in p38.
Statistical analysis
Descriptive statistics was used to determine group means and the standard errors of the means (the mean ± s.e.m.). Differences between two groups were tested for statistical significance using Student's t-test and differences among three or more groups were tested by analysis of variance (anova) with Dunnett's post-hoc t-test. A value of P ≤ 0·05 was considered statistically significant.
Results
Onset and severity of arthritis differ in the two PG-specific TCR-Tg lines
Previous studies have shown that mice of the original (first) 5/4E8 PG epitope-specific TCR-Tg (TCR-TgA) mice are highly susceptible to PGIA [16]. After finishing the back-cross of the second 5/4E8 PG epitope-specific TCR-Tg strain (TCR-TgB) into BALB/c background we immunized the two TCR-Tg strains side by side with rhG1. The TCR-TgA line responded to rhG1 immunizations as expected: arthritis had already developed a few days after the second immunization, and reached maximal severity scores with 100% incidence within 2 weeks after the second immunization (Fig. 1a,b). Surprisingly, the clinical phenotype and disease characteristics TCR-TgB were more similar to those found in WT BALB/c mice: the onset was delayed and the arthritis was less severe when compared to TCR-TgA mice (Fig. 1a,b) [1,2,12]. These results were surprising, because approximately the same proportion (∼90 to 94%) of CD4+ T cells in both lines expressed the TCR Vβ4 chain.
Fig. 1.
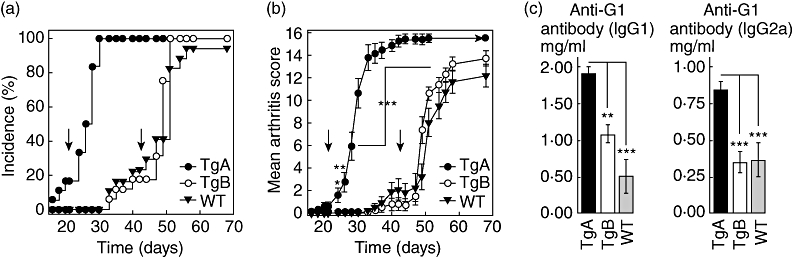
Comparison of (a) onset, incidence and (b) severity of arthritis of G1 domain of cartilage proteoglycan (PG)-induced arthritis (GIA) in T cell receptor transgenic (TCR-TgA), TCR-TgB and wild-type (WT) BALB/c mice (n = 18 mice in each group) and (c) antibody levels measured at the end of experiment (on day 70). WT and 5/4E8 epitope-specific TCR-TgA and TCR-TgB mice (all in BALB/c background) were immunized with rhG1 in dimethyldioctadecylammonium bromide (DDA) adjuvant three times. Mice were assessed for arthritis onset and severity three times a week from day 18. Vertical arrows indicate the second and third immunizations. All TCR-TgA mice were arthritic at the time of the third injection, and peripheral small joints (paws) were deformed and stiff by day 38–42. Significant differences (TCR-TgA versus TCR-TgB) are indicated (*P ≤ 0·05; **P ≤ 0·01; ***P ≤ 0·001).
To determine whether the differences in clinical phenotype were associated with differences in serum parameters, we assessed the serum levels of antibodies and cytokines using antigen-specific ELISA and CBA, respectively. Anti-G1 domain-specific antibodies were hardly detectable before the second immunization in either transgenic line (data not shown). The second immunization (day 21) induced significant anti-G1 IgG1 antibody secretion in TCR-TgA mice (65 ± 45 µg/ml), whereas the level of this antibody isotype remained almost undetectable in TCR-TgB mice (14 ± 9·8 µg/ml). TCR-TgA mice also exhibited higher serum levels of anti-G1 IgG2a antibodies compared to TCR-TgB mice (439 ± 364 µg/ml versus 176·1 ± 149 µg/ml). While the relatively high deviations and relatively low animal number did not allow us to perform a correct statistical analysis during the entire immunization period, we had a sufficient number of animals in all three genotypes at the end of the experiment (Fig. 1), showing significant differences in anti-G1 antibodies between TCR-TGA and TCR-TgB mice (Fig. 1c). This significantly higher antibody secretion observed in TCR-TgA mice was associated with a significantly higher B cell percentage compared to that in TCR-TgB mice during the whole immunization period (12·7–28·1% or 23·4–39·9% versus 5·5–13·9% or 16·8–23·6% in the joint draining lymph nodes or the spleen, respectively).
No significant differences were observed in the serum levels of IL-1β, IL-12p70 or IL-17 between the two TCR-Tg lines (data not shown). Serum IL-6 concentration was elevated in TCR-TgA mice throughout the entire experiment (in the range of 18–346 pg/ml). TCR-TgA mice also showed higher serum levels of IFN-γ (28 ± 8·0 pg/ml) and TNF-α (29 ± 6·4 pg/ml), when the first symptoms (acute phase) of arthritis appeared, and compared to serum levels of these two cytokines in the TCR-TgB line (7·0 ± 1·5 pg/ml and 14 ± 3·7 pg/ml, respectively).
