

SUMMARY
While disordered to ordered rearrangements are relatively common, the ability of proteins to switch from one ordered fold to a completely different fold is generally regarded as rare and few fold switches have been characterized. Here, in a designed system, we examine the mutational requirements for transitioning between folds and functions. We show that switching between monomeric 3α and 4β+α folds can occur in multiple ways with successive single amino acid changes at diverse residue positions, raising the likelihood that such transitions occur in the evolution of new folds. Even mutations on the periphery of the core can tip the balance between alternatively folded states. Ligand-binding studies illustrate that a new IgG-binding function can be gained well before the relevant 4β+α fold is appreciably populated in the unbound protein. The results provide new insights into the evolution of fold and function.
INTRODUCTION
Some proteins can adopt more than one folded state and have been termed “metamorphic” (Murzin, 2008). Prions are a classic example of the malleability of polypeptide chains, where conformational change from a benign, predominantly α-helical form to an infectious, β-strand rich state is driven by multimerization (Weissmann, 2005). However, there are a small but growing number of other naturally occurring examples such as lymphotactin (Tuinstra et al., 2008), Mad2 (Luo and Yu, 2008; Mapelli and Musacchio, 2007), and CLIC1 (Littler et al., 2004), suggesting that the phenomenon of fold switching may be more general. In these proteins, the equilibrium is shifted from one fold topology to another by changes in environmental factors such as salt conditions, the presence of a ligand, and redox state. Other studies, such as those on the Cro family of repressors (Roessler et al., 2008) and RfaH (Belogurov et al., 2009), support the idea that some folds may have resulted from switching an existing structure rather than evolving independently. Common features of such switchable folds are 1) flexible regions and diminished stability to allow large scale changes, 2) a fair degree of mutual exclusivity in the core regions, and 3) the generation of a new binding surface that stabilizes the alternative fold and expands function (Bryan and Orban, 2010). Further, theoretical studies predict that the sequences encoding certain protein folds are switchable to numerous other folds, and that protein fold-space may be more inter-connected than previously considered (Meyerguz et al., 2007).
In addition to natural examples of fold switches, protein design has been used to investigate the question of how high the amino acid sequence identity of two proteins can be while maintaining different fold topologies (Ambroggio and Kuhlman, 2006; Rose and Creamer, 1994). Some of the earlier studies in this area showed that sequence identities of 50% or more could be achieved but that aggregation became a problem at higher identities making biophysical characterization difficult (Blanco et al., 1999; Dalal and Regan, 2000). More recently, a binary system was designed where different fold topologies were obtained with very high (>85%) sequence identities (Alexander et al., 2007; Alexander et al., 2009; He et al., 2008). The starting points were two small 56 amino acid domains, termed GA and GB, from the multi-domain Streptococcus cell surface protein G (Fahnestock et al., 1986). The GA domain adopts a 3-α helix bundle (3α) structure (Johansson et al., 1997) and binds human serum albumin (HSA) (Falkenberg et al., 1992), whereas the GB domain has a 4β+α fold (Gronenborn et al., 1991) and binds IgG (Myhre and Kronvall, 1977). The albumin- and IgG-binding epitopes (Sauer-Eriksson et al., 1995; Lejon et al., 2004) were engineered into both domains, creating latent binding sites that could be exposed on fold switching. The GA and GB sequences were then co-evolved using site-directed mutagenesis and phage display, increasing identity at mutation tolerant sites using binary sequence space (i.e. only GA or GB amino acids) (Alexander et al., 2007). NMR structures were determined for 88% (He et al., 2008) and 95% (Alexander et al., 2009) identity protein pairs, while still maintaining different folds and functions.
The ability of proteins to switch folds is generally regarded as rare and relatively few have been characterized structurally. One possible reason there are not more reports is that the sequences of many such proteins may be inherently transient and rapidly evolve to their new functions and folds. The designed GA/GB system therefore provides an opportunity to examine the mutational requirements for transitioning between folds and functions. Here, we describe the three-dimensional structures for a series of high sequence identity GA/GB mutants, each being 98% identical to the next in the series. We show that the folds of these proteins switch between 3α and 4β+α with successive single amino acid changes at diverse residue positions, and that there is a near complete (≥95%) shift in the equilibrium between the two states. Thus, the pathway for fold switching is not unique, raising the probability of such events occurring. Moreover, ligand-binding studies on these high sequence identity mutants illustrate that changes in fold and function are not perfectly correlated. In our designed system, fold switching can be abrupt, occurring with a single amino acid mutation. However, the characteristics of a new binding function can be displayed well before the corresponding fold is appreciably populated in the unbound proteins. Overall, the results presented here provide new insights into how different folds can be closely connected in sequence space, and how new functions can evolve.
