

Abstract
Motivation: The continued progress in developing technological platforms, availability of many published experimental datasets, as well as different statistical methods to analyze those data have allowed approaching the same research question using various methods simultaneously. To get the best out of all these alternatives, we need to integrate their results in an unbiased manner. Prioritized gene lists are a common result presentation method in genomic data analysis applications. Thus, the rank aggregation methods can become a useful and general solution for the integration task.
Results: Standard rank aggregation methods are often ill-suited for biological settings where the gene lists are inherently noisy. As a remedy, we propose a novel robust rank aggregation (RRA) method. Our method detects genes that are ranked consistently better than expected under null hypothesis of uncorrelated inputs and assigns a significance score for each gene. The underlying probabilistic model makes the algorithm parameter free and robust to outliers, noise and errors. Significance scores also provide a rigorous way to keep only the statistically relevant genes in the final list. These properties make our approach robust and compelling for many settings.
Availability: All the methods are implemented as a GNU R package RobustRankAggreg, freely available at the Comprehensive R Archive Network http://cran.r-project.org/.
Contact: vilo@ut.ee
Supplementary information Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Data integration plays an important role in the analysis of high-throughput data. As the outputs of individual experiments can be rather noisy, it is essential to look for findings that are supported by several pieces of evidence to increase the signal and lessen the fraction of false positive findings. The structure of the data and specific research questions largely dictate the methods used in integration. Still, some very general techniques for aggregation of genomic data have been proposed such as the Bayesian networks (Troyanskaya et al., 2003) and the Kernel methods (Bie et al., 2007). In this work, we limit ourselves to the situation where all the data sources are represented as prioritized lists of genes. This is a very natural way to represent a result of a genome-wide study and, therefore, ubiquitous in different types of analyzes.
Finding a meaningful combination of different data sources is often a non-trivial task. For example, differences in measurement platforms and lab protocols can render gene expression levels incomparable. Hence, it might be better to analyze different datasets separately and then aggregate the resulting gene lists. Such a strategy has been employed to finding gene co-expression networks (Lee et al., 2004; Stuart et al., 2003) and for defining more robust sets of cancer-related genes (Griffith et al., 2006). Another example is the study by (Aerts et al., 2006), where the authors combined different kinds of data sources to find disease-related genes. For this they ranked genes by similarity to the disease genes in the Gene Ontology (GO), pathways, transcription factor binding sites, sequence and literature and subsequently aggregated these rankings to get the final result. Boulesteix and Slawski (2009) used an analogous methodology to combine results of differential expression analysis.
Combining preference lists into a single ranking is known as rank aggregation. This problem has a rich history in the fields of information retrieval (Dwork et al., 2001a) and theory of social choice (Copeland, 1951). In these contexts, the main focus is on the ‘correctness’ of the resulting ranking. For example, in elections it is critical that each vote gets counted and represented in the final result. However, finding the best ranking is often computationally very expensive, for example the one that minimizes the misplacement or so-called ‘bubble sort distance’, and thus applicable only for a relatively small class of problems. (Dwork et al., 2001b; Pihur et al., 2007).
Data from high-throughput genomic experiments usually contains a significant proportion of noise and thus ‘correctness’ is not right objective for these kinds of studies. For such settings, a good rank aggregation method must be robust enough to find a relevant ranking even in the presence of unreliable or irrelevant inputs. Since it possible that all rankings are meaningless in the context of a particular study, the method should also quantify the reliability or significance of the results. Finally, the method should be applicable also for partial or top rankings only, since full rankings are often unavailable, e.g. gene lists derived from articles or various tools and databases.
In this article, we propose a novel rank aggregation method based on order statistics that achieves all described objectives. Stuart et al. (2003) were the first to utilize order statistics in rank aggregation. The computational scheme for their method was further optimized by Aerts et al. (2006). This algorithm compares the actual rankings with the expected behavior of uncorrelated rankings, re-ranks the items and assigns significance scores. While being robust to noise, this method requires simulations to define significance thresholds (Aerts et al., 2006) and does not support incomplete lists.
