

Abstract
The p16INK4a and p15INK4b 5′ CpG island hypermethylation has been described as one of the most frequent mechanisms leading to inactivation of these tumor suppressor genes in hematological malignancies. The p16 and p15 promoter regions were studied using methylation-specific polymerase chain reaction in 53 CD30 non-Hodgkin’s lymphomas (25 anaplastic large-cell, 13 peripheral T cell, and 15 anaplastic diffuse large B cell) and 26 Hodgkin’s lymphomas, with the aim of comparing the methylation status of these tumor suppressor genes in anaplastic large-cell lymphomas and other related entities. p16 and p15 methylation was detected, respectively, in 28% and 60% of CD30 non-Hodgkin’s lymphomas and in 38% and 42% of Hodgkin’s neoplasms. This confirms the p16-methylated status in Hodgkin’s cases described in a single previous study and adds information concerning the p15 gene that was also found to be methylated in this lymphoma subtype. Methylation incidence within cases at diagnosis and at relapse suggests that it is an early event in anaplastic large-cell lymphomas, being involved in tumor progression in Hodgkin’s cases. Our results show that although p16 and/or p15 methylation is involved in non-Hodgkin’s and Hodgkin’s tumors that share morphological and phenotypic features, differences in incidence, pattern of methylation, and implication in tumor progression are observed.
p16 INK4a and p15INK4b are two members of the INK family of cyclin-dependent kinase inhibitors. Both molecules produce the functional inactivation of the Rb protein and the subsequent block of G1 phase through its binding to cyclin-dependent kinases 4 and 6 (CDK4 and CDK6).1,2
In recent years it has become apparent that the CpG islands of many cancer-associated genes are methylated in many types of human neoplasms.3 Hypermethylation of the 5′ CpG island of p16 has been reported in colon, bladder, breast, and lung carcinomas as well as in gliomas, melanomas, leukemias, and lymphomas.4-7 p15 promoter region hypermethylation has also been described but is almost exclusively involved in hematological malignancies.8 The studies that have analyzed p16 and p15 alterations in lymphomas describe a low proportion of cases with homozygous deletion and mutations, the 5′ CpG island hypermethylation of these tumor suppressor genes being the most frequent event in lymphoid neoplasms.9,10
Nevertheless, the incidence of both epigenetic alterations seems to vary with the lymphoma subtype and the technical approach used. p16 and/or p15 hypermethylation has been widely observed in mucosa-associated lymphoid tissue lymphomas, Burkitt’s lymphomas, and multiple myelomas and the highest percentages have been usually described in studies performed with very sensitive techniques such as methylation-specific polymerase chain reaction (MSP) or bisulfite genomic sequencing.8,11-13 Few cases of CD30 non-Hodgkin’s lymphomas (NHLs) have been previously studied11,14 and there is very little information with respect to p16 methylation status in Hodgkin’s lymphomas (HLs).15
The aim of this study was to determine the p16 and p15 methylation status in anaplastic large-cell lymphomas (ALCLs) and to compare it with that of other non-Hodgkin’s-related entities and HLs. These are malignancies characterized by having both morphological and immunophenotypical features in common with ALCL, such as anaplastic morphology or CD30 activation marker expression.16
To this end, ALCLs (T- and null-type), CD30-positive peripheral T cell lymphomas (CD30+ PTCLs), diffuse large B cell lymphomas (DLBCLs) with anaplastic cytology, and HLs were analyzed and the incidence and pattern of p16 and p15 promoter region methylation established. The possible role of this epigenetic alteration at disease onset and/or progression was also considered.
Materials and Methods
Samples
Methylation status of p16INK4a and p15INK4b genes was studied in 79 lymph node biopsies with the diagnosis of lymphoma according to the World Health Organization classification16 at the Pathology Department of the Fundación Jiménez Díaz (Universidad Autónoma, Madrid). The series comprised 53 NHLs and 26 HLs. The NHL cases included 25 ALCLs (T or null phenotype), 13 CD30+ PTCLs, and 15 anaplastic DLBCLs. The HL samples included 3 nodular lymphocytic predominance, 15 nodular sclerosis (3 nodular sclerosis-I and 12 nodular sclerosis-II) and 8 unclassifiable cases. Diagnoses were made on the basis of histological (routine methods on paraffin-embedded thin sections stained with hematoxylin and eosin and Giemsa) and complete immunohistochemical study. Thirteen of the 53 NHLs and 6 of the 26 HLs corresponded to relapsed samples, the rest being cases at diagnosis. Eighteen samples corresponding to reactive lymphadenopathies (12) and tonsils (6) were used as normal controls.