RESULTS
Description of fold topologies
The amino acid sequences of the four proteins described in this study, GA98, GB98, GB98-T25I, and GB98-T25I,L20A are shown in Figure 1A, highlighting the single residue positions of non-identity. Thus GA98 is converted to GB98 by mutating L45 to Y45, a T25I mutation changes GB98 to GB98-T25I, and GB98-T25I,L20A is generated through an amino acid change at position 20 of GB98-T25I. Although the sequences of these proteins are nearly identical, the single amino acid mutation from one protein to the next leads to significant differences in NMR spectra, reflecting large-scale alterations in 3D structure. Spectra in the first and third panels of Figure 1B have cross-peak patterns typical of other 3α folded proteins in this series, while the spectra in the second and fourth panels are characteristic of a 4β+α fold topology. To more quantitatively describe the affect of these mutations, NMR assignments were obtained for these proteins using heteronuclear triple resonance NMR spectroscopy and 3D structures were determined. All four proteins are monomeric under NMR conditions from size exclusion chromatography and light scattering measurements. GA98, GB98, GB98-T25I, and GB98-T25I,L20A have a melting temperature (Tm) of 37°C, 35°C, 36°C, and 46°C, respectively. Due to the relatively low stabilities, NMR spectra were recorded at 5°C. Figure 1C shows representative structures from each NMR ensemble, highlighting the amino acid differences between proteins. Complete structure statistics are summarized in Table 1 and the NMR ensembles for all four proteins are shown in Figure 2. The Protein Data Bank/BioMagResBank accession codes for GA98, GB98, GB98-T25I, and GB98-T25I,L20A are 2LHC/17839, 2LHD/17840, 2LHG/17843, and 2LHE/17841, respectively.
Figure 1.

Single amino acid mutations leading to fold switching. (A) Alignment of amino acid sequences for the four proteins in this study, highlighting the positions at which changes lead to switching between 3α and 4β+α folds. (B) 2D 15N-HSQC spectra for GA98 (left), GB98 (center left), GB98-T25I (center right), and GB98-T25I,L20A (right). Viewing NMR spectra from left to right, large differences are observed from one spectrum to the next as the three successive single site mutations, L45Y, T25I, and L20A, are made (see also Figure S1). (C) Representative structures from the NMR ensembles of GA98, GB98, GB98-T25I, and GB98-T25I,L20A. Residues mutated are highlighted.
Table 1.
Summary of structure statistics
| GA98 | GB98 | GB98-T25Ia | GB98-T25I,L20A | |
|---|---|---|---|---|
| A. Experimental restraints | ||||
| NOE restraints | ||||
| All NOEs | 816 | 648 | 1067 | |
| Intraresidue | 507 | 395 | 627 | |
| Sequential (|i-j| =1) | 165 | 119 | 214 | |
| Medium-range (1<|i-j|≤5) | 78 | 43 | 57 | |
| Long-range (|i-j|>5) | 66 | 91 | 169 | |
| Hydrogen bond restraints | 50 | 62 | 62 | |
| Dihedral angle restraints | 72 | 64 | 64 | |
| Total NOE restraints | 938 | 774 | 1193 | |
| CS-Rosetta input | ||||
| 13Cα shifts | 39 | |||
| 13Cβ shifts | 18 | |||
| 13C′ shifts | 43 | |||
| 15N shifts | 47 | |||
| 1HN shifts | 47 | |||
| 1Hα shifts | 42 | |||
| B. RMSDs to the mean structure (Å) | ||||
| Over all residuesb | ||||
| Backbone atoms | 0.35 ± 0.10 | 0.81 ± 0.19 | 1.00 ± 0.27 | 0.61 ± 0.13 |
| Heavy atoms | 1.15 ± 0.16 | 1.66 ± 0.23 | 1.71 ± 0.35 | 1.44 ± 0.23 |
| Secondary structuresc | ||||
| Backbone atoms | 0.31 ± 0.09 | 0.55 ± 0.14 | 0.96 ± 0.26 | 0.32 ± 0.07 |
| Heavy atoms | 1.12 ± 0.15 | 1.24 ± 0.16 | 1.72 ± 0.35 | 0.94 ± 0.13 |
| C. Measures of structure quality | ||||
| Ramachandran distribution | ||||
| Most favored, % | 85.1 ± 3.9 | 75.6 ± 3.7 | 95.2 ± 2.1 | 77.7 ± 3.1 |
| Additionally allowed, % | 12.5 ± 3.7 | 21.6 ± 4.5 | 4.8 ± 2.1 | 20.3 ± 3.7 |
| Generously allowed, % | 0.6 ± 0.9 | 1.9 ± 1.4 | 0.0 ± 0.0 | 1.4 ± 1.4 |
| Disallowed, % | 1.8 ± 1.3 | 0.9 ± 1.0 | 0.0 ± 0.0 | 0.6 ± 1.1 |
| Bad contacts/100 residues | 3.3 ± 1.1 | 1.0 ± 1.1 | 0.5 ± 0.7 | 1.5 ± 1.2 |
| Overall dihedral G factor | 0.08 ± 0.03 | −0.04 ± 0.03 | 0.41 ± 0.03 | 0.01 ± 0.02 |
CS-Rosetta model based on assigned chemical shifts.
Residues 1–56 for GB98 and GB98-T25I, L20A. Residues 9–51 for GA98 and GB98-T25I.
The secondary structure elements used were as follows: GB98 and GB98-T25I, L20A, residues 1–8, 13–20, 23–36, 42–46, and 51–55; GA98, residues 9–23, 27–34, and 39–51; and GB98-T25I, residues 9–23, 28–34, and 39–52.
Figure 2.