To overcome these limitations, we propose a new algorithm that is both computationally efficient and statistically stable. For each item, the algorithm looks at how the item is positioned in the ranked lists and compares this to the baseline case where all the preference lists are randomly shuffled. As a result, we assign a P-value for all items, showing how much better it is positioned in the ranked lists than expected by chance. This P-value is used both for re-ranking the items and deciding their significance.
We also extended this methodology to the case where only top elements in the lists are available. Compared with the common approach of counting the appearances of each item (Griffith et al., 2006; Lee et al., 2004), our method is clearly an improvement. It also takes into account the positional information and assigns significance levels to the findings according to a theoretical model.
All methods described in this article, including average rank and the method by Aerts et al. (2006), are implemented as a GNU R package RobustRankAggreg.
2 ROBUST RANK AGGREGATION
Let m be the number of objects and n be the number of preference lists. For instance, n might be the number of experiments and m the number of genes studied. Moreover, assume that in each survey genes are ordered according to their impact so that the ones most likely to behave in a certain way are in the beginning of the list. Then the rank of a gene is just the position in this ordering. If we divide ranks by the maximal rank value m, we obtain normalized ranks with the maximal value of 1. For each gene let the corresponding rank vector r=(r1,…, rn) be such that rj denotes the normalized rank of the gene in the j-th preference list.
A general strategy for obtaining an aggregated preference list is to score each rank vector somehow and order the genes based on the scores. Note that relative ranks can be used even if the preference lists contain slightly different sets of genes. This is quite a common scenario. For example, we might want to aggregate lists from different microarray platforms where each of them measures a unique subset of genes. To take this into account, we have to adjust the maximal rank m according to the number of elements in each list in the normalization step and use a scoring method that produces comparable scores even if the length of rank vector varies.
Classical rank aggregation methods try to maximize the coherence of the final ranking with all the preferences. However, maximal coherence with all preferences is not always the best aggregation criterion, especially if some preferences are unreliable. Some ranking algorithms resolve this issue assuming that we can estimate correctness probability for each rank or preference list see, for example Li et al. (2011). For most biological studies, this type of extra information is not available. Also, each ranking is most probably informative only on a subset of relevant genes, since it reflects only one aspect of the underlying biological question. Therefore, it is not reasonable to view an entire ranking as informative or non-informative but rather make a distinction at the gene level. This can be achieved via fixing a null model, which describes distribution of ranks when all studies produce irrelevant results, and estimate statistical significance.
The simplest possible null model assumes that all studies are non-informative and produce randomly ordered gene lists. When genes are ranked based solely on measurement data, the null model is equivalent to a permutation test, where measurements in each study are randomly relabeled before the analysis step.
2.1 Scoring rank vectors using order statistics
In most cases, we are interested in finding genes that are ranked highly in many preference lists ignoring a (small) fraction of non-informative studies. Thus, we can assume that all informative normalized ranks come from a distribution, which is strongly skewed toward zero and our task is to detect these distributions.
For any normalized rank vector r, let r1,…, rn be a reordering of r such that r1≤…≤rn. Then we can ask how probable it is to obtain 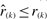 when the rank vector
when the rank vector 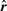 is generated by the null model, i.e. all ranks
is generated by the null model, i.e. all ranks 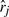 are sampled from uniform distribution.
are sampled from uniform distribution.
Let βk,n(r) denote the probability that 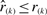 . Then under the null model the probability that the order statistic
. Then under the null model the probability that the order statistic 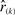 is smaller or equal to x can be expressed as a binomial probability
is smaller or equal to x can be expressed as a binomial probability
| (1) |
since at least k normalized rankings must be in the range [0, x]. Alternatively, βk,n(x) can be expressed through a beta distribution, as 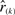 is the order statistic of n independent random variables uniformly distributed over the range [0, 1].
is the order statistic of n independent random variables uniformly distributed over the range [0, 1].
Since the number of informative ranks is not known, we define the final score for the rank vector r as the minimum of P-values
| (2) |
and order all rank vectors according to their ρ scores. Illustration of how the scores are calculated on two example genes is in the Figure 1.