Comparison of T cell activation markers and co-stimulatory molecules in TCR-TgA and TCR-TgB mice during the course of immunization
We compared the expression of activation markers on the cell surface of CD4+ T cells in the peripheral LNs and spleen to clarify whether there was any difference in the activation level of T cells in the two TCR-Tg lines. We detected a rapidly accumulating CD25highCD4+ T cell population after the first rhG1 injection, and the size of this cell population remained large in the peripheral LNs and spleens of TCR-TgA mice (Fig. 2a). In contrast, the ratio of CD25highCD4+ T cell population in TCR-TgB mice immunized with rhG1 remained as small as in naive mice during the entire experiment (Fig. 2a). The expression of the CD44 molecule (a marker of activated/memory T cells) in the CD4+ T cell population showed a similar pattern to that of CD25 in TCR-TgA mice (Fig. 2b), but was less consistent and remained significantly lower in TCR-TgB at almost all time-points tested (Fig. 2b).
Fig. 2.
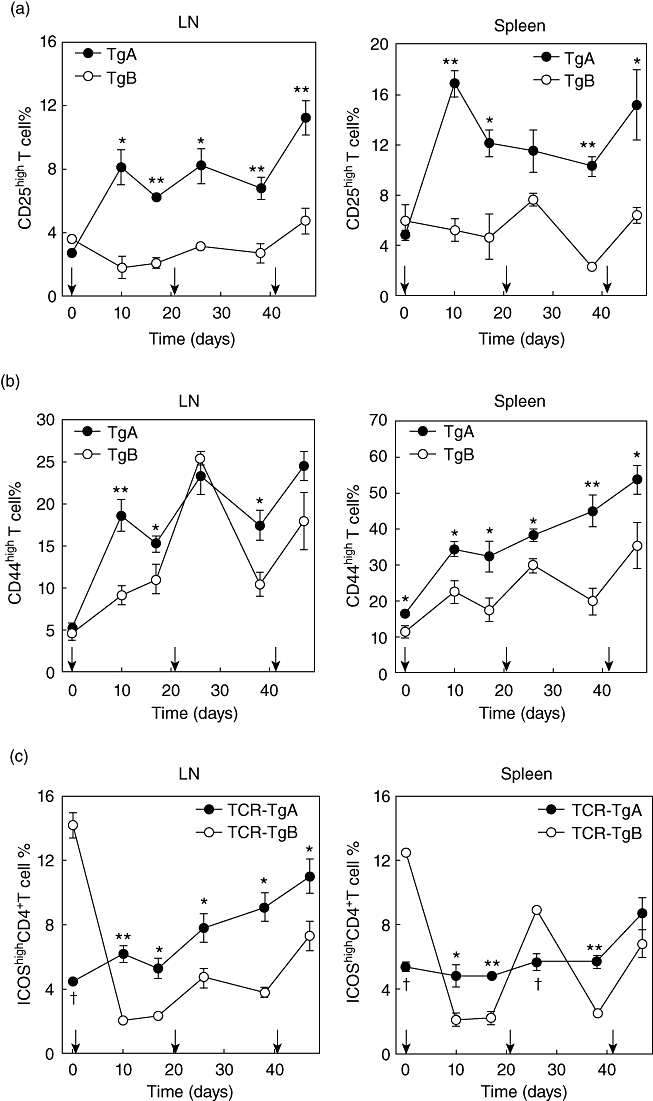
Characterization of T cell activation markers and co-stimulatory molecules by flow cytometry. Changes in the percentage of activated CD4+ T cells during rhG1 immunization of proteoglycan T cell receptor transgenic (PG-TCR-Tg) mice. Diagrams show the percentages of CD25highCD4+ T cells (a) and CD44highCD4+ T cells (b) in the peripheral lymph nodes (LN) and spleens of TCR-TgA and TCR-TgB mice. Changes in the proportions of inducible T cell co-stimulator (ICOS)highCD4+ T cells during immunization (c). The line graphs show the percentages of ICOShighCD4+ T cells in peripheral LNs and spleens of TCR-TgA and TCR-TgB mice. In all diagrams the mean ± standard error of the mean values at each time-point were obtained from four animals. Vertical arrows on the x-axes depict time-points of rhG1 immunizations. Significantly higher (*P ≤ 0·05; **P ≤ 0·01) or lower (†P ≤ 0·05) values (TCR-TgA versus TCR-TgB) are indicated.