Immunohistochemistry
Immunohistochemical analysis was performed with a complete panel of antibodies comprising pan-hematopoietic (CD45), cell lineage (CD3, CD4, CD8, C20, CD79a, CD1a, CD68), activation (CD30, CD15, EMA), cytotoxicity (CD57, TIA-1), proliferation (MIB1), and anti-p53 protein markers. The CD45, CD3, CD8, CD20, CD79a, CD68, CD30, and EMA antibodies were provided by DAKO (Glostrup, Denmark); CD4 and p53 by Novocastra Laboratories (Newcastle on Tyne, UK); CD1a, TIA-1, and MIB1 by Immunotech (Marseille Cedex, France); CD57 by Zymed Laboratories (San Francisco, CA); and CD15 by Becton (San José, CA). Sections of 4 to 6 μm from paraffin-embedded tissue blocks were placed onto Vectabond (Vector Laboratories, Burlingame, CA)-coated slides and dried at 60°C overnight. Antibodies were applied after a high-pressure cooking pretreatment for antigen retrieval in a 10-mmol/L citrate buffer, pH 6.0. Staining was achieved semiautomatically (Techmate 500, DAKO) using a streptavidin-biotin variant and diaminobenzidine was used as chromagen.
Sodium Bisulfite DNA Treatment
DNA was isolated from frozen samples using the DNAzol reagent kit as specified by the manufacturers (Molecular Research Center Inc., Cincinnati, OH). Genomic DNA (2 μg) was denatured for 15 minutes at 37°C in 0.3 mol/L of NaOH in a final volume of 80 μl. One μl of freshly prepared solution containing 4.5 mol/L of sodium bisulfite (Sigma, Steinheim, Germany) and 2 mmol/L of hydroquinone (Sigma) at pH 5.0 was added and samples were incubated under mineral oil for at least 16 hours at 55°C. Bisulfite-modified DNA was purified on Wizard DNA clean-up columns (Promega, Madison, WI), according to the manufacturer’s specifications, and eluted in 100 μl of water. The DNA was treated with 0.3 mol/L of NaOH for 15 minutes at 37°C, precipitated with ethanol, and resuspended in 40 μl of water.
MSP
Bisulfite-modified DNA was amplified using the previously described primer pairs p16M, p16U, p15M, and p15U, which yield fragments of 150, 151, 148, and 154 bp, respectively.17 As tumor cells are always admixed with reactive cells in uncultured lymphomas, the polymerase chain reactions (PCRs) using primers specific for unmethylated DNA provided an internal control of DNA integrity after bisulfite treatment.
PCRs using p16M and p16U primer sets were performed under the same conditions. The PCR mix reactions contained 1× PCR buffer (10 mmol/L Tris-HCl, pH 8.3, 50 mmol/L KCl), 1 mmol/L MgCl2, 200 μmol/L each dNTP, 30 pmol of each primer, 1 U of AmpliTaq Gold (Perkin-Elmer Corp., Norwalk, CT), and ∼250 ng of modified DNA as template in a final volume of 50 μl. Temperature conditions were as follows: 10 minutes at 94°C to activate the enzyme (hot start), 40 cycles of 30 seconds at 94°C, 30 seconds at 63°C, and 30 seconds at 72°, and 5 minutes at 72°C for final extension. p15 gene PCR conditions were identical to those used for the p16 gene except for the annealing temperature (61°C both primer sets, p15M and p15U).
DNA from the Raji cell line with hypermethylated p16 gene18 was used as the p16M-PCR-positive control and as the p16U-PCR-negative control. The KG1A cell line, which carries the unmethylated p16 gene and the methylated p15 gene,13 provided the p16U-PCR-negative control and the p15M-PCR-positive control. The HL60 cell line with known p15-unmethylated status13 served as the positive control for the p15U-PCR and as the negative control for the p15M-PCR. All cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). A tube without DNA was included for each set of reactions and all samples were analyzed at least twice after two bisulfite treatments.
The sensitivity of the assay in our laboratory was determined diluting DNA from Raji and KG1A cell lines with DNA from reactive lymphoid tissue (tonsil) with known unmethylated status for p16 and p15 tumor suppressor genes. The dilutions (1/10, 1/100, 1/500, 1/1000, and 1/5000) were treated with bisulfite and PCR amplified.
Bisulfite Genomic Sequencing
Direct sequence analysis of 28 PCR products obtained by MSP with the methylation-specific primer sets was performed. The PCR products corresponded to 13 lymphomas showing p16 methylation (7 NHLs and 6 HLs) and to 15 lymphomas exhibiting p15 methylation (10 NHLs and 5 HLs). The selected cases were representative of the different lymphoma subtypes included in this series.
Sequencing reactions were performed bidirectionally (with sense and antisense p16M primers and sense and antisense p15M primers)17 using the dRhodamine terminator cycle sequencing kit (Applied Biosystems, Foster City, CA) and the ABI PRISM 310 automatic sequencer (Applied Biosystems). Twenty-five cycles were performed in the sequencing reactions consisting of 30 seconds at 96°C, 15 seconds at 50°C, and 4 minutes at 60°C. Sequences enabled MSP results to be validated and precise information on the methylation status of the CpG dinucleotides covered in the amplified DNA fragments (11 CpG sites at p16 and 12 CpG sites at p15) to be obtained. The U conversion of all of the cytosines located outside of the CpG sites (and therefore not methylated) was checked to validate the technique, excluding false-positives because of incomplete DNA reaction with bisulfite.