Fig. 1.

Visual description of RRA. (A) Shows an example of 20 ranked lists, with the positions of two genes highlighted. The first gene is placed to the top of the lists and the second distributed uniformly. (B) Shows in detail how βk,n scores change and how the ρ score is found.
As a minimum of P-values, ρ score itself is not a P-value. Hence, it must be corrected against bias coming from multiple hypothesis testing to determine statistical significance. Exact P-values can be computed for each ρ score. However, the corresponding algorithm relies on the same methods as Aerts et al. (2006) and thus inherits all weaknesses. In particular, it is computationally expensive and becomes numerically unstable for rather small examples. A full description of the method is given in the Supplementary Material.
Alternatively, we can use Bonferroni correction for each ρ score separately to find an upper bound on the associated P-value. For that we must multiply each ρ score with the number of input lists. Since the number of input rankings is usually in the order of 10–100, the resulting correction is not overly stringent. Indeed, exact numerical comparisons between Bonferroni correction and actual P-values given in the Supplementary Material show upper bounds are very close to exact values. Thus, the Bonferroni correction is a good trade-off between efficiency and precision in most cases.
After the correction, we can decide whether the ranking of a particular gene is statistically significant. To find all significant genes, we have to correct derived (estimates of) P-values further for multiple testing. This can be done we with standard methods.
From the computational viewpoint, the Robust Rank Aggregation (RRA) method is inexpensive. Its complexity is linear with respect to the input size. Therefore, its applicability is not restricted by computational considerations.
2.2 Aggregation of partial preference lists
In many cases, studies provide only partial rankings. For example, instead of all genes the list contains only the differentially expressed ones. Also, small fluctuations in measurements can cause large changes in the bottom part of such lists. Thus, it might be beneficial to keep only genes with statistically significant changes.
There are two ways to extend our basic algorithm. First, we can obtain a conservative approximation of the P-values βn,k(r) and ρ scores. Due to missing ranks we cannot correctly determine r(k). However, if we replace all missing values with the maximal relative Rank 1, the resulting order statistics r(k) can only increase. Hence, by inserting these estimates of r(k) into the Formula (1) we get upper bounds for all P-values βn,k(r). This approach is generally applicable and suitable in most of the cases.
Alternatively, if we know the specific mechanism for eliminating ranks and the length of preference lists varies substantially, we can incorporate data deletion directly into the null model. This provides more accurate and less conservative estimates of P-values. As a drawback, the results must be computed with dynamic programming, which is slightly less efficient. Further details of this algorithm are shown in the Supplementary Material
3 RESULTS
3.1 Simulation study
We used simulations to investigate basic properties of our method. Namely, we generated rankings with planted elements that were preferentially ranked at the top. To add noise we included rankings where the order of elements was completely random. Such data allows us to study two aspects. First, we can study whether the resulting rankings separate planted elements from the others. Second, we can determine how many planted elements are revealed and compare it with the performance of alternative methods.
We compared our method with two alternatives: average rank and the method by Stuart et al. (2003). To calculate the scores for Stuart method, we used the optimized algorithm from Aerts et al. (2006). All three methods also provide significance scores for final ranks showing how much higher a gene is placed in the inputs than expected. Further details are given in Section 4.
All rankings consisted of 1000 elements out of which 5% were preferentially ranked at the top. We achieved that by associating randomly generated values to the elements and ordering them according to these. All values were drawn form normal distribution with unit variance. For planted elements, we chose the mean value from the exponential distribution with λ=1/2 and used the standard normal distribution for remaining elements.
For the first test, we generated 10 such lists. As can be seen from Figure 2A, all three methods can order the lists very well, placing elements from the positive class at the top of the aggregated list. Corresponding area under curve (AUC) scores are 0.997, 0.995 and 0.979 for Stuart method, RRA and rank average. However, the results start to differ if we study the number of elements deemed significant by each method using the false discovery rate of 0.05 as threshold (Fig. 2A). RRA method outperformed rank average even with the data that does not contain any noise in the form of random lists.