NMR structures of the designed proteins GA98, GB98, GB98-T25I, and GB98-T25I,L20A. (A) Comparison of the 3α structures for GA98 (blue) and GB98-T25I (gold). The NMR ensemble for GA98 consists of 20 final structures. The GB98-T25I structure was determined using CS-Rosetta and main chain chemical shift assignments, and is represented by an ensemble of 10 final structures (see also Table S1). (B) Comparison of the 4β+α structures for GB98 (blue) and GB98-T25I,L20A (gold). Both NMR ensembles are of 20 final structures (see also Table S2).
GA98 (3α)
The GA98 fold has a 3α helical bundle topology similar to previously determined 3D structures of the original parental GA sequence (He et al., 2006), GA88 (He et al., 2008), and GA95 (Alexander et al., 2009). Therefore, as the GA sequences are co-evolved with GB sequences to high identity levels, the 3α structures of the designed proteins do not veer significantly from the wild-type fold topology. A comparison of the pairwise backbone RMSD’s is shown in Table S1. The N-terminus from residues 1–8 and the C-terminus from residues 52–56 are disordered with helices from residues 9–23, residues 27–34, and residues 39–51. Overall, the hydrophobic core interactions in GA98 are similar to those in GA95 and GA88. Core residues in GA98 are: A12, A16, and A20 (α1-helix); I33 and A36 (α2-helix); and V42, K46, and I49 (α3-helix). The only difference in amino acid sequence in going from GA95 to GA98 is mutation of I30 to F30. Where I30 contributes to the core of GA95 and is ~17% solvent accessible, F30 has limited interactions with the GA98 core through its β-methylene group and has a solvent exposed aromatic ring (Figure 3A). The I30F mutation is therefore expected to be destabilizing based on the GA98 structure. Indeed, the structural data is consistent with stability measurements, which show that an I30F mutation destabilizes the 3α fold by ~1.5 kcal/mol (Alexander et al., 2009).
Figure 3.
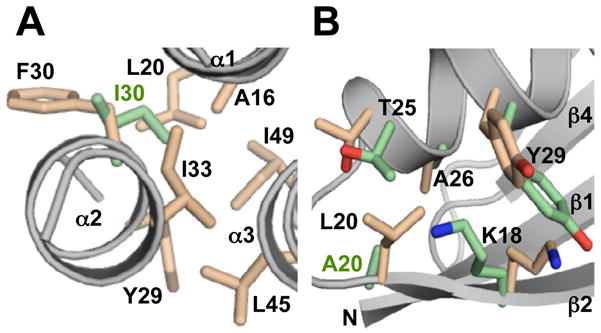
Structural changes in going from 95% to 98% sequence identity. (A) GA95 to GA98. A representative structure from the NMR ensemble for GA98, highlighting some of the hydrophobic core residues (pale orange) and the position of F30 relative to the core. By comparison, the position of I30 in GA95 is also shown (green). The main chain is in gray. (B) GB95 to GB98. A representative NMR structure of GB98 highlighting the A20L mutation site region in going from GB95 to GB98. GB98 side chain positions (pale orange) are compared with the corresponding GB95 conformations (green).
GB98 (4β+α)
A single amino acid change in GA98, L45Y, generates the GB98 sequence and the fold of this polypeptide chain is 4β+α rather than 3α. The differences between the GA98 and GB98 structures are striking. Residues that were at the unstructured ends of the GA98 sequence form the central β-strands, β1 and β4, of the GB98 fold. Residues in the α1- and α3-helices of GA98 correspond with the loop-β2-loop and loop-β3-loop-earlyβ4 regions of GB98, respectively. Comparison of the structure of GB98 with previous structures of the parent GB sequence (Gallagher et al., 1994), GB88 (He et al., 2008), and GB95 (Alexander et al., 2009) indicates very similar overall 4β+α fold topologies (Table S2). A single amino acid change from an alanine at position 20 to a leucine increases the identity from GB95 to GB98. The sterically more demanding leucine at position 20 is accommodated in the 4β+α fold with slight adjustments of the backbone and neighboring side chain conformations (Figure 3B). Nevertheless, the incorporation of the branched side chain at L20 does lead to increased steric interactions with adjacent residues, consistent with a decrease in Tm of GB98 by 12°C relative to GB95 (Alexander et al., 2009).
GB98-T25I (3α)
For GB98-T25I, de novo structure determination using extensive NOE restraints was not possible due to low sample solubility (<100 μM), which prevented complete assignments particularly of side chain resonances. However, assignment of a significant proportion of main chain 15N, 1HN, 1Hα, 13Cα, and 13C′ resonances (Table 1), as well as some 13Cβ signals, showed three distinct helical regions by consensus chemical shift index analysis (Wishart and Sykes, 1994). Comparison of backbone 1HN and 15N shifts showed a much closer match with those of GA98 rather than GB98 (Figure S1), indicating that fold switching from a 4β+α to 3α conformation had occurred with the T25I mutation. Further, a 3D structure was calculated from these experimental chemical shifts using the CS-Rosetta algorithm (Shen et al., 2008). We previously used CS-Rosetta to determine structures for GA88, GB88, GA95, and GB95, and found that these structures compared well (backbone RMSDs ~1.0–1.5 Å) with the structures calculated from mostly NOE restraint data (Shen et al., 2010). The CS-Rosetta structure of GB98-T25I shows a 3α fold with α-helices at residues 9–23, 28–34, and 39–52 and disordered regions at the N- and C-termini similar to GA98 (Figure 1C and 2A). The main differences from GA98 therefore are a slightly shorter α2-helix and an α3-helix that is longer by one residue.
