

Abstract
Retinoid acid receptors are DNA-binding proteins mediating the biological effects of ligands through transcriptional activation. It is known that the activity of the 26S proteasome is important for nuclear receptor-activated gene transcription. However, the molecular mechanism by which the 26S proteasome participates in this process is not well understood. Here we report that the proteasome activity is essential for ligand-dependent interaction of RAR with its co-regulators such as SRC, p300 and RXR. We also determined that the proteasome activity is required for the association of liganded RAR to the genomic DNA and, consequently, for the recruitment of the coactivator complex to the retinoic acid responsive elements. Moreover, the requirement of proteasome activity for the activator activity of RAR is determined by the promoter context. Our study suggests that the 26S proteasome regulates directly the activity of RAR as an activator.
Key words: gene regulation, transcriptional coactivator, nuclear receptor, the 26S proteasome, genomic association
Introduction
In vertebrates, the proper distribution and metabolism of vitamin A is essential for normal embryonic development and growth.1 Deficiency in vitamin A during early embryogenesis leads to congenital malformations affecting patterning and the development of many organ systems.2 The diversified biological functions of vitamin A are mediated by multiple levels of effectors including RAR, the retinoic acid receptor, and RXR, the retinoid X receptor.3
RAR and RXR are ligand-inducible transcription factors, regulating the transcription of an array of retinoid responsive genes through a bimodal mode.4 As a heterodimer, RAR and RXR bind constitutively to retinoic acid response elements (RARE) located within the regulatory region of retinoid responsive genes regardless of ligand.5 In the absence of ligand, DNA-bound RAR and RXR heterodimer acts as a repressor of transcription by associating with the NCoR corepressor complex, but upon ligand induction, it acts as an activator by recruiting SRC and p300 coactivator complexes. As a result, NCoR is present at the RARE in the absence of ligand, whereas SRC and p300 are detected at RARE-regulated promoters following ligand induction.6,7 Thus, some retinoid responsive promoters are classified as pre-set or poised promoters, since Pol II and TBP bind to the TATA box constitutively.7
The transcriptional coactivator p300, initially identified as an E1A-associated protein, contains an intrinsic histone acetyltransferase (HAT) activity and multiple interaction surfaces for association with many transcription factors, activators and components of basal transcription machinery.8,9 The function of p300 is critical for a broad array of biological processes including development, growth and cellular differentiation.10,11 Embryonic development is very sensitive to p300 gene dosage and cells derived from p300 knockout embryos are defective in retinoid signaling.12 In addition, p300 also functions as a tumor suppressor and mutations in the p300 gene have been detected in many epithelial cancers.13–15
The 26S proteasome pathway is one of the major proteolysis systems of the cell. It contains a 20S core particle capped at both ends by the 19S regulatory particles, which recognize and deliver ubiquitinated proteins to the 20S proteasome.16 Many transcriptional activators, nuclear receptors and coactivators are subject to modification by ubiquitination or degradation through the proteasome pathway.17–23 Previously, we reported that histone deacetylase inhibitor sodium butyrate enhances p300 degradation through the 26S proteasome, which may account for some of the negative effects of butyrate on glucocorticoid-induced transcriptional activation.24 We also reported that the histone deacetylase inhibitor-induced p300 degradation is mediated through the increase of gene expression of the B56γ3 regulatory subunit of protein phosphatase 2A, shedding light on the molecular basis for the negative effects of histone deacetylase inhibitors on p300 function.25 In addition, p300 is also a substrate of the cytoplasmic ubiquitin-proteasome system.26
The ubiquitin system plays a central role in diverse cellular processes including protein homeostasis, DNA repair and immune function.27 Dysfunction of this system leads to various pathological conditions such as cancer, neurodegenerative diseases and immunological disorders.28 In yeast, inhibition of the proteasome activity represses the expression of about 5% of all active genes.29 The effects of the 26S proteasome on gene transcription are mediated through either turnover of transcription factors or facilitation of transcription elongation.20,30,31 It is known that the 26S proteasome activity is important for RAR-mediated transcriptional activation.20 In addition, microinjection of an antibody against the 19S proteasome or pretreatment of cells with the proteasome inhibitor MG132 blocks ligand induced transcriptional activation of RARβ gene.32 However, the precise role of the 26S proteasome in RAR-mediated transactivation remains unclear.
In this study, we determined that the proteasome activity is essential for protein-protein interaction of RAR with its co-regulators, such as SRC, p300 and RXR, for the promoter occupancy of liganded RAR and, consequently, for the recruitment of the coactivator complex to the retinoid responsive promoters. In addition, the requirement of proteasome activity for the binding of liganded RAR to RARE is determined by the promoter context.
Results
The 26S proteasome activity is important for RAR mediate transcriptional activation.
It is known that retinoic acid (RA) induced transcriptional activation requires the 26S proteasome activity.32 However, it is not clear whether the proteasome activity is mediated solely through the action of ligand activated RAR or also through RXR, since all-trans RA used in previous studies can be metabolized into 9-cis RA and thus has affinity to both RAR and RXR. To decipher the role of the 26S proteasome in the function of RAR per se with respect to RARE-dependent gene activation, we employed TTNPB {4-[(E)-2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid}, a potent retinoic acid analog selective for all RAR subtypes.33 We also used mouse pluripotent embryonic carcinoma P19 cells in which the transcription of RARβ gene is rapidly induced by RA and the recruitment of p300 coactivator complex to the RARE region of the promoter is mediated by liganded RARα with RXRα acting as a silent partner.7,34
We first used a luciferase reporter containing the RARE segment of RARβ2 promoter to examine the role of the 26S proteasome activity in TTNPB-induced transactivation. P19 cells were transfected with the RARβ2 reporter and induced with the RAR selective ligand in the presence or absence of MG132, a reversible proteasome inhibitor,16 and then harvested for the luciferase assays. Consistent with previous reports, the RAR selective ligand induced transcriptional activation of the reporter by about 50-fold; this transactivation was significantly inhibited, about 85%, by the addition of proteasome inhibitor MG132 (Fig. 1A).