Because co-stimulatory molecules profoundly influence T cell activation and signalling, we examined CD28, CTLA-4, ICOS and PD-1 expression in CD4+ T cells in both TCR-Tg mouse lines. No significant differences were observed in CD28high, CTLA-4high or PD-1high CD4+ T cell percentages (data not shown). Naive TCR-TgB mice exhibited three to four times more ICOShighCD4+ T cells compared to naive TCR-TgA mice (Fig. 2c). However, during immunization the proportion of ICOShighCD4+ T cells showed a constantly increasing trend in TCR-TgA mice, especially in the peripheral LNs. In contrast, the percentage of ICOShighCD4+ cells in TCR-TgB mice decreased to a very low level after the first rhG1 injection and remained at this significantly reduced level at most time points tested (Fig. 2c). Interestingly, a peak of both CD44 in LNs (Fig. 2b) and ICOS in spleen (Fig. 2c) expression was observed in CD4+ T cells in TCR-TgB mice 5 days after the second rhG1 injection, which may indicate a temporary (short-term) activation of these preactivated CD4+ T cells following the antigen injection.
Accelerated in vitro apoptosis in TCR-TgB CD4+ T cells
Impaired AICD of arthritogenic T cells is considered to be one of the underlying mechanisms in the development of arthritis, and we have observed previously spontaneous arthritis in aging TCR-TgA mice [17]. Therefore, we hypothesized that the phenotypic differences between the two lines might be attributed to differences in T cell signalling and apoptosis. Accelerated apoptosis of CD4+ T cells in TCR-TgB mice could explain the phenotype of attenuated arthritis. Therefore, we cultured purified CD4+ T cells with 5/4E8 peptide pretreated irradiated A20 antigen-presenting cells for 3 days in vitro and measured the antigen stimulation-induced apoptotic T cell death (Fig. 3). The percentage of early apoptotic (annexin V+/7-AAD–) T cells was significantly higher in the cell cultures harvested from TCR-TgB mice during the entire immunization period (Fig. 3b,c), whereas the corresponding CD4+ T cell cultures of TCR-TgA mice contained significantly more viable (non-apoptotic annexin V-/7-AAD–) T cells (Fig. 3a,c).
Fig. 3.
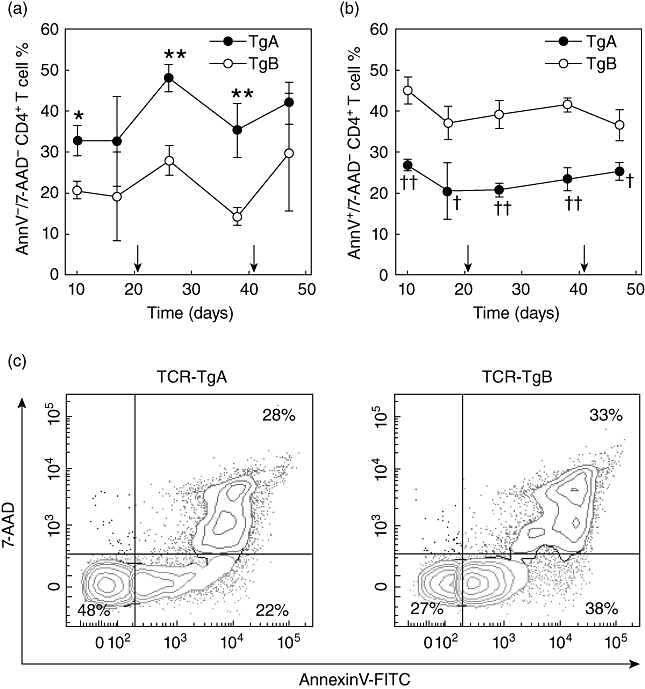
Comparison of in vitro apoptosis (activation-induced cell death) of T cells isolated from the spleens of TCR-TgA and TCR-TgB mice. Purified T cells were cultured with irradiated A20 cells pretreated with 5/4E8 synthetic peptide for 72 h. Apoptosis was assessed by annexin V/7-amino-actinomycin (7-AAD) staining using flow cytometry. Diagrams show (a) the percentages of viable (annexin V-/7-AAD–) and (b) early apoptotic (annexin V+/7-AAD–) CD4+ T cells. Arrows on the x-axis indicate the second and third rhG1 immunizations. Significantly higher *P < 0·05; **P < 0·01 or lower †P < 0·05; ††P < 0·01 values (TCR-TgA versus TCR-TgB) are indicated (n = 3 animals at each time-point). (c) Representative flow cytometric contour plots show annexin V/7-AAD staining of in vitro-stimulated T cells of TCR-TgA and TCR-TgB mice. The percentages of viable (annexin V-/7-AAD–, lower left quadrant, green), early apoptotic (annexin V+/7-AAD–, lower right quadrant, orange) and late apoptotic (annexin V+/7-AAD+, upper right quadrant, red) cells are indicated. Samples were collected 10 days after the second immunization.