At low contouring of the GB98-T25I 15N HSQC spectrum, a minor species (~5%) was also detected that has cross-peaks consistent with the alternative 4β+α fold. The minor component was detected reproducibly in different sample preparations that were purified on fresh columns, free of possible contaminants. The NMR peaks of the minor species could not be assigned directly due to the low intensity levels of these signals. However, the pattern of 15N HSQC peaks observed is very similar (but not identical) to that of GB98 (Figure 4). Indeed the differences from the GB98 spectrum are consistent with the incorporation of the T25I mutation. Two main lines of evidence indicate that the minor 4β+α species is in equilibrium with the major 3α species. Notably, changing the buffer from 100 mM KPi, pH 7.0 to 50 mM Tris, pH 8.0 leads to only the major 3α conformer within NMR detection limits - this process is reversible. Thus the minor 4β+α state cannot be an impurity or result from a covalent modification. Other evidence of equilibrium between the major and minor components comes from ligand binding experiments (described below). The data therefore indicate that both the 3α and 4β+α folds are detectably populated for the GB98-T25I sequence and that these two species are in slow exchange on the NMR time scale. This is similar to the case of lymphotactin where two different folds with identical amino acid sequences are in equilibrium and can be detected simultaneously in HSQC spectra (Tuinstra et al., 2008). Attempts to observe crosspeaks due to exchange between the two GB98-T25I conformations either in NOESY or zz-exchange experiments were unsuccessful, presumably due to the low relative population of the minor 4β+α state as well as the low solubility of the protein.
Figure 4.

15N HSQC spectra of GB98-T25I at low contour level. (A) Peaks due to the major 3α state of GB98-T25I are labeled in black. Other unlabeled low intensity peaks indicate the presence of a minor species. (B) Overlay with the 15N HSQC spectrum of GB98 (in red).
GB98-T251/L20A (4β+α)
The structure of GB98-T25I,L20A, the final mutant in this series, was determined using an extensive set of NOE and torsion angle restraints. The NMR structures show that mutation of a single amino acid, L20A, in the GB98-T25I sequence leads to switching from a predominantly (~95%) 3α fold in GB98-T25I to a 4β+α fold in GB98-T25I,L20A. The GB98-T25I,L20A structure is similar to other 4β+α structures in this series (Figure 2B, Table S2).
Ligand binding
NMR spectroscopy was used to measure IgG- and HSA-binding to GA and GB high identity proteins and the results are summarized in Table 2, Figure 5, and Figure S2. The dissociation constant (KD) obtained by NMR for binding between GA88 and HSA was also compared with the KD from isothermal titration calorimetry and found to be in good agreement (Figure S3). The 4β+α folded proteins, GB98 and GB98-T25I,L20A, bind to the wild-type ligand, IgG, with high affinity (KD<1 μM). The other designed GB proteins with 4β+α folds, ranging from GB77 to GB95, also bind to IgG tightly. None of the GB proteins, including GB98 and GB98-T25I,L20A, show any detectable binding to the GA ligand, HSA.
Table 2.
Ligand binding dataa
| Mutant | GA88 | GA95 | GA98 | GB98 | GB98-T25I | GB98-T25I,L20A |
|---|---|---|---|---|---|---|
| Fold | 3α | 3α | 3α | 4β+α | 3α (95%) 4β+α (5%) |
4β+α |
| KD(HSA) | 37 (31)b | 30 | 244 | n.b. | n.b. | n.b. |
| KD(IgG) | n.b. | n.b. | 62 | <1c | 15 | <1c |
Dissociation constants are in micromolar units. The experimental error is estimated to be ±30%. All binding measurements were done at 15°C except those for GB98-T25I, which were done at 5°C. n.b., no binding detected.
Value in parentheses is from isothermal titration calorimetry (see also Figure S3).
All GB protein was bound even at the lowest IgG concentrations used.
Figure 5.

Ligand binding curves (see also Figure S2). (A) HSA binding curves for GA88 (filled circles), GA95 (open squares), and GA98 (filled triangles). (B) IgG binding curves for GB98-T25I (filled circles) and GA98 (open squares).
In contrast, the GA proteins do not bind their wild-type ligand, HSA, as robustly with binding affinity to HSA decreasing as identity to GB is increased. Thus, GA88 binds HSA with a dissociation constant of 31–37 μM while GA98 has a considerably weaker affinity with a KD of 244 μM. In addition to decreased affinity for HSA, the GA proteins acquire affinity for the GB ligand, IgG, when the sequence identity is increased to 98%. Therefore GA98 is bi-functional with weak binding affinity to its cognate ligand HSA and approximately four-fold tighter binding to IgG (KD 62 μM).