Methylation Status and Outcome
Detailed clinical data could be retrieved from 30 NHLs taken at diagnosis. These cases were scored according the International Prognostic Index (IPI)19 and included in the low, low-intermediate, intermediate-high, and high risk of mortality categories. Despite the small number of cases, a correlative study of the p16 or p15 methylation status and the IPI index was attempted to establish the importance of these phenomena’s predicting prognosis. The chi-square of Pearson test was used to this end.
Results
p16 Methylation
Methylation of the p16 gene 5′ CpG island was found in 50% (6 of 12) of the null ALCLs, 15% (2 of 13) of the T-cell ALCLs, 23% (3 of 13) of the PTCLs, and 27% (4 of 15) of the DLBCLs (Figure 1)▶ . Grouping T cell lymphomas (T-ALCLs and PTCLs) gave a value of 19% (5 of 26). The frequency of this alteration considering the total NHLs was 28% (15 of 53). Among HLs, 38% (10 of 26) of the cases exhibited p16 methylation. Samples at diagnosis and relapse were separately considered to establish the relevance of the p16 hypermethylation in tumor onset and progression. Twenty-seven percent (11 of 40) of NHLs at diagnosis and 31% (4 of 13) at relapse had p16 CpG island methylation. Twenty-five percent (5 of 20) of HLs at diagnosis and five of the six available relapses (83%) were positive. These results are summarized in Table 1▶ .
Figure 1.

MSP of p16 5′ CpG island. Primer sets for amplification are designated as unmethylated (U) and methylated (M). *, Molecular weight marker PhiX-HinfI; Raji, positive control for M primer set and negative control for U primer set; KG1A, positive control for U primer set and negative control for M primer set. Electrophoresis corresponds to the amplification of bisulfite-treated DNA from DLBCL (lines 1 to 3) and null ALCL (lines 4 and 5). Line 6, no DNA control. Null ALCL in lines 4 and 5 showed methylation. Amplification with the U primers was observed in all of the primary lymphoma cases, which served as internal control of the DNA integrity after bisulfite treatment.
Table 1.
Incidence of the p16 CpG Island Methylation in the Different Lymphoma Subtypes
| Subtype | n | Total (%) | At diagnosis (%) | At relapse (%) | ||
|---|---|---|---|---|---|---|
| CD30 NHL | ||||||
| ALCL | Null | 12 | 6 (50) | 4/9 (44) | 2/3 | |
| T cell | 13 | 2 (15) | 2/10 (20) | 0/3 | ||
| PTCL | 13 | 3 (23) | (19) | 3/11 (27) | 0/2 | |
| DLBCL | 15 | 4 (27) | 2/10 (20) | 2/5 | ||
| Total NHL | 53 | 15 (28) | 11/40 (27) | 4/13 (31) | ||
| HL | 26 | 10 (38) | 5/20 (25) | 5/6 (83) |
n, Number of cases studied; NHL, non-Hodgkin’s lymphomas; ALCL, anaplastic large-cell lymphomas; PTCL, peripheral T cell lymphomas; DLBCL, diffuse large B cell lymphomas; HL, Hodgkin’s lymphomas.
p15 Methylation
Hypermethylation of the p15 gene promoter region was found in 58% (7 of 12) of the null ALCLs, 61% (8 of 13) of the T-cell ALCLs, 77% (10 of 13) of the PTCLs, and 47% (7 of 15) of the DLBCLs. Grouping T cell neoplasms (T-ALCLs and PTCLs) the incidence was 69% (18 of 26). The total frequency in the NHLs was 60% (32 of 53), compared with 42% (11 of 26) in the HLs. The frequency of p15-methylated NHLs at diagnosis was 60% (24 of 40), almost identical to that obtained in relapsed cases (61%, 8 of 13). Within HLs, 35% (7 of 20) of the cases at diagnosis and 67% (4 of 6) at relapse showed aberrant methylation. p15 methylation results are shown in Table 2▶ and Figure 2▶ .
Table 2.
Incidence of the p15 CpG Island Methylation in the Different Lymphoma Subtypes
| Subtype | n | Total (%) | At diagnosis (%) | At relapse (%) | ||
|---|---|---|---|---|---|---|
| CD30 NHL | ||||||
| ALCL | null | 12 | 7 (58) | 5/9 (56) | 2/3 | |
| T-cell | 13 | 8 (61) | 6/10 (60) | 2/3 | ||
| PTCL | 13 | 10 (77) | (69) | 8/11 (72) | 2/2 | |
| DLBCL | 15 | 7 (47) | 5/10 (50) | 2/5 | ||
| Total NHL | 53 | 32 (60) | 24/40 (60) | 8/13 (61) | ||
| HL | 26 | 11 (42) | 7/20 (35) | 4/6 (67) |
n, Number of cases studied; NHL, non-Hodgkin’s lymphomas; ALCL, anaplastic large-cell lymphomas; PTCL, peripheral T cell lymphomas; DLBCL, diffuse large B cell lymphomas; HL, Hodgkin’s lymphomas.