Higher TCR expression and TCR signalling strength in CD4+ T cells in TCR-TgB compared to TCR-TgA mice
AICD is controlled by TCR signal strength together with co-stimulatory signals [21]. Although the two transgenic lines expressed TCRs specific for the same epitope (same transgenic construct), they were generated independently. Therefore, it was possible that the TCR expression on CD4+ T cells from the two transgenic lines was different. We compared the cell surface expression of TCR Vβ4, CD3 and CD4 in transgenic T cells using a flow cytometric saturation binding test (Fig. 4) [22]. Surprisingly, TCR-TgB mice expressed approximately twice as much TCR Vβ4 and CD3 as TCR-TgA mice, but the amount of CD4 was similar in both strains (Fig. 4). Thus, the amount of CD4 expression did not correlate with other components of the TCR complex such as Vβ4 and CD3. To confirm this difference, we determined the copy numbers of the TCR-Tg Vβ4 and Vα1·1 chains by qPCR in the genomic DNA isolated from both TCR-Tg lines. Interestingly, there were seven copies of Vα1·1 but only three copies of Vβ4 of the transgene in homozygous TCR-TgA mice, whereas there were six copies of both TCR chains in homozygous TCR-TgB animals. Because TCR is always expressed as a heterodimer on the surface of peripheral T cells, these qPCR results confirmed the results of our flow cytometric analysis, demonstrating that TCR-TgA mice indeed expressed half as much TCR as TCR-TgB mice.
Fig. 4.
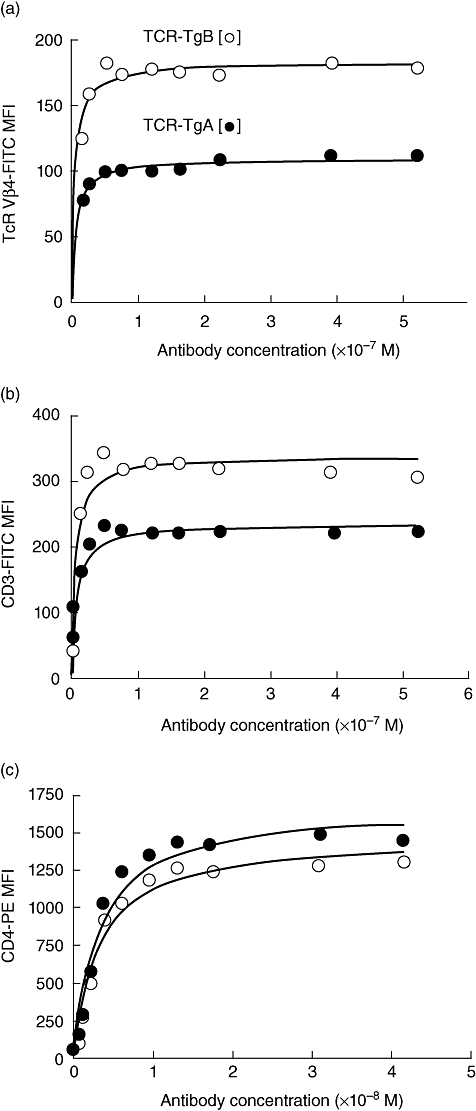
Comparison of cell surface expression of transgenic T cell receptor (Tg-TCR) (Vβ4 chain), CD3 and CD4 on T cells isolated from naive TCR transgenic (TCR-TgA) and TCR-TgB mice. Saturation binding test used fluorescently labelled anti-TCR Vβ4 (a), anti-CD3 (b) and anti-CD4 (c) monoclonal antibodies. Filled and open circles represent titration data from TCR-TgA and TCR-TgB mice, respectively. Significantly more anti-TCR Vβ4 and anti-CD3 antibodies bound to the T cells of TCR-TgB mice at and above saturation levels of the antibodies, indicating elevated surface expression of the transgenic TCR in the TCR-TgB line.
The higher TCR expression, associated with the delayed onset and decreased severity of arthritis, prompted us to determine if AICD of CD4+ T cells increased in TCR-TgB mice. More specifically, we asked whether the significantly higher expression of TCR and CD3 in TCR-TgB mice correlated with the phosphorylation levels of ZAP-70, ERK1/2 and p38, three key parameters of T cell signal strength. To this end, purified CD4+ T cells were co-cultured with A20 cells in the presence of the 5/4E8 peptide for 1 h (Fig. 5). We found that the TCR signal-elicited phosphorylation of ZAP-70 and p38 was significantly higher in TCR-TgB than in TCR-TgA mice, whereas ERK1/2 phosphorylation was similar (Fig. 5a,b).
Fig. 5.