Figure 1.
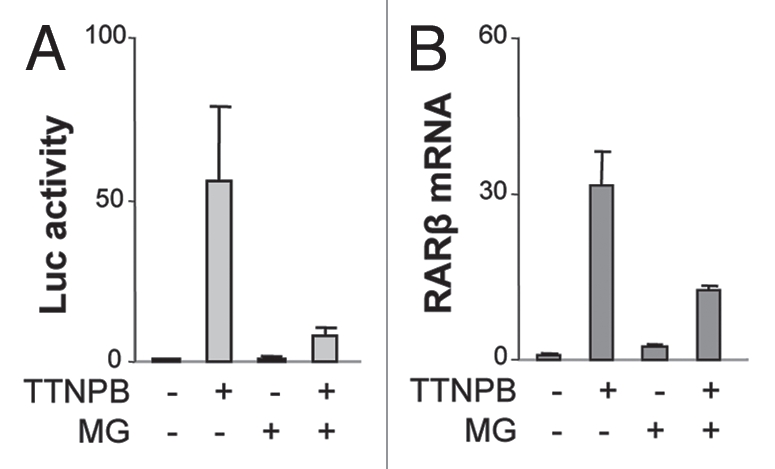
RARE-dependent transcriptional activation depends on the 26S proteasome activity. (A) Cells were transfected with a RARE reporter and then treated with TTNPB (1 µM) in the presence or absence of MG132 (MG, 5 µM) for 16 h. Shown are fold inductions of the luciferase activities in relation to the untreated control. β-galactotsidase activity was used as an internal control. Error bars represent the standard deviations of three independent experiments. (B) Real-time RT-PC R analysis of the levels of endogenous RARβ mRNA following 16 h of ligand induction in the presence or absence of MG132, which are presented as fold variations compared to the untreated control. 18S rRNA was used as an internal control. Error bars represent the standard deviations of triplicates of one representative experiment.
We next examined the role of the 26S proteasome in the expression of endogenous RARβ gene, since proteasome inhibitors can reduce luciferase activity in tissue culture cells.35 Real-time RT-PCR analysis revealed that treatment of the P19 cells with the RAR selective ligand for 16 h increased the transcript level of RARβ gene by about 30-fold, whereas inhibition of the 26S proteasome activity with MG132 reduced the accumulation of RARβ transcripts by about 60% (Fig. 1B). Thus, the 26S proteasome participates in RARE-dependent gene expression through the regulation of RAR as an activator.
Short term of RAR selective ligand induction does not affect RAR stability.
To determine if the RAR selective ligand targets RAR degradation through the 26S proteasome pathway, we also examined the impact of the RAR selective ligand on protein turnover of RARα and its co-regulators. The cells were induced with ligand in the presence or absence of MG132 for 4, 8 or 16 h and subjected to quantitative western analysis. As shown in Figure 2A and B, the steady-state levels of RARα protein remained constant following treatment with the RAR selective ligand, regardless of proteasome inhibition for up to 16 h. Similarly, the steady-state levels of RXRα, SRC-1 and p300 were not decreased by these treatments (Fig. 2A and B). In contrast, the levels of NCoR protein increased significantly by about two-fold, compared to the untreated control cells following 4 or 8 h of proteasome inhibition (Fig. 2A and B).
Figure 2.

Short term of ligand induction does not affect the stability of RARα. (A) Equal amounts of whole cell extracts (50 µg) were used for western blot analysis of the endogenous RARα, RXRα, SRC-1, p300 and NcoR proteins following treatments with TTNPB (1 µM) for 4, 8 and 16 h in the presence or absence of MG132 (MG, 5 µM). The blots were then stripped and re-probed with a γ-tubulin antibody for internal control. (B) Quantitative analysis of the blots as in (A) is expressed as fold variations compared with the untreated controls after being normalized to the loading controls. Error bars represent standard deviations of at least three independent experiments (*p < 0.05). (C) After pulse labeling, the cells were chased in the presence or absence of ligand for 4–24 h. The endogenous RARα proteins were immunopurified, separated by SDS-PA GE and quantified by PhosphorImager. The apparent half-life of endogenous RARα is 23 ± 5 hours in the absence of ligand and 25 ± 4 h in the presence of ligand (n = 3). (D) The cells were treated with cycloheximide (10 µg/ml) in the presence or absence of ligand for 4–24 h and harvested for western analysis of the endogenous RXRα. The apparent half-life of endogenous RXRα is 37 ± 2 h in the absence of ligand and 40 ± 3 h in the presence of ligand (n = 3).
We next determined that the stability of RARα protein in response to the treatment with RAR selective ligand and proteasome inhibitor. In agreement with the literature, RARα is a fairly stable protein with an apparent half-life of about 23 h in the absence of selective ligand as determined by a pulse-chase protocol (Fig. 2C). Interestingly, the apparent half-life of RARα was not significantly affected by ligand induction per se, and was about 25 h (Fig. 2C). RXRα is a more stable protein; therefore, we used a cycloheximide protocol instead to determine the apparent half-life of RXRα. Cells were treated with cycloheximide, which inhibits ribosome translocation,36 for 4–24 h and then harvested for quantitative western analysis of RXRα. As shown in Figure 2D, the apparent half-life of RXRα is about 40 h in the absence of RAR selective ligand. Again, the apparent half-life of RXR was not significantly affected by ligand induction and was about 37 h (Fig. 2D). Taken together, these data suggest that the 26S proteasome activity may participate in the activation of retinoid-responsive genes through the regulation of RAR as an activator rather than through the mechanisms of RAR degradation or protein turnover.
RAR selective ligand enhances RAR ubiquitination.