Fig. 2.

Results from a simulation study. (A) Shows significance scores calculated with different methods on the 10 lists, which contained 50 planted elements. The number of true and false positives was computed on FDR level of 0.05. Both methods based on order statistics (RRA and Stuart) separate planted elements from noise better than average rank. Still, the Stuart method produces many false positives and thus cannot be used for deciding the significance of genes. (B) Shows ROC curves of different methods on noisy data (10 lists with signal, 30 random). Methods based on order statistics outperform the average rank considerably. (C) Shows the number of true positives given at different levels of noise. At each level, we simulated 10 datasets. RRA shows much higher resistance to noise than an average rank. The Stuart method was excluded from (C) as it failed to identify planted elements from noise.
Stuart method has problems with significance scoring. It produces probability values for each element and the original article treats them as P, but actually they are not (Aerts et al., 2006). Using them as P-values produces many false positives.
In the second test, we studied resistance against noise. Figure 2B shows the receiver-operating characteristic (ROC) curve for the three methods on data that contained 10 lists with signal as described before and 30 randomly ordered lists. We can see that both RRA and Stuart method perform considerably better than average rank in ordering the lists, showing their robustness to this type of noise.
We also tested the performance on recognizing planted elements given various levels of noise (Fig. 2C). We generated 10 lists with signal and added different numbers of randomly shuffled lists. In each case, we counted the number of true positive predictions on FDR level of 0.05. Because of the problems with significance scores, we omitted the Stuart method. Results show that RRA performs consistently better than average rank and gives meaningful results even if the levels of noise are relatively high.
Finally, we studied how well the RRA method works if we consider only ℓ top ranking elements. To answer this question, we simulated datasets with 10 lists with planted elements as described before. We cut out different proportions of top elements and counted the number of statistically significant results as given by RRA. The outcome is in Figure 3. As before 5% of the elements were planted. A full list allowed us to identify about three-fourths of them. This number started to fall only when we removed >95% of the elements in the bottom of rankings. But, even with top-1% lists, we can on average correctly identify about half of the planted elements. It must be noted that the statistics are still correct using top-ℓ lists. The number of false positives was well within the allowed limits.
Fig. 3.

The proportion of planted elements that were correctly identified by RRA given different numbers of top elements available in input rankings. The gray line shows the proportion of planted elements in the inputs. We can see that the number of correctly identified elements starts to drop only after almost the whole list is dropped. Therefore, by using partial instead of full rankings we usually lose very little information.
In summary, we can conclude that RRA is usable also in situations where complete rankings are unavailable.
3.2 Predicting pathway members with knock-out data
Gene expression measurements before and after some transcription factor knockouts reveal important information about genomic pathways. However, we cannot infer the pathway members directly from this data. We can only identify putative targets for these factors. To identify pathway members, we must join target lists of all factors related to a particular pathway. However, these target lists are inherently noisy, since a knockout often affects genes that do not correspond to the pathway. By robust rank aggregation, we can filter out genes that are not specific for the process.
We used yeast data by Hu et al. (2007) to validate this approach. The data consist of experiments where the transcription factors are knocked out one by one. Reimand et al. (2010) analyzed the data and published the lists of genes where expression was most affected by each knockout. We used these lists to predict members of the GO groups. For each GO group, we identified the transcription factors related to the group and extracted corresponding gene lists. Next we used our methods to compile a list of statistically significant targets. As a quality control, we used Fisher's exact test on each list to estimate the enrichment of the known members of the functional group. Figure 4 depicts results for cell cycle and response to chemical stimulus.
Fig. 4.

Predicting genes to a GO category based on the knockouts of its transcription factors. A gene name on the x-axis corresponds to a knockout and each bubble represents the Fisher's exact test P-value, showing the enrichment of the knock-out affected genes in the GO category. The horizontal line shows the same enrichment P-value for the aggregated list. The size of the bubble corresponds to the number of regulated genes in the knockout and the color shows if the P-value is significant. The P-values show that the aggregated list is more enriched in the genes related to the corresponding process than most of the inputs.