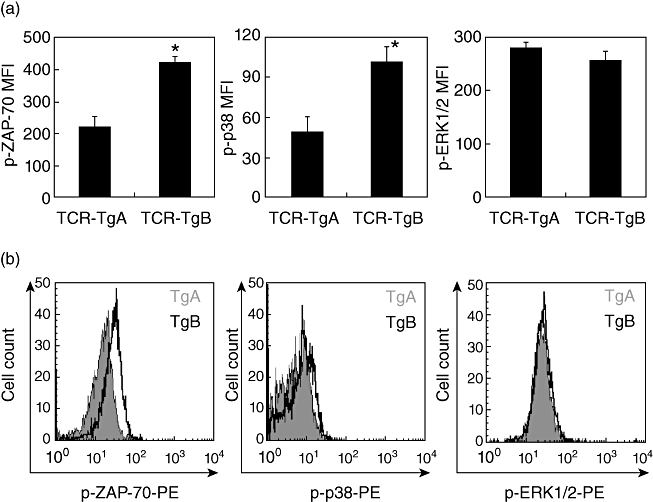
In vitro comparison of T cell receptor (TCR) signalling in T cells isolated from the spleens of naive TCR-TgA and TCR-TgB mice. (a) Irradiated A20 cells were pre-incubated with 5/4E8 synthetic peptide overnight, washed and cultured with TCR-TgA and TCR-TgB CD4+ T cells for 1 h. Phosphorylation status of zeta-chain-associated protein kinase 70 (ZAP-70), p38 and extracellular regulated kinase (ERK)1/2 was detected with phospho-specific antibodies using flow cytometry. Results shown are the mean ± standard error of the mean from six animals in each group. Significant differences are indicated (*P < 0·05). (b) Representative flow cytometric histograms illustrate the differences between TCR-TgA (grey, filled histograms) and TCR-TgB (black, open histograms) in the phosphorylation levels of ZAP-70, p38 and ERK1/2.
Discussion
Co-operation between autoreactive (Th1 and Th17) CD4+ T cells and antibody-secreting (and antigen-presenting) B cells is indispensable for the initiation and progression of systemic autoimmune arthritis. Both PGIA and GIA, similar to RA, are T cell-dependent B cell-mediated autoimmune diseases [1,2,4,12]. Adoptive transfer experiments have proved that neither T nor B cells alone were sufficient for the transfer of the disease, and that only co-injection of both cell types was successful [6,9]. Therefore, T–B cell co-operation is definitely required for the development of PGIA and GIA. In this TCR-Tg study we used the GIA model by immunizing mice with rhG1 protein, and stimulating/activating CD4+ T cells of TCR-Tg mice with their 5/4E8 epitope-specific synthetic peptide. We observed that rhG1 provoked severe arthritis in TCR-TgA mice, whereas a milder form of the disease with significantly delayed onset developed in TCR-TgB mice, which was comparable to that described in WT BALB/c mice [12]. This was an unexpected result, because both TCR-TgA and TCR-TgB mice possess the same 5/4E8 (PG)-specific (monoclonal) TCR expressing CD4+ T cell repertoire.
From the laboratory parameters analysed, elevated levels of serum anti-G1 antibodies (IgG1/IgG2a, cross-reactive with the G1 domain of mouse PG) in the TCR-TgA line, shortly after the second immunization but before the appearance of visible inflammatory symptoms, suggest that these antibodies may have contributed to the accelerated disease development. The higher B cell percentage in TCR-TgA mice was consistent with the higher serum anti-G1 antibody levels. Previous studies have indicated that the concentrations of antigen-specific antibodies in sera, especially autoantibodies, correlate well with the severity of the disease in WT BALB/c mice, and that such antibodies can already be detected long before the appearance of the first inflammatory symptoms [1,2,9,12]. However, despite the higher B cell numbers at most time-points tested during the immunization period, the serum antibody levels in TCR-TgA mice never reached the levels measured in WT BALB/c controls (data not shown). One possible explanation for the lower autoantibody concentration in TCR-Tg mice is the highly restricted TCR repertoire, specific for only one epitope. None the less, the role of B cells in the development of the disease is unquestionable, even in this epitope-restricted TCR-Tg model [12].
Secretion of antigen-specific antibodies, however, is only part of B cell function because B cells are important antigen-presenting cells in PGIA [3,23–25] and most probably in GIA as well. Therefore, specific co-operation between antigen-primed T cells and antigen-specific B cells appears to be a critical component of arthritis induction. The higher number of B cells in TCR-TgA mice could present the antigen to T cells more effectively, leading to accelerated T cell activation. Indeed, the highest expression of the T cell activation markers during the immunization with rhG1 protein was observed on TCR-TgA CD4+ T cells (Fig. 2).