To define the role of the 26S proteasome in the post-translational modification of RAR, we examined the ubiquitination of RARα and RXRα upon ligand induction with an in vivo ubiquitination assay,37 since RARα and RXRα are known substrates of ubiquitin conjugation and degraded through the 26S proteasome pathway.38,39 Cells were first treated with MG132 for 4 h in the absence or presence of RAR selective ligand. The endogenous RARα or RXRα protein was immunopurified with specific antibodies against RARα or RXRα and then subjected to western analysis. The RARα or RXRα blots were first probed with an ubiquitin antibody to examine the ubiquitination status of these proteins and then re-probed with specific antibodies against RARα and RXRα for internal controls.
As shown in Figure 3A and B, the ubiquitin signal of the RARα immunoprecipitates became readily detectable following 4 h of proteasome inhibition in the absence of ligand, about five-fold in comparison to the untreated control. Following the addition of the RAR selective ligand, the level of RARα ubiquitination increased significantly, more than three-fold when compared with the MG132 only treatment (Fig. 3A and B). The level of RXRα ubiquitination was also readily detectable following 4 h of proteasome inhibition, about six-fold in relation to the untreated control (Fig. 3C and D). However, the degree of RXRα ubiquitination was not significantly affected by the addition of RAR selective ligand (Fig. 3C and D). Collectively, these data suggest that the RAR selective ligand play a specific role in RAR ubiquitination and the 26S proteasome regulates the activator activity of RAR through an ubiquitin-dependent pathway.
Figure 3.

RAR selective ligand enhances RARα ubiquitination. (A) An in vivo ubiquitin assay was used to assess the levels of RARα ubiquitin conjugation following 4 h of treatment with MG132 (MG, 5 µM) in the presence or the absence of TTNPB (1 µM). The blots were first probed with an ubiquitin specific antibody, and then stripped and re-probed for RARα as internal controls. (B) Quantification of the western blots as in (A) is presented as average fold variation of the ubiquitin signal normalized to the levels of immunopurified RARα following ligand induction and MG132 treatments in comparison to the MG132 only control. Error bars are the standard deviations of three independent experimental repeats (*p < 0.05). (C and D) The experimental procedures were as (A and B) except that the ubiquitination of RXRα protein was studied.
The 26S proteasome activity is important for ligand-dependent interaction of RAR with its co-regulators.
To determine the impact of proteasome on ligand-induced RAR conformation changes, we examined the association of RAR with its coactivators since one important aspect of RAR acting as activator is to recruit coactivators to the RARE regions of genomic DNA through protein-protein interaction. First, cells were treated with RAR selective ligand in the presence or absence of MG132 for 4 h and the endogenous SRC-1 were immunoprecipitated with a specific antibody against SRC-1 and subjected to western analysis. The blots were first probed with an antibody against RARα to detect the endogenous RARα co-immunoprecipitated with SRC-1 and then they were striped and re-probed for RARα as an internal control. As shown in Figure 4A, the association of RARα with SRC-1 depends on RAR selective ligand. This ligand-dependent interaction of RARα with SRC-1 was disrupted following 4 h of MG132 administration, compared with the ligand alone treatment.
Figure 4.
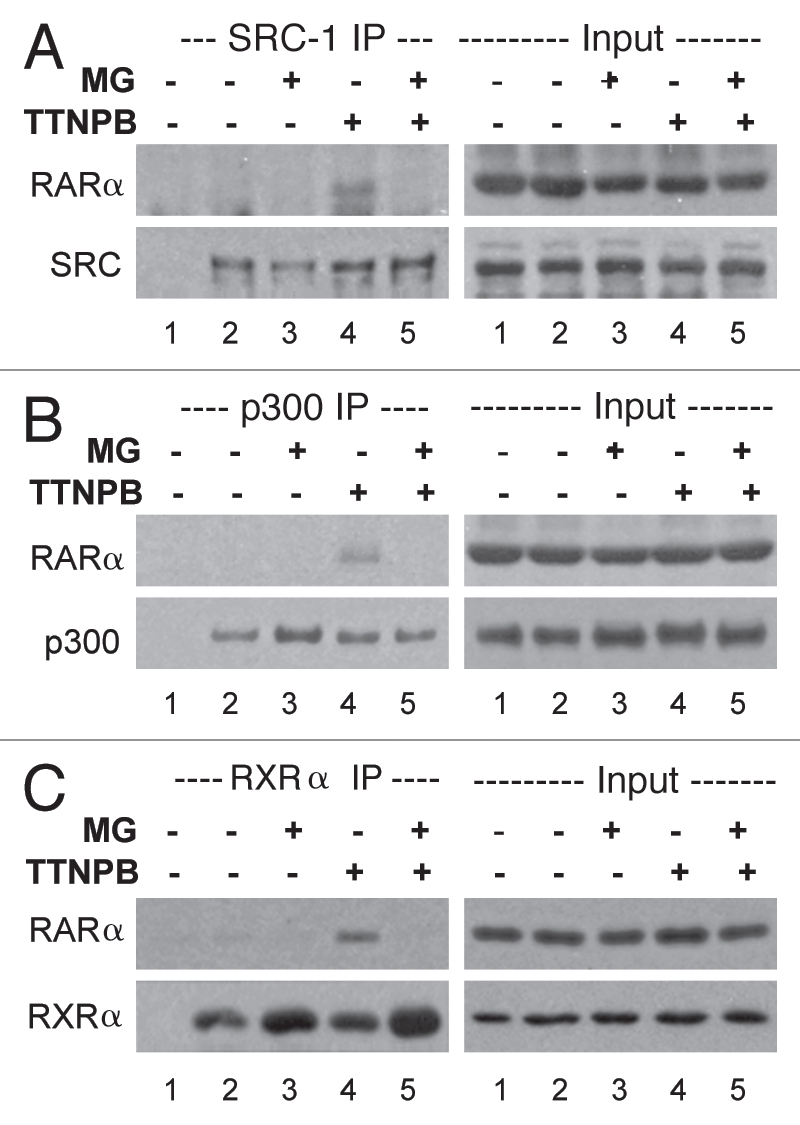
The 26S proteasome activity is important for ligand-dependent interaction of RARα with its coregulators. (A) Co-immunoprecipitation of endogenous RARα with SRC-1 was examined following 4 h of TTNPB induction (1 µM) in the presence or absence of MG132 (MG, 5 µM). The western blots were first probed with a RARα antibody, and then stripped and re-probed for SRC-1. Lane 1 is a negative immunoprecipitation control. 10% of the input extracts were also subject to western analysis as internal controls. Shown are the representatives of three independent experiments. (B) Co-immunoprecipitation of endogenous RARα with p300 following the same treatments. Western blots were first probed with a RARα antibody and then re-probed for p300. (C) Co-immunoprecipitation of endogenous RARα with RXRα was also examined following the same treatment. The western blots were first probed with a RARα antibody then re-probed for RXRα.