In both cases, aggregated lists are more enriched with genes belonging to the GO category of interest than most of the individual lists. Although, a particular knockout can outperform the aggregated list, the impact of less informative lists is limited. Generally, we do not know in advance which experiment is most informative and thus aggregation is a good way to consolidate data.
Results with response to chemical stimulus demonstrate the utility of robustness of our rank aggregation method. Only 6 of 39 of the input rankings were enriched with genes related to the category. Still, our method recognized the signal among all that noise and returned a list with significant enrichment.
3.3 MEM project
Our method is already used in the Multi Experiment Matrix (MEM) web server (Adler et al., 2009), which searches for co-expressed genes over large collections of microarray data. Given a gene name, the web server performs a co-expression query in hundreds of datasets and then aggregates the results using the RRA algorithm.
To demonstrate the usefulness of rank aggregation in this setting we performed the following study. The goal was to find transcription factor targets by searching for genes that are co-expressed with it. As a gold standard, we used targets found by ChIP-seq study Chen et al. (2008). The study covered 15 embryonic stem cell-related transcription regulators. We performed the co-expression search with these 15 genes on 12 ES-related mouse datasets and recorded both individual and aggregated search results. For each of those we calculated ROC AUC scores showing prediction accuracy. We used both RRA and Stuart method for this. The AUC values are close to one if the prediction for the gold standard is good and ∼0.5 if the gold standard cannot be predicted. The results can be seen in Figure 5.
Fig. 5.

AUC scores when predicting transcription factor targets based on gene co-expression. The gray dots represent the individual results and black dots and plus signs aggregated results with RRA and Stuart method. These values show that in the presence of a signal in the inputs, aggregation methods pick it up and outperform most of the inputs. When the signal is low in the input (AUC ∼0.5), aggregated results are not considerably better. The results for RRA and Stuart method are almost identical, since they use very similar criteria for aggregation.
Generally, the aggregated list outperforms most of the input lists but not all. For CTF, MYCS and SMAD1 queries, results of the RRA algorithm are in the middle of pack. However, in all those cases AUC scores for input lists are dispersed ∼0.5, i.e. there is no signal. This explains the poor performance of our algorithm: it can only amplify the signal if it is present in the input. So if we could tell in advance what input is the most relevant, then we could completely omit the aggregation step. However, pinpointing good lists is not feasible in most cases. Even in our example, the best individual dataset is different for each transcription factor. Hence, it is safer to include all the data and use the aggregated results.
3.4 Comparison with other methods
The RRA algorithm has four key features: it is robust to noise, it can handle incomplete rankings, it assigns a significance score to each element in the resulting ranking, and it is also efficient to compute. All these features are important in practice; in particular, if one wants to build a tool for interactive data analysis.
To our knowledge, other available methods cannot match these properties. Classical rank aggregation methods are often sensitive to noise (as illustrated in Fig. 2) and do not identify the relevant part of the aggregated ranking. In addition, some of them can be computationally very expensive (Dwork et al., 2001b).
The Stuart method is a good alternative to RRA, as it is robust to noise (Fig. 2A and B). The rankings produced by Stuart and RRA algorithms are very similar in terms of discrimination. The Stuart algorithm yields ROC AUC scores that are comparable if not a bit better than RRA (Figs 2 and 5). This is an expected result, as the statistics are very similar while Stuart's method takes more information into account. However, the scores of the Stuart method are not directly P-values (Aerts et al., 2006) and closed form solutions are not known. Hence, extensive simulations are needed to assess the significance of aggregated ranks. These results are not easily pre-calculable, since we need simulations for each shape of the rank matrix. As a consequence, the Stuart method with proper thresholding is hundreds if not thousands times slower than the RRA algorithm.
The Stuart method does not handle missing ranks. This can be resolved with a small modification shown in the Supplementary Material. Finally, the algorithm can become numerically unstable when the number of lists is bigger. In our experience, it happens around 40–50 input lists (see Supplementary Material for examples), but depending on the data this number can be also smaller.