The GB98-T25I mutant is similar to GA98 in that it also binds IgG, despite adopting a predominantly 3α fold in the unbound state. However, binding to IgG is approximately four-fold tighter than seen for GA98, and there is no detectable binding to HSA. The tighter binding to IgG corresponds with a higher level of the alternative 4β+α state in GB98-T25I than in GA98. In GB98-T25I, the minor 4β+α state is detectable in the HSQC spectrum (Figure 4) at a level of ~5% whereas the 4β+α conformer cannot be observed in the HSQC spectrum of GA98. Based on the relative IgG-binding constants for GA98 (KD ~ 62 μM) and GB98-T25I (KD ~ 15 μM), the 4β+α state is therefore estimated to be at a level of approximately 1% in GA98. Thus, the binding results support the assumption that IgG-binding only occurs through the 4β+α fold, where the engineered IgG-binding epitope is revealed. Ligand-induced fold switching would then presumably occur through IgG-binding to the weakly populated 4β+α state, driving the equilibrium from the 3α to the 4β+α conformer. Further experiments to test this assumption are in progress.
DISCUSSION
Structural basis for fold switching
The three mutation sites described here that are responsible for fold switching are not localized in one specific part of the 3α or 4β+α structures but rather are spread over several different structural elements. In the 3α fold the switching loci correspond to amino acid changes in the α3-helix (L45Y), the α1-α2 loop (T25I), and the α1-helix (L20A), while in the 4β+α fold these mutations are in the β3-strand, the α-helix, and the β2-strand, respectively. Most of the mutation sites, with the exception of L20 in the 3α fold, occur on the periphery of the 3α or 4β+α cores. As such, the resulting changes in stability are generally small (in an absolute sense) but nonetheless can have a significant impact on the fold outcome due to the low stability (ΔGu ~ 1.5–2.5 kcal/mol) of the high identity proteins. The extent to which each state is populated will depend on the relative energy levels of the 3α and 4β+α conformations. The large shifts in equilibrium between these two states can best be understood by considering the effects of the mutations on the two folds, destabilizing one structure while simultaneously stabilizing the alternative conformer in this binary system.
In GA98, the L45Y mutation destabilizes the 3α conformation in the following way. The L45 side chain is not buried in the core but rather packs against it, making stabilizing hydrophobic contacts with the core residues I33 and I49 as well as with the more exposed L32 and Y29 side chains (Figure 3A and 6A). When L45 is mutated to Y45, these relatively small stabilizing interactions are mostly lost, since the more rigid tyrosine side chain with its fewer rotational degrees of freedom cannot pack as efficiently against the 3α core as a leucine. At the same time, the L45Y mutation also stabilizes the 4β+α conformation of GB98 through a favorable hydrophobic interaction between the aromatic rings of Y45 in the β3-strand and the core F52 residue in the β4-strand (Figure 6B). Further stabilizing hydrogen bond interactions from Y45-ηOH to the D47-carboxylate and to the hydroxyl group of Y3 in the β1-strand are also likely based on the GB98 NMR ensemble of structures. Thus, the destabilization of the 3α conformation is small but significant due to the low stability of GA98. The relative gain in stability of the 4β+α fold is enough to shift the equilibrium to this state almost completely (~99%).
Figure 6.

Structural basis for switching between 3α and 4β+α folds. (A) A representative NMR structure of GA98 showing L45 and nearest neighbors (pale orange) described in the text. The side chain conformation of Y45 (green) in the GB98-T25I CS-Rosetta structure is superimposed for comparison purposes. (B) GB98 NMR structure highlighting Y45 and adjacent amino acids. (C) NMR structure of GB98 showing T25 and surrounding residues (pale orange). The I25 side chain (green) in the NMR structure of GB98-T25I,L20A is superimposed for comparison. (D) CS-Rosetta structure of GB98-T25I highlighting I25 and neighboring hydrophobic contacts (pale orange). The corresponding position of T25 in GA98 (green) is superimposed.
Introduction of a T25I mutation into GB98 produces GB98-T25I, which populates mostly (~95%) the 3α state with a small amount (~5%) of the 4β+α conformer. Inspection of the GB98 structure gives insights into the destabilizing influence of the T25I mutation on the 4β+α fold. The T25 residue in GB98 is located near the N-terminus of the α-helix, and has closest proximity to the L20, V21, and D22 side-chains (Figure 6C) with a likely H-bonding interaction between the T25-γOH group and the carboxylate of D22. Loss of this H-bond in a T25I mutant will therefore destabilize the 4β+α fold. This is consistent with earlier stability studies on GB77, which showed that a T25I mutation decreased the Tm by 4.4°C (Alexander et al., 2009). However, the destabilizing effect of the T25I mutation is probably larger in GB98 due to additional unfavorable steric interactions that are likely to exist between the adjacent branched side chains of I25 and L20.