We next examined the interaction of RARα with endogenous p300 by using a specific antibody against p300 with the same co-immunoprecipitation approach. As shown in Figure 4B, the interaction of RARα with p300 also depends on ligand induction. Similarly, the ligand-dependent interaction of RARα with p300 was also disrupted following 4 h of proteasome inhibition (Fig. 4B). In addition, the association of RARα with RXRα was also disrupted following 4 h of MG132 addition compared to the TTNPB alone treatment (Fig. 4C). Thus, the activity of the 26S proteasome may be important for of RARα to interact with its co-regulators during ligand-induced transcriptional activation.
The 26S proteasome is critical for ligand-dependent occupancy of RARβ2 promoter.
To delineate the role the 26S proteasome in the activator activity of RAR, we employed the RARβ2 promoter as a model system, as it contains a classic DR5 RARE in the vicinity of TATA box (Fig. 5A). Cells were induced with ligand in the presence or absence of MG132 for 1, 2 and 4 h since the association of RARα with its coactivators was disrupted following 4 h of proteasome inhibition (Fig. 4). A time course real-time RT-PCR analysis revealed that RARβ gene transcription was rapidly induced up to 15-fold by the addition of RAR selective ligand for 4 h, whereas inhibition of the 26S proteasome activity impaired the increase of RARβ transcripts by about 50% (Fig. 5B). Most intriguingly, the decrease in RARβ mRNA levels was observed only after 2 h of co-treatment with MG132 (Fig. 5B).
Figure 5.

The 26S proteasome activity is essential for ligand-dependent occupancy of RARβ2 promoter. (A) Schematic presentation of the relation of the DR5 RARE and TATA box at the promoter. (B) Real-time RT-PC R analysis of the levels of RARβ mRNA in cells treated with TTNPB (1 µM) in the presence or absence of MG132 (MG, 5 µM) for 1, 2 and 4 h. Quantification is presented as fold variations compared to the untreated control. 18S rRNA was used as an internal control. Error bars represent the standard deviations of triplicates from one representative experiment. (C) ChIP analysis of the occupancy of RARβ2 promoter by NCoR, RARα, RXRα, SRC-1, p300 and Pol II following 1, 2 and 4 h of ligand induction in the presence or absence of MG132. Lane 1 is the negative ChIP control. The input DNA was also analyzed in parallel. (D–F) Quantitative analysis of the association of NCoR, RARα and RXRα to the RARE is expressed as fold variations compared with the untreated controls after being normalized to the input controls. Error bars represent standard deviations of five independent experiments (**p < 0.01). (G) Following pretreatments with MG132 for 2 and 3 h and together with ligand for an additional hour, the occupancy of RARα and RXRα was examined by ChIP analysis.
We next examined the occupancy of the RARβ2 promoter by NCoR, because co-repressor release is a prerequisite for transcriptional activation of the retinoid responsive genes.37 Consistent with previous reports, NCoR was detected at the RARβ2 promoter in the absence of ligand and was immediately released from the RARE upon ligand induction, as shown by the time course chromatin immunoprecipitation (ChIP) analysis (Fig. 5C). Interestingly, inhibition of proteasome activity did not disrupt the ligand-independent association of NCoR to the promoter; rather, following MG132 treatment, this association increased to a comparable level with the increase of NCoR protein (compare Fig. 5C and D with Fig. 2A and B).
We also examined the binding of RARα and RXRα to the RARβ2 promoter by using the same time course ChIP analysis. In line with the literature, the association of RARα and RXRα to the RARE was constitutive, regardless of ligand (Fig. 5C, E and F). Moreover, treatment of the cells with MG132 in the absence of ligand did not perturb the ligand-independent occupancy of RARE by RARα and RXRα (Fig. 5C, E and F). However, in the presence of RAR selective ligand, MG132 treatment impeded significantly the binding of RARα and RXRα to the RARE (Fig. 5C, E and F). Most intriguingly, this impediment only became significant after 2 h of MG132 administration, which is concomitant with the decrease in RARβ gene transcripts (Fig. 5B, C, E and F). Following 4 h of treatment, the RARE-bound RARα and RXRα was reduced by about 90% (Fig. 5C, E and F).
To further assess the impact of the 26S proteasome on the coactivator occupancy of the RARβ2 promoter, we also performed the time course ChIP analysis with specific antibodies against SRC-1, p300 and Pol II. As shown in Figure 5C, SRC-1 was detected at the RARE following 1 h of ligand induction, whereas the association of p300 to the RARE was observed later, following 2 h of induction. However, the ligand-dependent recruitment of SRC-1 and p300 was disrupted in a time-dependent manner when the cells were treated with proteasome inhibitor MG132 (Fig. 5C). Again, the loss of SRC-1 and p300 recruitment was seen after 4 h of treatment, coinciding with the perturbation of RAR and RXR occupancy at the promoter and the decrease in RARβ gene transcripts (Fig. 5B, C, E and F). However, the association of Pol II at the promoter was not affected by the MG132 treatment, regardless of ligand (Fig. 5C).