Figure 2.
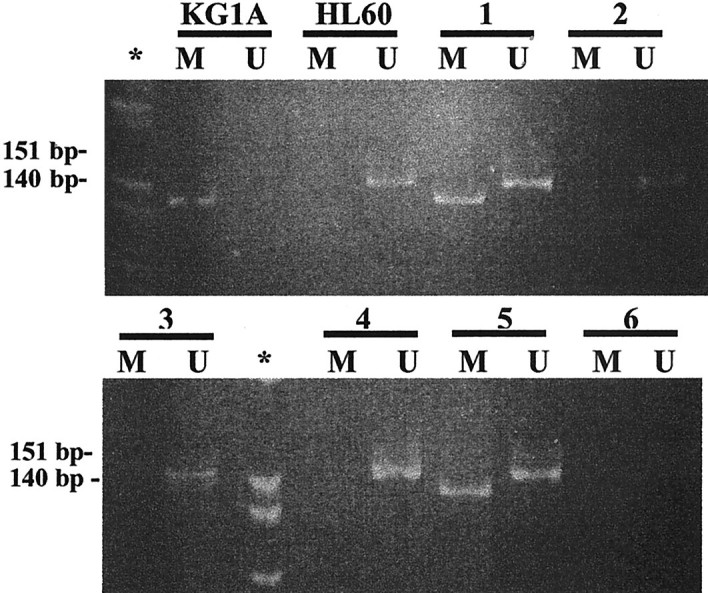
MSP of p15 5′ CpG island. Primer sets for amplification are designated as unmethylated (U) and methylated (M). *, Molecular weight marker PhiX-HinfI; KG1A, positive control for M primer set and negative control for U primer set; HL60, positive control for U primer set and negative control for M primer set. Electrophoresis corresponds to the amplification of bisulfite-treated DNA from HL (lines 1 to 5). Line 6, no DNA control. HL in lines 1 and 5 showed methylation. Amplification with the U primers was observed in all of the primary lymphoma cases, which served as internal control of the DNA integrity after bisulfite treatment.
All 18 reactive samples displayed unmethylated p15 and p16 genes, and the analysis of the DNA from the cell lines used as positive and negative controls yielded the expected results. The sensitivity of the MSP assay was 1/1000 for both p15 and p16 genes (data not shown).
Selective and Concomitant p16 and p15 Methylation
Thirty-eight NHLs showed p16 and/or p15 methylation. Among them, 6 had selective p16 methylation, 23 showed selective p15 methylation, and 9 had concurrent hypermethylation of both genes. The three classes accounted for 16%, 60%, and 24%, respectively, of the total number of methylated NHLs. A more detailed analysis within the different NHL subtypes revealed that p15-only involvement was frequent among lymphomas showing T cell phenotype: 78% (7 of 9) of the methylated T-cell ALCLs and 73% (8 of 11) of the methylated PTCLs.
Methylation of p16 and/or p15 genes was detected in 15 HLs. Twenty-seven percent (4 of 15) displayed selective methylation of p16 gene whereas 33% (5 of 15) showed selective methylation of p15. The concomitant alteration of both loci was found in 40% (6 of 15).
Methylation Patterns
p16 Methylation Pattern
Seven of the 15 p16-methylated NHLs [3 ALCLs (2 null, 1 T-cell type), 3 DLBCLs, and 1 PTCL] and 6 of the 10 p16-methylated HLs were sequenced. All samples displayed methylation at the 11 CpG sites contained in the amplified fragment (Figures 3 and 4)▶ ▶ .
Figure 3.