In contrast, the T25I mutation stabilizes the 3α fold of GB98-T25I. While CS-Rosetta only models the positions of the side chains shown in Figure 6D, their conformations are similar to those obtained in related NMR structures and provide a useful guide for discussing mutational effects. In the CS-Rosetta structure of GB98-T25I, the side chains in the region of the T25I mutation have an average heavy atom RMSD of 1.6±0.5 Å in the ensemble. These side chain arrangements have average RMSDs of 1.5–1.9 Å when compared with their corresponding positions in both NMR (He et al., 2008; Alexander et al., 2009) and CS-Rosetta (Shen et al., 2010) structures of GA88 and GA95. Thus, I25 is located in the loop between the α1- and α2-helices of the GB98-T25I 3α fold and examination of the CS-Rosetta structure suggests that I25 contributes more extensively to the hydrophobic core than a threonine residue. A similar stabilizing conformation is also seen for I25 in GA88 (He et al., 2008). The putatively stabilizing hydrophobic interactions involving I25 and neighboring residues L20, A23, I49, and F52 (Figure 6D), counteract the known destabilizing influence of having Y45 in the α3-helix such that this sequence adopts a predominantly 3α conformation. The GB98-T25I mutant is the only case so far where both folds are detectable by NMR, and therefore the 4β+α and 3α conformations of this amino acid sequence must be the closest in energy of the four mutants in this series.
The third fold switch involves an L20A mutation in the α1-helix of GB98-T25I. L20 is completely buried in the hydrophobic core of the 3α conformation of GB98-T25I (Figure 6D). Conversion to alanine at this position decreases packing interactions with other neighboring residues contributing to the core such as A16, I25, and I49, thereby destabilizing the 3α fold. Indeed this is the most destabilizing of the three mutations in this study because the L20A mutation cannot be tolerated even in more stable 3α mutants such as GA77 (Tm 77°C) and leads to an unfolded protein (Alexander et al., 2009). In the 4β+α conformer, mutation to the smaller A20 residue lowers the energy of this state by removing unfavorable steric contacts that would otherwise exist between the proximal L20 and I25 side chains. Thus, the equilibrium is shifted almost completely (>95%) to the 4β+α fold with an L20A substitution. GB98-T25I,L20A is the most stable mutant in this series (Tm ~ 46°C) and therefore must have the largest energy gap between 4β+α and 3α states of any of the proteins studied here.
Gain and loss of function
The ligand binding study provides insights into how folds and functions can evolve. In particular, a new IgG-binding function is gained in GA98 before complete loss of the original HSA-binding function. Bi-functional mutants such as GA98 therefore serve as transitory species between distinct functional states. Moreover, the new IgG-binding function of GA98 is detectable before there is significant population of the corresponding 4β+α fold. This stems from the intrinsically tight binding of IgG to GB sequences adopting the 4β+α conformation. In this way, even low levels of the 4β+α fold in the unbound states of GA98 (~1%) and GB98-T25I (~5%) can lead to IgG-binding with KD values of 62 and 15 μM, respectively (Table 2). Thus, increased equilibrium amounts of the 4β+α state in samples with predominantly 3α conformers correlate with a gain of IgG-binding function.
In contrast, the baseline HSA-affinity of GA88 is at least 30-fold weaker than IgG affinity to the 4β+α proteins GB98 and GB98-T25I,L20A. Much of this loss in affinity is due to the mutation of A52F, which apparently alters the contact with HSA. Further loss of HSA-binding function occurs in GA98 and GB98-T25I even though 3α levels are still high. The drop in HSA affinity from GA95 to GA98 is primarily due to a decrease in protein stability as the only amino acid change (I30F) is located away from the HSA-binding epitope (Lejon et al., 2004; He et al., 2007) at the α2-α3 surface (Figure 3A). In the case of GB98-T25I, the complete loss of HSA-binding function is mainly due to the presence of a tyrosine residue at position 45 (Figure 1C). Even in other more stable 3α mutants, changing leucine to tyrosine at residue 45 was found to abolish HSA binding. This is consistent with the observation that L45 is centrally located in the binding interface between HSA and a variant of wild-type GA (He et al., 2007).
The results here demonstrate that the mode for fold switching is not unique but can occur in multiple ways, thus increasing the probability for such events. These large-scale structural changes can occur through a series of single amino acid substitutions, once a suitable high sequence identity background has been reached. The present study uses only binary sequence space (either GA or GB amino acids) and is not exhaustive, so it is likely that other switch mutants also exist. Expansion to the complete range of amino acids may further increase the number of single amino acid switch mutants, and could also potentially lead to other folds and functions. Indeed, recent theoretical studies suggest that the high identity GA/GB sequences may be capable of adopting numerous other fold topologies (Cao and Elber, 2010).
It is possible that the in vitro directed evolution of the GA/GB system may reflect some aspects of the in vivo evolution of the GA and GB domains in protein G. Protein G is a multi-domain protein with 2–3 copies of each of the GA and GB domains. One plausible hypothesis based on our results is that a duplicated HSA-binding GA domain evolved the IgG-binding function through fold switching. In such a multi-domain system, the likelihood that this could occur seems high because functionality would be gained with no loss of fitness (due to the multiple copies). Other multi-domain proteins may therefore provide further examples where fold switching has occurred.
EXPERIMENTAL PROCEDURES
Mutagenesis
Mutants were prepared with a QuikChange (Stratagene) kit using the manufacturer’s protocol.