To determine whether the 26S proteasome is required for the maintenance of the activator activity of RAR in general or specifically required for the process of ligand activation, we also designed a MG132 pretreatment protocol, as the association of liganded RAR to RARE promoter was impeded only after 2 h of MG132 addition (Fig. 5C). Cells were first treated with MG132 alone for 2 or 3 h, and then together with RAR selective ligand for an additional hour. As shown in Figure 5G, loss of liganded RARα or its silent partner RXRα at the RARE correlates with the hours of MG132 treatment, rather than with ligand induction. Taken together, these data suggest that the 26S proteasome activity is important for the binding of the receptor to the RARE in response to ligand induction possibly through modulation of the receptor conformation change or ubiquitination.
The role of promoter context in liganded RAR occupancy.
To define the determinants for the role of proteasome in the occupancy of liganded RAR at RARE, we employed another RA target gene, Cyp26A1.38 The Cyp26A1 gene contains two well defined DR5 RARE, one (R1) is close to the TATA box and another (R2) about 2 kb upstream (Fig. 6A). Cells were treated with ligand in the presence or absence of MG132 for 1 and 4 h and subjected to real-time RT-PCR analysis. As shown in Figure 6B, the Cyp26A1 transcripts were robustly induced by RAR selective ligand, about 60-fold by 4 h, whereas inhibition of the 26S proteasome activity impaired the accumulation of Cyp26A1 transcripts by about 70%. Most interestingly, the decrease in Cyp26A1 mRNA level was observed only at 4 h of co-treatment with MG132 (Fig. 6B).
Figure 6.

The 26S proteasome activity is essential for ligand-dependent occupancy of R1, but not R2 region of Cyp26A1 promoter. (A) Schematic presentation of the R1 and R2 regions relative to the TATA box at Cyp26A1 promoter. (B) Real-time RT-PC R analysis of the levels of Cyp26A1 mRNA in cells treated with TTNPB (1 µM) in the presence or absence of MG132 (MG, 5 µM) for 1 and 4 h. Quantification is presented as fold variations compared to the untreated control. Error bars represent the standard deviations of the triplicates from one representative experiment. (C) ChIP analysis of the occupancy of Cyp26A1 regulatory region by RARα and RXRα following 1 and 4 h of ligand induction in the presence or absence of MG132. Lane 1 is the negative ChIP control. The input DNA was also analyzed in parallel.
Consistent with previous reports, the occupancy of RARα and RXRα was detected at the R1 and R2 region of the Cyp26A1 promoter, regardless of ligand, as shown by ChIP analysis (Fig. 6C). In the absence of RAR selective ligand, inhibition of proteasome activity did not disrupt the association of RARα and RXRα to the R1 or R2 region (Fig. 6C). However, in the presence of ligand, MG132 treatment impeded significantly the binding of RARα and RXRα to the R1 region but not the R2 region (Fig. 6C). Again, this impediment of R1 occupancy only became apparent after 4 h of MG132 administration, concomitant with the decrease in Cyp26A1 gene transcripts (Fig. 6B and C). Taken together, these data suggest that the proximity of RARE to the TATA box, or the looping of distal RARE to the proximal promoter, may be a factor for the involvement of proteasome in the binding of liganded receptor to the RARE.
Proteasome inhibitor PS-341 has the same effects as MG132 on the occupancy of RARE by liganded RAR.
To determine the specificity of proteasome activity in the occupancy of liganded RAR at RARE, we also employed another potent proteasome inhibitor, PS-341, which is well tolerated in clinical trials.39 Western analysis demonstrated that treatment with 10 or 20 nM of PS-341 for up to 8 h did not affect the steady-state levels of RARα and RXRα (Fig. 7A). We then examined the effects of PS-341 on RARE dependent gene expression. Cells were induced with ligand in the presence or absence of PS-341 for 1 and 4 h because the occupancy of RARα and RXRα at the RARβ2 or R1 region of Cyp26A1 was disrupted following 4 h of MG132 treatments (Figs. 5 and 6). PS-341 reduced the level of RARβ mRNA by about 50% (similar to the reduction obtained with MG132) and this reduction only became evident after 4 h of PS-341 administration (compare Fig. 7B with 5B). In addition, the effect of PS-341 on the transcript level of Cyp16A1 gene was also similar to the effect obtained with MG132 (compare Fig. 7B with 6B). Interestingly, Cyp26A1 transcripts appear to be more sensitive to MG132 and PS-341 than RARβ transcripts (Figs. 5B, 6B and 7B).
Figure 7.

Effects of proteasome inhibitor PS -341 on RAR-mediated transcriptional activation (A) Cells were treated with TTNPB (1 µM) in the presence or absence of PS-341 (10 or 20 nM) for 4 or 8 h and then subjected to western analysis of RARα and RXRα protein. The blots were then stripped and re-probed for γ-tubulin as an internal control. (B) Real-time RT-PCR analysis of the levels of RARβ and Cyp26A1 mRNA in cells treated with TTNPB and PS-341 (10 nM) for 1 and 4 h. Quantification is presented as fold variations compared to the untreated control. Error bars represent the standard deviations of the triplicates from one representative experiment. (C) ChIP analysis of the occupancy of RARβ2 and R1 and R2 region of Cyp26A1 by RARα in cells treated with TTNPB and PS-341. Lane 1 is the negative ChIP control. The input DNA was also analyzed in parallel. (D) The binding of RXRα was also analyzed by the ChIP assay.
We next examined the effects of PS-341 on RARE occupancy. Similar to MG132, PS-341 did not affect the binding of unliganded RARα to the RARβ2 or the R1 region of Cyp26A1, but disrupted the RARE occupancy of RARα in the presence of the RAR selective ligand (Fig. 7C). Again, this impediment only became apparent after 4 h of PS-341 administration, coinciding with the decrease in gene specific transcripts (Fig. 7B and C). In addition, the effects of PS341 on the binding of liganded RARα to the R2 region of Cyp26A1 and on the occupancy of RXRα at these RARE regions were also similar to the effects of MG132 (Fig. 7C and D). Collectively, these data demonstrated that the proteasome activity is essential for the binding of liganded RARα to RARE in the vicinity of TATA box.