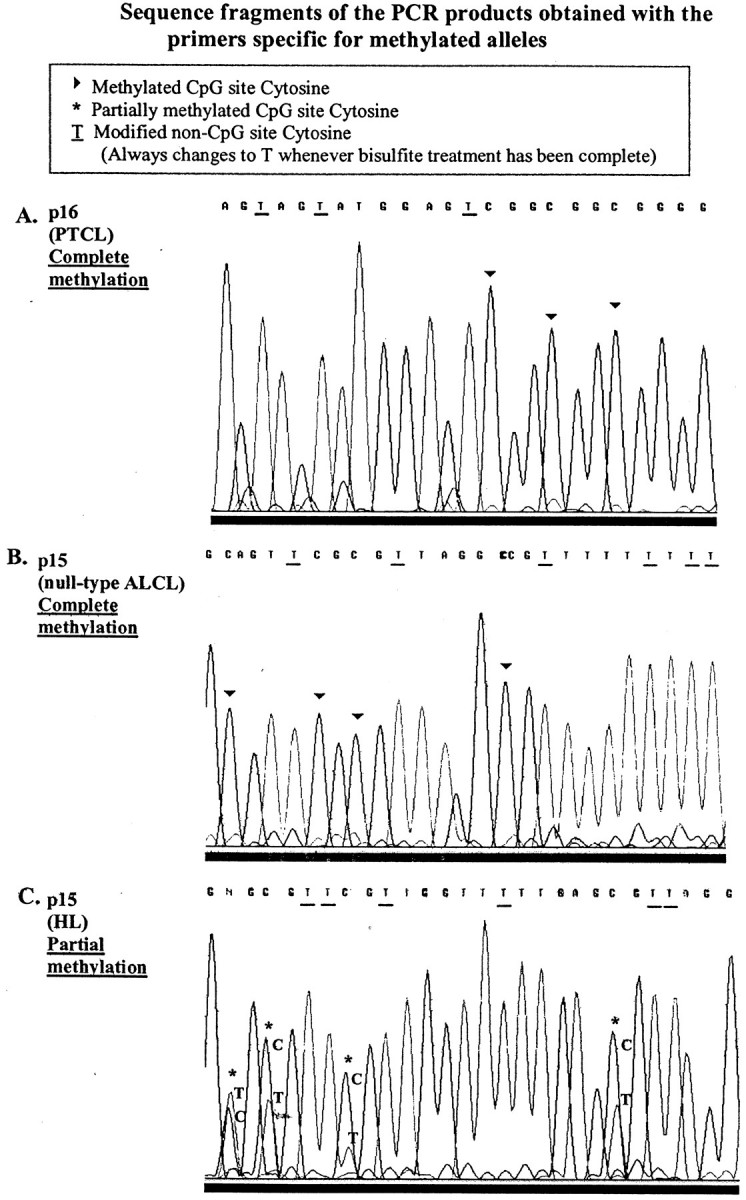
Fragments of the automated DNA sequencing traces corresponding to methylated cases. A: PTCL showing p16 complete methylation. B: Null-type ALCL exhibiting complete p15 methylation. C: HL displaying p15 partial methylation; double peaks of C and T indicating the presence of methylated and unmethylated alleles at the corresponding CpG dinucleotide were observed.
Figure 4.

Methylation pattern representation of the p15 and p16 CpG island-amplified fragments. Black oval, methylated CpG site; white oval, unmethylated CpG site; half black/half white oval, partially methylated CpG site. A: All the 11 CpG sites included in the p16-amplified fragment showed complete methylation in all of the sequenced NHLs and HLs. B1: The 12 CpG sites comprised in the p15-amplified fragment showed complete methylation in NHLs. B2: The p15 methylation pattern was heterogeneous in HL because variation in the state of the CpG sites was observed not only in the same sample but also between cases (each row represents a HL case).
p15 Methylation Pattern
Ten of the 32 p15-methylated NHLs [4 ALCLs (3 null, 1 T-cell type), 2 DLBCLs, and 1 PTCL] and 5 of the 11 positive HLs were sequenced. The NHLs displayed complete methylation of the p15-amplified fragment. In contrast, the p15 methylation pattern in HL was heterogeneous: methylated and unmethylated CpG sites were observed, as well as sites where a double peak of thymine and cytosine coexisted, revealing the existence of both methylated and unmethylated alleles at the corresponding CpG dinucleotide (Figure 3)▶ . In addition to the different status of the CpG sites within a given sample, heterogeneity among the different lymphomas was also observed, because no repetitive pattern of methylation was detected (Figure 4)▶ .
Methylation Status and Outcome
Among the 30 NHLs in which the IPI value was assessed, 68% (15 of 22) of the p16-unmethylated cases corresponded to the low- and low-intermediate risk groups whereas 87% (7 of 8) of the p16-methylated cases corresponded to the high-intermediate and high-risk groups. These differences were found to be significant (P = 0.036). The distribution of IPI scores among the p15-unmethylated and -methylated cases was similar.
Discussion
The unmethylated status of p15 and p16 genes in normal tissues such as hematopoietic cells, kidney, lung, or urinary bladder has been demonstrated in different studies.5,8,12,17 In addition, it has also been reported that p15 and p16 hypermethylation is associated with a loss of transcription that is restored by treatment with demethylating drugs.12,20 None of the reactive lymphoid samples analyzed here showed abnormal methylation of these tumor suppressor genes supporting the fact that anomalous methylation of p15 and p16 is a de novo event in lymphomas related to the tumoral process.
There is growing evidence that epigenetic silencing rather than mutational events is a common mechanism for loss of suppressor gene function, but the timing of methylation has yet to be resolved to determine whether it is responsible for initiating tumoral process or it is just a consequence of previous genetic alterations.