Protein Expression and Purification
GA and GB mutants were cloned into a vector encoding an N-terminal subtilisin-prodomain fusion tag system (Profinity eXact, Bio-Rad) described previously (Ruan et al., 2004). E. coli BL21(DE3) cells were transformed with this vector, and cells were grown at 25°C to a density of 0.6–0.8 OD600 in M9 minimal media for 13C and 15N-labeling. Protein expression was induced with 1 mM IPTG and the cells were grown a further 6 h at 25°C. The culture was then centrifuged, the cells suspended in 100 mM KPi buffer (pH 7.0), and sonicated. The cleared cell extract was loaded onto a 5 mL eXact column at 5 mL/min and then washed extensively with 100 mM potassium phosphate buffer (pH 7.0). The pure target protein was cleaved and eluted with an injection of 6 mL of 10 mM sodium azide, 100 mM potassium phosphate (pH 7.0) at 0.5 mL/min. Purified samples were then concentrated for NMR analysis.
NMR Spectroscopy
Isotope-labeled samples were prepared at concentrations of 0.05–0.3 mM for NMR analysis in 100 mM potassium phosphate buffer (pH 7.0) containing 5% D2O. NMR spectra were acquired on a Bruker AVANCE 600 MHz spectrometer with cryoprobe. Spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed with Sparky (Goddard and Kneller, 2004). Backbone resonances were assigned with HNCA, HN(CO)CA, HN(CA)CO, HNCO, HNCACB, and CBCA(CO)NH experiments. Aliphatic side chain assignments were obtained with (H)C(CO)NH-TOCSY and H(CCO)NH-TOCSY spectra while aromatic resonances were assigned with 2D CBHD/CBHE and NOESY spectra. NOEs were measured using 3D 15N NOESY and aliphatic and aromatic 3D 13C NOESY spectra.
Ligand Binding
Free and bound states were in slow exchange on the NMR timescale and hence peaks due to the high molecular weight bound states were broadened beyond detection. Therefore binding was determined by measuring the decay in peak intensity of amide protons in 15N HSQC spectra of GA or GB mutants as a function of ligand concentration. In a typical binding experiment, the 15N-labeled protein was approximately 50 μM and ligand concentrations ranging from 0.1–8.0 molar equivalents were used depending on binding affinity. Control experiments were also carried out to determine how much of the peak intensity decay was due to an increase in solution viscosity from added IgG or HSA. This was done by adding increasing concentrations of IgG or HSA to a known non-binder and measuring the decrease in amide peak intensities. Thus, the viscosity affect of IgG was determined by adding IgG to the non-binder, 15N-labeled PSD-1, a variant of wild-type GA (He et al., 2006). Similarly, HSA was added to the non-binder, 15N-labeled wild-type GB, to determine the HSA viscosity affect. Amide intensity decay curves due to binding were then corrected for the viscosity affect.
Structure Calculations and Analysis
NMR structures were determined for GA98, GB98, and GB98-T25I,L20A using CNS 1.1 (Brunger et al., 1998). Assignment of NOEs was assisted with an in-house program, NOEID. Interproton NOE distance restraints were classified as strong (1.8–3.0 Å), medium (1.8–4.0 Å), weak (1.8–5.0 Å), and very weak (2.8–6.0 Å). TALOS (Cornilescu et al., 1999) was used to provide backbone dihedral restraints from chemical shift data. Hydrogen bond restraints were incorporated in the latter stages of refinement. The final ensemble of 20 structures was chosen based on low total energy, no NOE distance violations >0.3 Å, no dihedral angle violations >5°, and other parameters shown in Table 1. Structures were analyzed with PROCHECK-NMR (Laskowski et al., 1996), PyMol (Delano, 2002), and MOLMOL (Koradi et al., 1996). The standard CS-Rosetta3.2 protocol (Shen et al., 2008) was used to determine the GB98-T25I structure based on chemical shifts. One thousand CS-Rosetta models were generated and the 10 lowest energy models clustered with a backbone RMSD of 1.00±0.27 Å.
Supplementary Material
Acknowledgments
We thank John Moult, Ron Elber, Nick Grishin, and Jeff Skolnick for helpful discussions; Yang Shen for assistance with CS-Rosetta; and Jane Ladner for laser light scattering analysis. This work was supported by National Institutes of Health Grant GM62154 and a grant from the W. M. Keck Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander PA, He Y, Chen Y, Orban J, Bryan PN. The design and characterization of two proteins with 88% sequence identity but different structure and function. Proc Natl Acad Sci USA. 2007;104:11963–11968. doi: 10.1073/pnas.0700922104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander PA, He Y, Chen Y, Orban J, Bryan PN. A minimal sequence code for switching protein structure and function. Proc Natl Acad Sci USA. 2009;106:21149–21154. doi: 10.1073/pnas.0906408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambroggio XI, Kuhlman B. Design of protein conformational switches. Curr Opin Struct Biol. 2006;16:525–530. doi: 10.1016/j.sbi.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Belogurov GA, Mooney RA, Svetlov V, Landick R, Artsimovitch I. Functional specialization of transcription elongation factors. Embo J. 2009;28:112–122. doi: 10.1038/emboj.2008.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco FJ, Angrand I, Serrano L. Exploring the conformational properties of the sequence space between two proteins with different folds: an experimental study. J Mol Biol. 1999;285:741–753. doi: 10.1006/jmbi.1998.2333. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse KR, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D (Biol Crystallogr) 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Bryan PN, Orban J. Proteins that switch folds. Curr Opin Struct Biol. 2010;20:482–488. doi: 10.1016/j.sbi.