As a final test, we used updated Stuart's method on the knock-out data by making some adjustments for it to work in this setting. Out of 15 cases, where we reached a significant result, in 11 cases the RRA had the same or better enrichment score (see Supplementary Material for details) showing the utility of RRA approach.
In summary, the unique properties of RRA make it suitable for a variety of practical situations for which there are no good alternatives available. Our tests show that even in the cases where some alternatives could be used, our method gives comparable results.
4 MATERIALS AND METHODS
4.1 Rank aggregation methods used in comparisons
We used average rank and the method by Stuart et al. (2003) for evaluating RRA. To assign significance scores for average ranks, note that the resulting rank follows normal distribution with mean 0.5, and SD 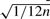 , provided that samples come from uniform distribution.
, provided that samples come from uniform distribution.
The method by Stuart et al. (2003) outputs in our notation a probability 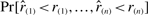 where
where 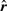 is a sample from standard uniform distribution as an output score. We tried to use this probability as a significance P-value, as was done in the original article, although permutation tests would have been more appropriate to assign significance scores. Since the formulas used in the original article are computationally very demanding, we used the algorithm proposed by Aerts et al. (2006). Both these methods are also implemented in the associated GNU R package RobustRankAggreg.
is a sample from standard uniform distribution as an output score. We tried to use this probability as a significance P-value, as was done in the original article, although permutation tests would have been more appropriate to assign significance scores. Since the formulas used in the original article are computationally very demanding, we used the algorithm proposed by Aerts et al. (2006). Both these methods are also implemented in the associated GNU R package RobustRankAggreg.
4.2 Pathway prediction
For pathway prediction, we used the statistical analysis results from Reimand et al. (2010) that are available in ArrayExpress (accession: E-MTAB-109). We used false discovery rate of 0.05 as the significance cutoff when defining the lists. The response of the chemical stimulus gene list was downloaded as a GO slim category from Saccharomyces Genome Database (Hong et al., 2008) and cell cycle genes from a review by de Lichtenberg et al. (2005).
4.3 Stem cell study using MEM
A gold standard for transcription factor targets were obtained from the study by Chen et al. (2008). The article defined an association score between each gene and a transcription factor that varied from 0 to 1. As scores showed a clear bimodal distribution, we considered all genes with a score >0.6 to be targets of the corresponding transcription factor.
In the MEM webtool (http://biit.cs.ut.ee/mem), we selected 12 datasets on mouse Affymetrix platform 430 2.0. Since the ChIP-seq study was performed on mouse embryonic stem (ES) cells, we selected only the datasets that mentioned ES cell in their description. The list of datasets is available in the Supplementary Material.
We queried each of the transcription factors, for which we had binding information, separately. The MEM webtool performed a similarity search on each dataset using correlation distance between the transcription factor and other genes. The resulting lists of correlated genes were used in aggregation and assessing the AUC.
To combine the data from ChIP-seq with co-expression queries, we translated all the results into Ensembl gene identifiers using g:Convert (Reimand et al., 2007).
5 DISCUSSION
5.1 Gene expression meta-analysis
The accumulation of gene expression studies has created a situation where one can pose new questions about the co-expression and functional role of genes by integrating the already existing data (Wren, 2009). It is also easier to answer some biological questions by reanalyzing and combining old datasets rather than redoing experiments. For example, meta-analysis of gene expression data has been successfully used for studying cancer and its subtyping (Rhodes et al., 2002; Wirapati et al., 2008).
There are two general strategies for meta-analysis. The first involves rigorous normalization, integration and analysis of the raw expression datasets. In general, this is the preferred type; however, it might not be always practical. The main problem is the availability of the data. Many older publications do not have the data uploaded to public repositories, such as ArrayExpress (Parkinson et al., 2009) or GEO (Barrett et al., 2009). Even from the data that is available in these repositories, about two-thirds might be unusable for meta-analysis due to poor quality or missing raw files (Larsson and Sandberg, 2006). Another problem is caused by the usage of different microarray platforms. Each platform uses different technology to measure the expression and features its own set of probes and genes. Therefore, proper integration of gene expression values over platforms is very difficult, although, doable in special cases.