Emerging data show that both genetic and epigenetic phenomenon interact in tumorigenesis so that hypermethylation events seem to predispose to genetic changes and genetic modifications could predispose to epigenetic alteration in malignant cells.21,22
In the present series p15 and p16 aberrant methylation was a frequent event in both NHLs and HLs (NHL: p16, 28%; p15, 60%; HL: p16, 38%; p15, 42%). These high frequencies, consistent with previous studies using similar technical approaches,12,15 highlight the importance of CpG island hypermethylation in cancer whatever the initiating events are. Although the molecular mechanisms involved in this process remain to be established, new findings suggest that methylated DNA-binding proteins and histone deacetylases participate in the transcriptional silencing of hypermethylated genes in cancer.23,24
It has been proposed that anomalous p16 methylation is more closely associated with tumor onset than tumor progression.25-27 Nevertheless, p16 alterations have been associated with aggressive transformation from indolent lymphoma subtypes14,28,29 and its loss of expression has been related to tumoral progression in melanomas or gliomas.5,30
In the present series, the frequency of p16 and p15 hypermethylation in CD30 NHL at diagnosis and at relapse was similar (p16, 27% versus 31%; p15, 60% versus 61%). These results suggest that p16 and/or p15 methylation within high-grade lymphomas (such as those analyzed here) is an early event that may be related to tumor onset and may determine the aggressive behavior of the neoplasm. The early inactivation of p16 and/or p15 in tumor evolution through promoter hypermethylation may be an indirect example of epigenetic predisposition to genetic change. The early disruption of the cell cycle G1/S checkpoint may predispose cells to subsequent genetic alterations contributing to uncontrolled proliferation and cell immortality. A more direct evidence of epigenetic predisposition to genetic change is the association of microsatellite instability with hMLH1 gene promoter hypermethylation in colon, endometrial, and gastric carcinomas.21
HLs behaved differently because a lower incidence of methylation was observed in samples at diagnosis than in patients at relapse (p16, 25% versus 83%; p15, 35% versus 67%). When HL relapses there is generally an increase in the number of malignant cells compared to the diagnostic sample. It could be argued that the increase in the methylation incidence within the relapsed cases could be a result of the assay sensitivity, becoming detectable previously undetectable methylation. Although in most HL cases the Reed-Sternberg cells vary between 1 to 5%, the above-mentioned possibility can be reasonably discarded considering the high sensitivity of our MSP assay (1/1000). Therefore, although the available number of relapses was limited, the significant increase of p16 and p15 methylation suggests that these epigenetic alterations may play an important role in HL disease progression. In these tumors early genetic changes could predispose to subsequent epigenetic alterations during tumor evolution.
Selective methylation of p15 was the most frequent form of methylation among the CD30 NHLs (60% of methylated NHL cases). This suggests that p15 inactivation has an important role in high-grade NHL oncogenesis and that its loss of function is not merely the result of its proximity to p16. This circumstance has also been observed in human acute myeloid and lymphoid leukemias that show p15 methylation in the absence of p16 methylation8 and also in mouse radiation-induced lymphoid leukemias.31
p16 or p15 selective inactivation appeared at a similar frequency in HLs, the concomitant hypermethylation of both genes being the predominant condition in these neoplasms. The alteration of the two tumor suppressor genes could have a cooperative effect on HL development but, because this circumstance was observed equally in diagnostic and relapsed cases, it does not seem to be particularly related to disease progression.
Sequencing of MSP products revealed the complete methylation of the p16 amplified fragment in NHLs and HLs of this series. There are no data concerning the p16 CpG island methylation pattern in HL but Baur and colleagues12 have also reported an almost complete methylation of 42 CpG sites in the promoter region of this gene in NHLs.
p15 methylation was complete in NHLs and heterogeneous in HLs. The heterogeneity of the p15 methylation pattern in HL could arise in two ways. It is possible that all of the alleles of a given sample are densely methylated but the exact location of the sparse unmethylated CpG sites varies from allele to allele. Another possibility is that there are two types of alleles, densely methylated and scarcely methylated, although sufficient to be amplified with the M primer pairs. Both explanations could account for the double peaks of cytosine and adenine observed in many of the CpG dinucleotides.
The Sternberg-Reed (SR) cells of HL are sparse and coexist with a profuse background of B and T mature cells. Some recent studies support the hypothesis that HL tumoral population includes a subgroup of morphologically normal cells.32 The detection of alleles with different p15 methylation patterns could correspond to the presence of tumoral populations at different stages of clonal progression undergoing increasing methylation at the p15 CpG Island.
Despite the low number of cases an association between p16 methylation and poor prognosis according to IPI score was found in the NHLs. This supports the proposed significance of p16 inactivation as a factor predicting poor prognosis in high-grade lymphomas.33,34
Some of the findings of the present study are worth emphasizing. p15 and/or p16 hypermethylation was detected in a high percentage of ALCLs and HLs. Nevertheless, although these alterations might be an early event in ALCLs (as also occurs in the rest of the high-grade CD30 lymphomas considered), they seem to be related to tumoral progression in HLs. In addition, the p15 methylation pattern was complete in ALCL and heterogeneous in HL. These data are relevant because they reinforce the distinction between the two entities that share morphological and immunophenotypical features and contribute to a better knowledge of their biology.