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B, Elber R. Computational exploration of the network of sequence flow between protein structures. Proteins. 2010;78:985–1003. doi: 10.1002/prot.22622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Dalal S, Regan L. Understanding the sequence determinants of conformational switching using protein design. Protein Sci. 2000;9:1651–1659. doi: 10.1110/ps.9.9.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- Fahnestock SR, Alexander P, Nagle J, Filpula D. Gene for an immunoglobulin-binding protein from a Group G Streptococcus. J Bacteriol. 1986;167:870–880. doi: 10.1128/jb.167.3.870-880.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenberg C, Bjorck L, Akerstrom B. Localization of the binding site for streptococcal protein G on human serum albumin. Identification of a 5.5-kilodalton protein G binding albumin fragment. Biochemistry. 1992;31:1451–1457. doi: 10.1021/bi00120a023. [DOI] [PubMed] [Google Scholar]
- Gallagher TD, Alexander P, Bryan P, Gilliland G. Two crystal structures of the B1 Immunoglobulin-binding domain of Streptococcal Protein G and comparison with NMR. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY. Vol. 3. University of California; San Francisco: 2004. [Google Scholar]
- Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, Clore GM. A novel, highly stable fold of the immunoglobulin binding domain of Streptococcal Protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- He Y, Chen Y, Alexander P, Bryan PN, Orban J. NMR structures of two designed proteins with high sequence identity but different fold and function. Proc Natl Acad Sci USA. 2008;105:14412–14417. doi: 10.1073/pnas.0805857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Chen Y, Rozak DA, Bryan PN, Orban J. An artificially evolved albumin binding module facilitates chemical shift epitope mapping of GA domain interactions with phylogenetically diverse albumins. Protein Sci. 2007;16:1490–1494. doi: 10.1110/ps.072799507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Rozak DA, Sari N, Chen Y, Bryan P, Orban J. Structure, dynamics, and stability variation in bacterial albumin binding modules: implications for species specificity. Biochemistry. 2006;45:10102–10109. doi: 10.1021/bi060409m. [DOI] [PubMed] [Google Scholar]
- Johansson MU, de Chateau M, Wikstrom M, Forsen S, Drakenberg T, Bjorck L. Solution structure of the albumin-binding GA module: a versatile bacterial protein domain. J Mol Biol. 1997;266:859–865. doi: 10.1006/jmbi.1996.0856. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmann JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lejon S, Frick IM, Bjorck L, Wikstrom M, Svensson S. Crystal structure and biological implications of a bacterial albumin binding module in complex with human serum albumin. J Biol Chem. 2004;279:42924–42928. doi: 10.1074/jbc.M406957200. [DOI] [PubMed] [Google Scholar]
- Littler DR, Harrop SJ, Fairlie WD, Brown LJ, Pankhurst GJ, Pankhurst S, DeMaere MZ, Campbell TJ, Bauskin AR, Tonini R, et al. The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transition. J Biol Chem. 2004;279:9298–9305. doi: 10.1074/jbc.M308444200. [DOI] [PubMed] [Google Scholar]
- Luo X, Yu H. Protein metamorphosis: the two-state behavior of Mad2. Structure. 2008;16:1616–1625. doi: 10.1016/j.str.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Musacchio A. MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr Opin Struct Biol. 2007;17:716–725. doi: 10.1016/j.sbi.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Meyerguz L, Kleinberg J, Elber R. The network of sequence flow between protein structures. Proc Natl Acad Sci USA. 2007;104:11627–11632. doi: 10.1073/pnas.0701393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG. Biochemistry. Metamorphic proteins Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- Myhre EB, Kronvall G. Heterogeneity of nonimmune immunoglobulin Fc reactivity among gram-positive cocci: description of three major types of receptors for human immunoglobulin. G Infect Immun. 1977;17:475–482. doi: 10.1128/iai.17.3.475-482.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler CG, Hall BM, Anderson WJ, Ingram WM, Roberts SA, Montfort WR, Cordes MH. Transitive homology-guided structural studies lead to discovery of Cro proteins with 40% sequence identity but different folds. Proc Natl Acad Sci USA. 2008;105:2343–2348. doi: 10.1073/pnas.0711589105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose GD, Creamer TP. Protein folding: predicting predicting. Proteins. 1994;19:1–3. doi: 10.1002/prot.340190102. [DOI] [PubMed] [Google Scholar]
- Ruan B, Fisher KE, Alexander PA, Doroshko V, Bryan PN. Engineering subtilisin into a fluoride-triggered processing protease useful for one-step protein purification. Biochemistry. 2004;43:14539–14546. doi: 10.1021/bi048177j. [DOI] [PubMed] [Google Scholar]
- Sauer-Eriksson AE, Keywegt GJ, Uhlen M, Jones TA. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure. 1995;3:265–278. doi: 10.1016/s0969-2126(01)00157-5. [DOI] [PubMed] [Google Scholar]
- Shen Y, Bryan PN, He Y, Orban J, Baker D, Bax A. De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds. Protein Sci. 2010;19:349–356. doi: 10.1002/pro.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, et al. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci USA. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann C. Birth of a prion: spontaneous generation revisited. Cell. 2005;122:165–168. doi: 10.1016/j.cell.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.