As mentioned above, the CD4+ T cells of TCR-TgA mice, unlike those of TCR-TgB mice, expressed more activation markers, such as CD25 and CD44. Activation of CD4+ T cells is central for the initiation of both PGIA and GIA [7,12], and these T cells in TCR-TgB mice were not activated as strongly as TCR-TgA T cells by immunization. Therefore, the lower percentage of activated T cells in TCR-TgB mice could account for the delayed disease onset and less severe arthritis. An additional difference between the TCR-Tg lines was that a larger percentage of ICOShigh CD4+ T cells were present in TCR-TgA mice, especially in the peripheral LNs (Fig. 3). ICOS has been shown to be indispensable in collagen-induced arthritis and K/BxN arthritis models [26–28], and certain ICOS polymorphisms are associated with RA [29]. ICOS is expressed prominently by follicular T helper (Tfh) cells in lymphoid organs [30]. Tfh cells have been shown to provide critical support to autoreactive B cells in systemic autoimmunity [31] and in the initiation of arthritis in K/BxN mice [32]. Therefore, here, the lower percentage of ICOShigh CD4+ T cells (most probably Tfh) cells could also contribute to the diminished antibody production and the lower severity of autoimmune arthritis in TCR-TgB mice.
The most interesting observation in TCR-TgA and TCR-TgB mice was the differential susceptibility of their CD4+ T cells to activation-induced apoptosis. Upon stimulation with the 5/4E8 peptide, T cells isolated from TCR-TgB mice exhibited higher sensitivity to apoptosis compared to the CD4+ T cells from TCR-TgA mice. Altered T cell apoptosis has been found to be a critical factor in the development of autoimmune arthritis [17], and TCR signalling threshold and co-stimulatory signals have been shown to regulate AICD [33–35]. In our case, a saturation-binding test and qPCR analysis proved that CD4+ T cells from TCR-TgB mice expressed twice as much TCR and CD3 molecules than the T cells from the TCR-TgA line. Variance in the number of expressed TCRs on the cell surface may significantly influence signal strength through the TCR-initiated signalling pathway [33]. Indeed, when TCR-TgB CD4+ T cells were engaged with the 5/4E8 peptide, significantly higher levels of phosphorylation of the key downstream signalling molecules (ZAP-70 and p38) were detected than in TCR-TgA T cells. In other words, the higher the TCR expression, the stronger the signalling response to the 5/4E8 epitope became. Thus, a ‘too strong’ TCR signal could have led to extensive apoptosis of self-reactive T cells during immunization with an antigen (rhG1) containing the 5/4E8 epitope. The importance of TCR signal strength has also been underlined in a recent human study: RA patients showed altered ERK and ZAP-70 phosphorylation levels upon CD3/CD28 stimulation [36], implicating a potential human relevance for our results.
Based on these results, we propose that TCR signal strength controls arthritis susceptibility in the two lines of PG-specific TCR-Tg mice. The differences in the phosphorylation levels of the downstream effectors in TCR signalling offered a logical explanation for the differences in arthritis severity. The different TCR levels in the two transgenic lines resulted in significant alterations of TCR signal strength. A strong PG (5/4E8 epitope)-specific TCR-initiated signal in TCR-TgA mice led to optimal T cell activation and, compared to WT, generated a ‘super-arthritic’ phenotype. In contrast, an extremely ‘strong’ TCR signal, due probably to higher TCR expression, led to in vivo T cell apoptosis and diminished arthritis in TCR-TgB mice. Thus, the correlation between TCR signal strength and arthritis severity is not linear: a weaker signal (as seen in the TCR TgA mice) appears to be more ‘optimal’ for the activation of self-reactive T cells.
Acknowledgments
We thank our colleagues who contributed to the development of the TCR-Tg mice, especially Dr. Suzanne E. Berlo and Dr. Chris P. Broeren (University of Utrecht, Utrecht, the Netherlands). We also thank Dr Cynthia Detre (BIDMC Division of Immunology, Harvard Center for Life Sciences, Boston, MA, USA), who performed the saturation binding test and the colleagues who helped maintain the TCR-Tg colonies for research. This work was supported by grants from the National Institutes of Health (NIH/NIAMS R01 AR040310, R01 AR059356), and the Dutch Arthritis Association.
Disclosure
The authors do not have any conflicts of interest or any other disclosures.