In further studies the use of microdissection analysis or techniques consisting of fluorescence in situ hybridization after MSP on histological sections will allow us to accurately determine the cellular origin of the methylated alleles. The analysis of a higher number of cases including more relapsed patients with detailed clinical data will help to better define the utility of p15 and p16 methylation analysis in the disease follow-up and outcome prediction of non-Hodgkin’s and Hodgkin’s lymphomas. Although more studies are needed, these important applications as well as new therapeutic strategies using demethylating drugs will hopefully be validated in the near future.
Acknowledgments
We thank Jesús Gallego and Trinidad Carrizosa for their excellent technical assistance.
Footnotes
Address reprint requests to María J. García, Dept. Anatomía Patológica, Fundación Jiménez Díaz, Avda. Reyes Católicos, 2. 28040 Madrid, Spain. E-mail:crivas@fjd.es.
Supported by the Fundación Conchita Rábago and the Fundación Eugenio Rodríguez Pascual (to M. J. G.).
References
- 1.Serrano M, Hannon GJ, Beach D: A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366:704-707 [DOI] [PubMed] [Google Scholar]
- 2.Hannon GJ, Beach D: P15ink4b is a potential effector of TGF-B induced cycle arrest. Nature 1994, 371:257-261 [DOI] [PubMed] [Google Scholar]
- 3.Jones PA: DNA-methylation errors and cancer. Cancer Res 1996, 56:2463-2467 [PubMed] [Google Scholar]
- 4.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB: Inactivation of the CKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995, 55:4525-4530 [PubMed] [Google Scholar]
- 5.Reed JA, Loganzo F, Shea CR, Walker GJ, Flores JF, Glendening JM, Bogdany JK, Shiel MJ, Haluska FG, Fountain JW, Albino AP: Loss of expression of the p16/cyclin-dependent kinase inhibitor 2 tumor suppressor gene in melanocytic lesions correlates with invasive stage of tumor progression. Cancer Res 1995, 55:2713-2718 [PubMed] [Google Scholar]
- 6.Fueyo J, Gómez-Manzano C, Bruner JM, Saito Y, Zhang B, Zhang W, Levin VA, Yung WK, Kyritsis AP: Hypermethylation of the CpG island of p16/CDKN2 correlates with gene inactivation in gliomas. Oncogene 1996, 13:1615-1619 [PubMed] [Google Scholar]
- 7.Nakamura M, Sugita K, Inukai T, Goi K, Iijima K, Tezuka T, Kojika S, Shiraishi K, Miyamoto N, Karakida N, Kagami K, O-Koyama T, Mori T, Nakazawa S: p16/MTS1/INK4A gene is frequently inactivated by hypermethylation in childhood acute lymphoblastic leukemia with 11q23 translocation. Leukemia 1999, 13:884-890 [DOI] [PubMed] [Google Scholar]
- 8.Herman JG, Civin CI, Issa JJ, Collector MI, Sharkis SJ, Baylin SB: Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res 1997, 57:837-841 [PubMed] [Google Scholar]
- 9.Otsuki T, Clark HM, Wellmann A, Jaffe ES, Raffeld M: Involvement of CDKN2 (p16INK4A/MTS1) and p15INK4B/MTS2 in human leukemias and lymphomas. Cancer Res 1995, 55:1436-1440 [PubMed] [Google Scholar]
- 10.Drexler HG: Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 1998, 12:845-859 [DOI] [PubMed] [Google Scholar]
- 11.Martínez-Delgado B, Fernández-Piqueras J, García MJ, Gallego J, Rivas C, Robledo M, Benítez J: Hypermethylation of a 5′CpG island of p16 is a frequent event in non-Hodgkin’s lymphoma. Leukemia 1997, 11:425-428 [DOI] [PubMed] [Google Scholar]
- 12.Baur AS, Shaw PH, Burri N, Delacrétaz F, Bosman FT, Chaubert P: Frequent methylation silencing of p15INK4B (MTS2) and p16INK4A (MTS1) in B-cell and T-cell lymphomas. Blood 1999, 94:1773-1781 [PubMed] [Google Scholar]
- 13.Cameron EE, Baylin SB, Herman JG: P15INK4B CpG island methylation in primary acute leukemia is heterogeneous and suggests density as a critical factor for transcriptional silencing. Blood 1999, 94:2445-2451 [PubMed] [Google Scholar]
- 14.Villuendas R, Sánchez-Beato M, Martínez JC, Saez AI, Martínez-Delgado B, García JF, Mateo MS, Sánchez-Verde L, Benítez J, Martínez P, Piris MA: Loss of p16/INK4A protein expression in non-Hodgkin’s lymphomas is a frequent finding associated with tumor progression. Am J Pathol 1998, 153:887-897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García JF, Villuendas R, Algara P, Sáez AI, Sánchez-Verde L, Martínez-Montero JC, Martínez P, Piris MA: Loss of p16 protein expression associated with methylation of the p16INK4A gene is a frequent finding in Hodgkin’s Disease. Lab Invest 1999, 79:1453-1459 [PubMed] [Google Scholar]