Discussion
In this study, we examined the impact of the 26S proteasome on the expression of retinoid-responsive genes and determined the molecular basis for the role of the proteasome in retinoid-induced transcriptional activation. First, the 26S proteasome plays an important role in the regulation of RARα to serve as an activator. Second, the proteasome activity is essential for ligand-dependent interaction of RAR with its co-regulators such as SRC-1, p300 and RXR. Third, the proteasome activity is required for the occupancy of RARE by RARα and RXRα upon ligand induction; and, consequently, for the recruitment of coactivator complex in the vicinity of TATA box. Thus, the promoter context determines the involvement of the proteasome in the regulation of RARα as an activator.
The importance of the 26S proteasome in the activation of hormone-responsive genes was initially established for estrogen-induced gene activation.20 Subsequent studies have revealed the ubiquitination and degradation of several nuclear receptors as well as coactivators during the course of their nuclear activities.40 Proteins related to the 26S proteasome pathway have also been found at the promoter regions of hormone-responsive genes.32,41 Collectively, these studies appear to suggest that protein degradation is coupled with the activator activity of nuclear receptors and that the ubiquitin-proteasome pathway modulates transcription by promoting turnover of the nuclear receptor-transcription complex. However, it is not clear at which molecular level the 26S proteasome regulates the activator activities of nuclear receptors and whether the proteasome is required for the activator activity or just for receptor turnover.
In this study, we focused on the molecular basis for the involvement of the 26 proteasome pathway in RARα-mediated transcriptional activation. We used a RAR-selective ligand and two well established promoter systems in which ligand rapidly induces the expression of RARβ and Cyp16A1 genes. Our observations that ligand does not promptly enhance RAR turnover and that the 26S proteasome activity is required for the binding of liganded RAR to the genomic RARE (Figs. 2–7) are suggestive of a non-proteolytic role for the proteasome in RARα-mediated gene activation. Such a role for the proteasome has emerged recently.42
Ligand induced receptor degradation has been reported for many nuclear receptors including estrogen receptor, progesterone receptor, thyroid hormone receptors, RAR and RXR.17,18,43–45 The turnover of these receptors appears to have links with the proteasome pathway. In the case of RAR or RXR, the ligand-induced receptor degradation is often observed after extended ligand treatments.23,44–46 We did not detect negative effects of ligand on RARα protein following short periods of treatment (4–16 h; Fig. 2). In addition, RARα-mediated transactivation was significantly affected within 4 h of proteasome inhibition (Figs. 5–7). Hence, our data indicate a role for the proteasome in the involvement of RARα to function as an activator through a proteolysis-independent pathway. However, ligand promptly enhances the ubiquitination of RAR (Fig. 3), suggesting that the 26S proteasome may be also involved in the function of RAR through a proteolysis-dependent manner. What type of ubiquitination is engaged, where the ubiquitination occurs, or which E3 ligase is involved in this process, are some of the question that remain to be unanswered.
Besides nuclear receptors, the destruction of other activators has also been intimately linked to the transcriptional potential of these proteins.19,30,47 While the proteasome may play a dual role in transcriptional regulation through a proteolysis-dependent pathway, studies using yeast as model system have demonstrated that the proteasome pathway can also participate in the regulation of gene expression in a proteolysis-independent manner.48,49 Thus, it is possible that the 26S proteasome system targets the ubiquitinated receptors for degradation subsequent to transcriptional activation.
Transcriptional activation mediated by RAR depends on the cooperation of RXR to form a heterodimer at the RARE to recruit coactivator complex.50 Although, unliganded RAR is able to form a heterodimer with RXR as the core of the co-repressor complex at the RARE, the interaction of RAR with RXR in the absence of DNA is dependent on ligand induction.51 In addition, the dimerization interface of RAR with RXR determines the cooperative binding of the heterodimer to DNA.52 It has been proposed that ligand-controlled dimerization is important for RAR activation and depends on remodeling of the ligand domain of RAR.51 Our study shows that the proteasome pathway is involved in RARα activation process through modulating the interaction of RARα with its co-regulators and, consequently, with the formation of heterodimer on the genomic DNA (Figs. 4–7). Further mechanistic studies would be essential for elucidating the mechanisms underlying the complex dynamics of RAR genomic association, transcriptional activation and subsequent turnover.
In conclusion, our study shields new light on how the activator activity of RARα is regulated during ligand-induced transcriptional activation and provides molecular basis for the involvement of the 26 proteasome pathway in this process.
Materials and Methods
Cell culture and reagents.
Mouse embryonic carcinoma P19 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) with 10% fetal bovine serum and non-essential amino acids at 37°C with 5% of CO2. TTNPB and MG132 were purchased from Sigma-Aldrich. PS-341 and cycloheximide were from LC Laboratory. Antibodies against RARα, RARβ, RXRα, NCoR, SRC-1, p300, Pol II and ubiquitin were purchased from Santa Cruz Biotechnology. Protein A agarose/salmon sperm DNA was from Upstate.
Cell transfection and luciferase assay.
Transient transfections were performed with reporter plasmid by using ExGen 500.24 Luciferase assay was performed as previously described.53 The luciferase activities are expressed as fold induction relative to the untreated controls after being normalized to the β-galactosidase activity.
Quantitative real-time RT-PCR.
Total RNA was isolated by using RNeasy Mini Kit (Qiagen) and reverse transcribed by using High Capacity cDNA Archive Kit (Applied Biosystems). Quantitative real-time RT-PCR analysis was performed as previously described54 by using the 7500 Fast Real-Time PCR System (Applied Biosystems). Gene specific primers and TaqMan probes used for the amplification were obtained from Applied Biosystems. Quantification of mRNA levels was performed by using 18S rRNA as an internal control.
Whole cell extracts and immunoprecipitation.