The second option is to merge only the published results. This way we can avoid normalization problems caused by different platforms and also improve the data availability dramatically, since most of the microarray publications include a gene list or a signature of some sort. On the downside, this approach introduces many potential sources of noise arising from different preprocessing, normalization and statistical analysis steps (Cahan et al., 2007).
Commonly used methods for integrating published gene lists are usually very simple. Sometimes, the aggregated list is obtained by counting the number of times each gene occurs in the input lists (Griffith et al., 2006; Miller and Stamatoyannopoulos, 2010). Rank aggregation methods are an obvious choice and several of them have also been tried in this setting (DeConde et al., 2006; Pihur et al., 2008). Generally, these methods give reasonable results but there are different drawbacks. Counting-based methods do not naturally take into account the ranked nature of the gene lists, although, there are some workarounds. Many of the rank aggregation methods are computationally very expensive, making some questions intractable (Dwork et al., 2001b). Most of the classical methods also lack robustness, which is critical, since some input sources can be dubious due to differences in clinical and experimental setups, etc. The main problem with all the methods is the lack of a statistical model for assessing the relevance of the results.
Our RRA algorithm fits very well to this meta-analytic setting. Most importantly, it is based on a statistical model that naturally allows evaluating the significance of the results. In addition, RRA is easy to compute and robust, not restricting its use to certain subset of problems or requiring all data to be of top quality. The RRA algorithm can also handle variable gene content of different microarray platforms. By defining the rank vector for each gene based only on the datasets where it is present, we do not have to omit the genes that are not present in every platform.
6 CONCLUSIONS
Ranked lists of genes are a common output of a vast array of bioinformatics tools. Often, the same question can be answered using several datasets or algorithms producing multiple lists of genes and there is a need to combine the results. In this article, we present a novel rank aggregation algorithm RRA that is very well suited for such bioinformatic settings. The aggregation is based on the comparison of actual data with a null model that assumes random order of input lists. A P-value assigned to each element in the aggregated list described how much better it was ranked than expected. This provides basis for reordering and identifies significant elements. As the P-value calculation procedure takes into account only the best ranks for each factor, the method is very robust, which is important when using high-throughput data.
We validated the method both on simulated and biological data. The simulations showed that the algorithm can very well retrieve the positive factors planted into the input lists, even in the presence of noise. The method still managed to find some of the planted factors, even if over 75% of the input rankings did not contain any relevant information. We also showed that the method can work with partial rankings as well. In fact, we can omit very large parts of the input lists without influencing the results at all.
In the two case studies on biological data, we found that RRA can amplify the biological signal if it exists in the input data. The aggregated ranking displays stronger signal than most of the inputs lists. Even if some of the inputs may perform well individually, we are in practice often better off by taking into account all the possible information rather than trying to guess the most informative source. All the methods described here have been implemented and are available in additional GNU R package RobustRankAggreg.
Supplementary Material
ACKNOWLEDGEMENT
R.K. acknowledges the Tiger University Program of the Estonian Information Technology Foundation.
Funding: Tiger University Program of the Estonian Information Technology Foundation. EU FP6 and FP7 projects ENFIN (LSHG-CT- 2005-518254); ESNATS (HEALTH-F5-2008-201619); European Regional Development Fund through the Estonian Centre of Excellence in Computer Science project and Estonian Science Foundation (ETF7437, CIESCI).
Conflict of Interest: none declared.