References
- 1.Glant TT, Mikecz K, Arzoumanian A, Poole AR. Proteoglycan-induced arthritis in BALB/c mice. Clinical features and histopathology. Arthritis Rheum. 1987;30:201–12. doi: 10.1002/art.1780300211. [DOI] [PubMed] [Google Scholar]
- 2.Mikecz K, Glant TT, Poole AR. Immunity to cartilage proteoglycans in BALB/c mice with progressive polyarthritis and ankylosing spondylitis induced by injection of human cartilage proteoglycan. Arthritis Rheum. 1987;30:306–18. doi: 10.1002/art.1780300310. [DOI] [PubMed] [Google Scholar]
- 3.Glant TT, Buzas EI, Finnegan A, Negroiu G, Cs-Szabó G, Mikecz K. Critical role of glycosaminoglycan side chains of cartilage proteoglycan (aggrecan) in antigen recognition and presentation. J Immunol. 1998;160:3812–19. [PubMed] [Google Scholar]
- 4.Glant TT, Finnegan A, Mikecz K. Proteoglycan-induced arthritis: immune regulation, cellular mechanisms and genetics. Crit Rev Immunol. 2003;23:199–250. doi: 10.1615/critrevimmunol.v23.i3.20. [DOI] [PubMed] [Google Scholar]
- 5.Finnegan A, Mikecz K, Tao P, Glant TT. Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J Immunol. 1999;163:5383–90. [PubMed] [Google Scholar]
- 6.Bardos T, Mikecz K, Finnegan A, Zhang J, Glant TT. T and B cell recovery in arthritis adoptively transferred to SCID mice: antigen-specific activation is required for restoration of autopathogenic CD4+ Th1 cells in a syngeneic system. J Immunol. 2002;168:6013–21. doi: 10.4049/jimmunol.168.12.6013. [DOI] [PubMed] [Google Scholar]
- 7.Boldizsar F, Tarjanyi O, Nemeth P, Mikecz K, Glant TT. Th1/Th17 polarization and acquisition of an arthritogenic phenotype in arthritis-susceptible BALB/c, but not in MHC-matched, arthritis-resistant DBA/2 mice. Int Immunol. 2009;21:511–22. doi: 10.1093/intimm/dxp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee S, Webber C, Poole AR. The induction of arthritis in mice by the cartilage proteoglycan aggrecan: roles of CD4+ and CD8+ T cells. Cell Immunol. 1992;144:347–57. doi: 10.1016/0008-8749(92)90250-s. [DOI] [PubMed] [Google Scholar]
- 9.Mikecz K, Glant TT, Buzás E, Poole AR. Proteoglycan-induced polyarthritis and spondylitis adoptively transferred to naive (nonimmunized) BALB/c mice. Arthritis Rheum. 1990;33:866–76. doi: 10.1002/art.1780330614. [DOI] [PubMed] [Google Scholar]
- 10.Szanto S, Bárdos T, Szabo Z, et al. Induction of arthritis in HLA-DR4-humanized and HLA-DQ8-humanized mice by human cartilage proteoglycan aggrecan but only in the presence of an appropriate (non-MHC) genetic background. Arthritis Rheum. 2004;50:1984–95. doi: 10.1002/art.20285. [DOI] [PubMed] [Google Scholar]
- 11.Buzas E, Vegvari A, Murad YM, Finnegan A, Mikecz K, Glant TT. T-cell recognition of differentially tolerated epitopes of cartilage proteoglycan aggrecan in arthritis. Cell Immunol. 2005;235:98–108. doi: 10.1016/j.cellimm.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 12.Glant TT, Radacs M, Nagyeri G, et al. Proteoglycan-induced arthritis and recombinant human proteoglycan aggrecan G1 domain-induced arthritis in BALB/c mice resembling two types of rheumatoid arthritis. Arthritis Rheum. 2011;63:1312–21. doi: 10.1002/art.30261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murad YM, Szabo Z, Ludanyi K, Glant TT. Molecular manipulation with the arthritogenic epitopes of the G1 domain of human cartilage proteoglycan aggrecan. Clin Exp Immunol. 2005;142:303–11. doi: 10.1111/j.1365-2249.2005.02921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buzas EI, Brennan FR, Mikecz K, et al. A proteoglycan (aggrecan)-specific T cell hybridoma induces arthritis in BALB/c mice. J Immunol. 1995;155:2679–87. [PubMed] [Google Scholar]
- 15.Berlo SE, Van Kooten PJ, ten Brink CB, et al. Naive transgenic T cells expressing cartilage proteoglycan-specific TCR induce arthritis upon in vivo activation. J Autoimmun. 2005;25:172–80. doi: 10.1016/j.jaut.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 16.Berlo SE, Guichelaar T, ten Brink CB, et al. Increased arthritis susceptibility in cartilage proteoglycan-specific T cell receptor-transgenic mice. Arthritis Rheum. 2006;54:2423–33. doi: 10.1002/art.22013. [DOI] [PubMed] [Google Scholar]