Whole cell extracts were prepared by incubating the cells in whole cell extract buffer (10% glycerol, 50 mM Tris-HCl pH 7.6, 400 mM NaCl, 5 mM EDTA, 1 mM DTT, 1 mM PMSF and 1% NP-40) for 30 min at 4°C and then centrifuged at 14,000 rpm for 15 min at 4°C. Bradford assay (Bio-Rad) was used to determine the protein concentrations. For immunoprecipitation assay, equal amounts of protein extracts were diluted with dilution buffer (20 mM Tris-HCl pH 8.0, 0.5 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM PMSF and 1 mg/ml BSA) and incubated with antibodies specific to protein of interest for overnight at 4°C. Protein A agarose was added to the incubation for additional 2 h and the precipitates were then washed three times by PBS supplemented with 1% of Triton X-100.
Protein stability assays.
Cells were labeled for 4 h with 10 µCi/ml of 35S-methionine/cysteine (PerkinElmer) in methionine-free media and the cells were chased for additional 4–24 h in regular medium in the presence or absence of ligand. The cells were then harvested for the preparation of whole cell extracts and immunoprecipitation with a RARα antibody. The immunopurified RARα was separated by SDS PAGE. The gel was treated with amplify as manufacturer recommended (Amersham). The apparent half-life was determined by the remaining incorporated radioactivity of the immonoprecipitated RARα following various hours of chase. Quantification was performed by using a PhosphorImager (Molecular Dynamics). Alternatively, cells were treated with 10 µg/ml of cycloheximide in the presence or absence of ligand for 4–24 h and harvested for quantitative western analysis to determine the apparent half-life of RXRα.
Chromatin immunoprecipitation assay.
Cells were cross-linked with 1% formaldehyde for 15 min at 37°C, lysed with lysis buffer (1% SDS, 50 mM Tris-HCl pH 8.0, 10 mM EDTA and 0.1% protease inhibitor cocktail) and sonicated with Bioruptor (Diagenode). Equal amounts of DNA were diluted with dilution buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 and 0.1% protease inhibitor cocktail) for immunoprecipitation with specific antibodies overnight at 4°C. Protein A agarose/salmon sperm DNA were then added to the incubation for additional 2 h. The immune complexes were washed sequentially for 10 min with washing buffers A (0.1% SDS, 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA and 1% Triton X-100), buffer B (0.1% SDS, 20 mM Tris-HCl pH 8.0, 500 mM NaCl, 2 mM EDTA and 1% Triton X-100), buffer C (1% sodium deoxycholate, 20 mM Tris-HCl pH 8, 0.25 M LiCl, 1 mM EDTA and 1% NP-40) and two times with TE buffer (10 mM Tris-HCl pH 8.0 and 1 mM EDTA). The immunocomplexes were then extracted with elution buffer (1% SDS and 0.1 M NaHCO3) for 15 min at room temperature. Reverse cross linking were performed at 65°C for overnight. The precipitated DNA fragments were then purified using QIAquick PCR purification Kit (Qiagen) and amplified by PCR using the RARβ2 and Cyp26A1 primers.55,56
Acknowledgements
This work is supported by an operating grant from Natural Sciences and Engineering Research Council of Canada. During the course of this study, Q.L. held an investigator award from Canadian Institutes of Health Research and A.H. is a recipient of doctoral scholarship from Egyptian Ministry of Higher Education.
Abbreviations
- ChIP
chromatin immunoprecipitation
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- RAR
retinoic acid receptor
- RARE
retinoic acid response elements
- RXR
retinoid X receptor
References
- 1.Niederreither K, Dolle P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9:541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 2.Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92:189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 3.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 4.Lonard DM, O'Malley BW. Nuclear receptor coregulators: judges, juries and executioners of cellular regulation. Mol Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 5.Chambon P. The nuclear receptor superfamily: a personal retrospect on the first two decades. Mol Endocrinol. 2005;19:1418–1428. doi: 10.1210/me.2005-0125. [DOI] [PubMed] [Google Scholar]
- 6.Folkers GE, van der Burg B, van der Saag PT. Promoter architecture, cofactors and orphan receptors contribute to cell-specific activation of the retinoic acid receptor beta2 promoter. J Biol Chem. 1998;273:32200–32212. doi: 10.1074/jbc.273.48.32200. [DOI] [PubMed] [Google Scholar]
- 7.Pavri R, Lewis B, Kim TK, Dilworth FJ, Erdjument-Bromage H, Tempst P, et al. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell. 2005;18:83–96. doi: 10.1016/j.molcel.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 8.Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 9.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 10.Torchia J, Glass C, Rosenfeld MG. Co-activators and co-repressors in the integration of transcriptional responses. Curr Opin Cell Biol. 1998;10:373–383. doi: 10.1016/s0955-0674(98)80014-8. [DOI] [PubMed] [Google Scholar]
- 11.Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300 kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 12.Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 13.Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, Chong JM, et al. p300 gene alterations in colorectal and gastric carcinomas. Oncogene. 1996;12:1565–1569. [PubMed] [Google Scholar]
- 14.Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, et al. Mutations truncating the EP300 acetylase in human cancers. Nat Genet. 2000;24:300–303. doi: 10.1038/73536. [DOI] [PubMed] [Google Scholar]