- 16.Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD: The World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting—Airlie House, Virginia, November 1997. J Clin Oncol 1999, 17:3835-3849 [DOI] [PubMed] [Google Scholar]
- 17.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin S: Methylation-specific PCR: a novel assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996, 93:9821-9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klangby U, Okan I, Magnusson KP, Wendland M, Lind P, Wiman KG: P16/INK4a and p15/INK4b gene methylation and absence of p16/INK4a mRNA and protein expression in Burkitt’s lymphoma. Blood 1998, 91:1680-1687 [PubMed] [Google Scholar]
- 19.Shipp MA, Harrington DP, Anderson JR, Bonadonna G, Brittinger G, Cabanillas F, Canellos GP, Coiffer B, Connors JM, Cowan RA, Crowther D, Dahlberg S, Engelhard M, Fisher RI, Gisselbrech C, Horming SJ, Lepage E, Lister A, Meerwaldt JH, Montserrat E, Nisserr NI, Oken MM, Peterson BA, Tondini C, Velasques WS, Yeap BY: A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. N Engl J Med 1993, 329:987-994 [DOI] [PubMed] [Google Scholar]
- 20.Otterson GA, Khleif SN, Chen W, Coxon AB, Kaye FJ: CDKN2 gene silencing in lung cancer by DNA hypermethylation and kinetics of p16ink4 protein induction by 5-aza 2′dioxycytidine. Oncogene 1995, 11:1211-1216 [PubMed] [Google Scholar]
- 21.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB: Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998, 95:6870-6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeddeloh JA, Stokes TL, Richards EJ: Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet 1999, 22:94-97 [DOI] [PubMed] [Google Scholar]
- 23.Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP: Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet 1999, 23:62-66 [DOI] [PubMed] [Google Scholar]
- 24.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A: Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393:386-389 [DOI] [PubMed] [Google Scholar]
- 25.Ng MH, Chung YF, Lo KW, Wickman NW, Lee JC, Huang DP: Frequent hypermethylation of the p16 and p15 in multiple myeloma. Blood 1997, 89:2500-2506 [PubMed] [Google Scholar]
- 26.Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB, Herman JG: In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci 1999, 96:12754-12759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez M, Mateos MV, Garcia-Sanz R, Balanzategui A, Lopez-Perez R, Chillon MC, Gonzalez D, Alaejos I, San Miguel JF: De novo methylation of tumor suppressor gene p16/INK4a is a frequent finding in multiple myeloma patients at diagnosis. Leukemia 2000, 14:183-187 [DOI] [PubMed] [Google Scholar]
- 28.Pinyol M, Cobo F, Bea S, Jares P, Nayach I, Fernández PL, Montserrat E, Cardesa A, Campo E: P16INK4A gene inactivation by deletions, mutations and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin’s lymphomas. Blood 1998, 91:2977-2984 [PubMed] [Google Scholar]
- 29.Uchida T, Kinoshita T, Ohno T, Ohashi H, Nagai H, Saito H: Hypermethylation of p16INK4A gene promoter during the progression of plasma cell dyscrasia. Leukemia 2001, 15:157-165 [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa R, Furnari FB, Lin H, Arap W, Berger MS, Cavanee WK, Huang H-JS: Loss of p16INK4 expression is frequent in high-grade gliomas. Cancer Res 1995, 55:1941-1945 [PubMed] [Google Scholar]
- 31.Cleary HJ, Boulton E, Plumb M: Allelic loss and promoter hypermethylation of the p15INK4b gene features in mouse radiation-induced lymphoid—but not myeloid—leukaemias. Leukemia 1999, 13:2049-2052 [DOI] [PubMed] [Google Scholar]
- 32.Jansen MP, Hopman AH, Haesevoets AM, Gennotte IA, Bot FJ, Arends JW, Ramaekers FC, Schouten HC: Chromosomal abnormalities in Hodgkin’s disease are not restricted to Hodgkin/Reed-Sternberg cells. J Pathol 1998, 185:145-152 [DOI] [PubMed] [Google Scholar]
- 33.Gronbaek K, de Nully Brown P, Moller MB, Nedergaard T, Ralfkiaer E, Moller P, Zeuthen J, Guldberg P: Concurrent disruption of p16INK4a and the ARF-p53 pathway predicts poor prognosis in aggressive non-Hodgkin’s lymphoma. Leukemia 2000, 14:1727-1735 [DOI] [PubMed] [Google Scholar]
- 34.Moller MB, Kania PW, Ino Y, Gerdes AM, Nielsen O, Louis DN, Skjodt K, Pedersen NT: Frequent disruption of the RB1 pathway in diffuse large B cell lymphoma: prognostic significance of E2F-1 and p16INK4A. Leukemia 2000, 14:898-904 [DOI] [PubMed] [Google Scholar]