REFERENCES
- Adler P., et al. Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biol. 2009;10:R139. doi: 10.1186/gb-2009-10-12-r139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts S., et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- Barrett T., et al. Ncbi geo: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie T.D., et al. Kernel-based data fusion for gene prioritization. Bioinformatics. 2007;23:i125–i132. doi: 10.1093/bioinformatics/btm187. [DOI] [PubMed] [Google Scholar]
- Boulesteix A., Slawski M. Stability and aggregation of ranked gene lists. Brief. Bioinformatics. 2009;10:556. doi: 10.1093/bib/bbp034. [DOI] [PubMed] [Google Scholar]
- Cahan P., et al. Meta-analysis of microarray results: challenges, opportunities, and recommendations for standardization. Gene. 2007;401:12–18. doi: 10.1016/j.gene.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Copeland A. A reasonable social welfare function. Ann Arbor, MI: University of Michigan; 1951. Technical Report. [Google Scholar]
- DeConde R.P., et al. Combining results of microarray experiments: a rank aggregation approach. Stat. Appl. Genet. Mol. Biol. 2006;5 doi: 10.2202/1544-6115.1204. Article15. [DOI] [PubMed] [Google Scholar]
- de Lichtenberg U., et al. Comparison of computational methods for the identification of cell cycle-regulated genes. Bioinformatics. 2005;21:1164–1171. doi: 10.1093/bioinformatics/bti093. [DOI] [PubMed] [Google Scholar]
- Dwork C., et al. Proceedings of the 10th International Conference on World Wide Web. Hong Kong: 2001a. Rank aggregation methods for the web; pp. 613–622. [Google Scholar]
- Dwork C., et al. Proceedings of WWW10. Geneva, Switzerland: Association IW3C2; 2001b. Rank aggregation revisited; pp. 613–622. [Google Scholar]
- Griffith O.L., et al. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J. Clin. Oncol. 2006;24:5043–5051. doi: 10.1200/JCO.2006.06.7330. [DOI] [PubMed] [Google Scholar]
- Hong E.L., et al. Gene ontology annotations at sgd: new data sources and annotation methods. Nucleic Acids Res. 2008;36:D577–D581. doi: 10.1093/nar/gkm909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., et al. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 2007;39:683–687. doi: 10.1038/ng2012. [DOI] [PubMed] [Google Scholar]
- Larsson O., Sandberg R. Lack of correct data format and comparability limits future integrative microarray research. Nat. Biotechnol. 2006;24:1322–1323. doi: 10.1038/nbt1106-1322. [DOI] [PubMed] [Google Scholar]
- Lee H.K., et al. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004;14:1085–1094. doi: 10.1101/gr.1910904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., et al. A unified approach to ranking in probabilistic databases. VLDB J. 2011;20:249–275. [Google Scholar]
- Miller B.G., Stamatoyannopoulos J.A. Integrative meta-analysis of differential gene expression in acute myeloid leukemia. PLoS One. 2010;5:e9466. doi: 10.1371/journal.pone.0009466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson H., et al. Arrayexpress update–from an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Res. 2009;37:D868–D872. doi: 10.1093/nar/gkn889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihur V., et al. Weighted rank aggregation of cluster validation measures: a monte carlo cross-entropy approach. Bioinformatics. 2007;23:1607–1615. doi: 10.1093/bioinformatics/btm158. [DOI] [PubMed] [Google Scholar]
- Pihur V., et al. Finding common genes in multiple cancer types through meta-analysis of microarray experiments: a rank aggregation approach. Genomics. 2008;92:400–403. doi: 10.1016/j.ygeno.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Reimand J., et al. g:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., et al. Comprehensive reanalysis of transcription factor knockout expression data in Saccharomyces cerevisiae reveals many new targets. Nucleic Acids Res. 2010;38:4768–4777. doi: 10.1093/nar/gkq232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes D.R., et al. Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002;62:4427–4433. [PubMed] [Google Scholar]
- Stuart J.M., et al. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Troyanskaya O.G., et al. A Bayesian framework for combining heterogeneous data sources for gene function prediction (in saccharomyces cerevisiae) Proc. Natl Acad. Sci. USA. 2003;100:8348–8353. doi: 10.1073/pnas.0832373100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirapati P., et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:R65. doi: 10.1186/bcr2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren J.D. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics. 2009;25:1694–1701. doi: 10.1093/bioinformatics/btp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.