- 17.Boldizsar F, Kis-Toth K, Tarjanyi O, et al. Impaired activation-induced cell death promotes spontaneous arthritis in antigen (cartilage proteoglycan)-specific T cell receptor transgenic mice. Arthritis Rheum. 2010;62:2984–94. doi: 10.1002/art.27614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glant TT, Mikecz K. Proteoglycan aggrecan-induced arthritis. A murine autoimmune model of rheumatoid arthritis. Methods Mol Med. 2004;102:313–38. doi: 10.1385/1-59259-805-6:313. [DOI] [PubMed] [Google Scholar]
- 19.Brennan FR, Negroiu G, Buzas EI, Fülöp C, Mikecz K, Glant TT. Presentation of cartilage proteoglycan to a T cell hybridoma derived from a mouse with proteoglycan-induced arthritis. Clin Exp Immunol. 1995;100:104–10. doi: 10.1111/j.1365-2249.1995.tb03610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krutzik PO, Hale MB, Nolan GP. Characterization of the murine immunological signaling network with phosphospecific flow cytometry. J Immunol. 2005;175:2366–73. doi: 10.4049/jimmunol.175.4.2366. [DOI] [PubMed] [Google Scholar]
- 21.Hsu HC, Scott DK, Mountz JD. Impaired apoptosis and immune senescence – cause or effect? Immunol Rev. 2005;205:130–46. doi: 10.1111/j.0105-2896.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- 22.Wiczling P, Rosenzweig M, Vaickus L, Jusko WJ. Pharmacokinetics and pharmacodynamics of a chimeric/humanized anti-CD3 monoclonal antibody, otelixizumab (TRX4), in subjects with psoriasis and with type 1 diabetes mellitus. J Clin Pharmacol. 2010;50:494–506. doi: 10.1177/0091270009349376. [DOI] [PubMed] [Google Scholar]
- 23.O'Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174:3781–8. doi: 10.4049/jimmunol.174.6.3781. [DOI] [PubMed] [Google Scholar]
- 24.O'Neill SK, Glant TT, Finnegan A. The role of B cells in animal models of rheumatoid arthritis. Front Biosci. 2007;12:1722–36. doi: 10.2741/2184. [DOI] [PubMed] [Google Scholar]
- 25.O'Neill SK, Cao Y, Hamel KM, Doodes PD, Hutas G, Finnegan A. Expression of CD80/86 on B cells is essential for autoreactive T cell activation and the development of arthritis. J Immunol. 2007;179:5109–16. doi: 10.4049/jimmunol.179.8.5109. [DOI] [PubMed] [Google Scholar]
- 26.Wilson EH, Zaph C, Mohrs M, et al. B7RP-1-ICOS interactions are required for optimal infection-induced expansion of CD4+ Th1 and Th2 responses. J Immunol. 2006;177:2365–72. doi: 10.4049/jimmunol.177.4.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frey O, Meisel J, Hutloff A, et al. Inducible costimulator (ICOS) blockade inhibits accumulation of polyfunctional T helper 1/T helper 17 cells and mitigates autoimmune arthritis. Ann Rheum Dis. 2010;69:1495–501. doi: 10.1136/ard.2009.119164. [DOI] [PubMed] [Google Scholar]
- 28.Frey O, Reichel A, Bonhagen K, Morawietz L, Rauchhaus U, Kamradt T. Regulatory T cells control the transition from acute into chronic inflammation in glucose-6-phosphate isomerase-induced arthritis. Ann Rheum Dis. 2010;48:345–53. doi: 10.1136/ard.2009.123422. [DOI] [PubMed] [Google Scholar]
- 29.Kim YO, Kim HJ, Kim SK, Chung JH, Hong SJ. Association of the CD28/CTLA4/ICOS polymorphisms with susceptibility to rheumatoid arthritis. Clin Chem Lab Med. 2010;48:345–53. doi: 10.1515/CCLM.2010.074. [DOI] [PubMed] [Google Scholar]
- 30.Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192:1553–62. doi: 10.1084/jem.192.11.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linterman MA, Rigby RJ, Wong RK, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. 2009;206:561–76. doi: 10.1084/jem.20081886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Victoratos P, Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity. 2009;30:130–42. doi: 10.1016/j.immuni.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 33.She J, Matsui K, Terhorst C, Ju ST. Activation-induced apoptosis of mature T cells is dependent upon the level of surface TCR but not on the presence of the CD3 zeta ITAM. Int Immunol. 1998;10:1733–40. doi: 10.1093/intimm/10.11.1733. [DOI] [PubMed] [Google Scholar]
- 34.Metz DP, Farber DL, Taylor T, Bottomly K. Differential role of CTLA-4 in regulation of resting memory versus naive CD4 T cell activation. J Immunol. 1998;161:5855–61. [PubMed] [Google Scholar]
- 35.Zhang J, Xu X, Liu Y. Activation-induced cell death in T cells and autoimmunity. Cell Mol Immunol. 2004;1:186–92. [PubMed] [Google Scholar]
- 36.Singh K, Deshpande P, Pryshchep S, et al. ERK-dependent T cell receptor threshold calibration in rheumatoid arthritis. J Immunol. 2009;183:8258–67. doi: 10.4049/jimmunol.0901784. [DOI] [PMC free article] [PubMed] [Google Scholar]