- 15.Suganuma T, Kawabata M, Ohshima T, Ikeda MA. Growth suppression of human carcinoma cells by reintroduction of the p300 coactivator. Proc Natl Acad Sci USA. 2002;99:13073–13078. doi: 10.1073/pnas.192586699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 17.Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA. 1999;96:1858–1862. doi: 10.1073/pnas.96.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lange CA, Shen T, Horwitz KB. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA. 2000;97:1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molinari E, Gilman M, Natesan S. Proteasome-mediated degradation of transcriptional activators correlates with activation domain potency in vivo. EMBO J. 1999;18:6439–6447. doi: 10.1093/emboj/18.22.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell. 2000;5:939–948. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 21.Poizat C, Sartorelli V, Chung G, Kloner RA, Kedes L. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol Cell Biol. 2000;20:8643–8654. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gianni M, Kopf E, Bastien J, Oulad-Abdelghani M, Garattini E, Chambon P, et al. Downregulation of the phosphatidylinositol 3-kinase/Akt pathway is involved in retinoic acid-induced phosphorylation, degradation and transcriptional activity of retinoic acid receptor-gamma2. J Biol Chem. 2002;277:24859–24862. doi: 10.1074/jbc.C200230200. [DOI] [PubMed] [Google Scholar]
- 23.Kopf E, Plassat JL, Vivat V, de The H, Chambon P, Rochette-Egly C. Dimerization with retinoid X receptors and phosphorylation modulate the retinoic acid-induced degradation of retinoic acid receptors alpha and gamma through the ubiquitin-proteasome pathway. J Biol Chem. 2000;275:33280–33288. doi: 10.1074/jbc.M002840200. [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Su A, Chen J, Lefebvre YA, Hache RJ. Attenuation of glucocorticoid signaling through targeted degradation of p300 via the 26S proteasome pathway. Mol Endocrinol. 2002;16:2819–2827. doi: 10.1210/me.2002-0154. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, St-Germain JR, Li Q. B56 regulatory subunit of protein phosphatase 2A mediates valproic acid-induced p300 degradation. Mol Cell Biol. 2005;25:525–532. doi: 10.1128/MCB.25.2.525-532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Halappanavar S, Th'ng JP, Li Q. Ubiquitin-dependent distribution of the transcriptional coactivator p300 in cytoplasmic inclusion bodies. Epigenetics. 2007;2:92–99. doi: 10.4161/epi.2.2.4326. [DOI] [PubMed] [Google Scholar]
- 27.Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 29.Dembla-Rajpal N, Seipelt R, Wang Q, Rymond BC. Proteasome inhibition alters the transcription of multiple yeast genes. Biochim Biophys Acta. 2004;1680:34–45. doi: 10.1016/j.bbaexp.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Salghetti SE, Caudy AA, Chenoweth JG, Tansey WP. Regulation of transcriptional activation domain function by ubiquitin. Science. 2001;293:1651–1653. doi: 10.1126/science.1062079. [DOI] [PubMed] [Google Scholar]
- 31.Ferdous A, Gonzalez F, Sun L, Kodadek T, Johnston SA. The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol Cell. 2001;7:981–991. doi: 10.1016/s1097-2765(01)00250-7. [DOI] [PubMed] [Google Scholar]
- 32.Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- 33.Bissonnette RP, Brunner T, Lazarchik SB, Yoo NJ, Boehm MF, Green DR, et al. 9-cis retinoic acid inhibition of activation-induced apoptosis is mediated via regulation of fas ligand and requires retinoic acid receptor and retinoid X receptor activation. Mol Cell Biol. 1995;15:5576–5585. doi: 10.1128/mcb.15.10.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McBurney MW, Jones-Villeneuve EM, Edwards MK, Anderson PJ. Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature. 1982;299:165–167. doi: 10.1038/299165a0. [DOI] [PubMed] [Google Scholar]
- 35.Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol. 2002;22:4113–4123. doi: 10.1128/MCB.22.12.4113-4123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem. 1971;246:174–181. [PubMed] [Google Scholar]
- 37.Kao HY, Han CC, Komar AA, Evans RM. Co-repressor release but not ligand binding is a prerequisite for transcription activation by human retinoid acid receptoralpha ligand-binding domain. J Biol Chem. 2003;278:7366–7373. doi: 10.1074/jbc.M207569200. [DOI] [PubMed] [Google Scholar]
- 38.Loudig O, Maclean GA, Dore NL, Luu L, Petkovich M. Transcriptional co-operativity between distant retinoic acid response elements in regulation of Cyp26A1 inducibility. Biochem J. 2005;392:241–248. doi: 10.1042/BJ20050874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams J. Development of the proteasome inhibitor PS-341. Oncologist. 2002;7:9–16. doi: 10.1634/theoncologist.7-1-9. [DOI] [PubMed] [Google Scholar]
- 40.Nawaz Z, O'Malley BW. Urban renewal in the nucleus: is protein turnover by proteasomes absolutely required for nuclear receptor-regulated transcription? Mol Endocrinol. 2004;18:493–499. doi: 10.1210/me.2003-0388. [DOI] [PubMed] [Google Scholar]
- 41.Verma S, Ismail A, Gao X, Fu G, Li X, O'Malley BW, et al. The ubiquitin-conjugating enzyme UBCH7 acts as a coactivator for steroid hormone receptors. Mol Cell Biol. 2004;24:8716–8726. doi: 10.1128/MCB.24.19.8716-8726.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 43.Dace A, Zhao L, Park KS, Furuno T, Takamura N, Nakanishi M, et al. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proc Natl Acad Sci USA. 2000;97:8985–8990. doi: 10.1073/pnas.160257997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka T, Rodriguez de la Concepcion ML, De Luca LM. Involvement of all-trans-retinoic acid in the breakdown of retinoic acid receptors alpha and gamma through proteasomes in MCF-7 human breast cancer cells. Biochem Pharmacol. 2001;61:1347–1355. doi: 10.1016/s0006-2952(01)00600-1. [DOI] [PubMed] [Google Scholar]
- 45.Andela VB, Rosier RN. The proteosome inhibitor MG132 attenuates retinoic acid receptor trans-activation and enhances trans-repression of nuclear factor kappaB. Potential relevance to chemo-preventive interventions with retinoids. Mol Cancer. 2004;3:8. doi: 10.1186/1476-4598-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gianni M, Bauer A, Garattini E, Chambon P, Rochette-Egly C. Phosphorylation by p38MAPK and recruitment of SUG-1 are required for RA-induced RAR gamma degradation and transactivation. EMBO J. 2002;21:3760–3769. doi: 10.1093/emboj/cdf374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salghetti SE, Muratani M, Wijnen H, Futcher B, Tansey WP. Functional overlap of sequences that activate transcription and signal ubiquitin-mediated proteolysis. Proc Natl Acad Sci USA. 2000;97:3118–3123. doi: 10.1073/pnas.050007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferdous A, Sikder D, Gillette T, Nalley K, Kodadek T, Johnston SA. The role of the proteasomal ATPases and activator monoubiquitylation in regulating Gal4 binding to promoters. Genes Dev. 2007;21:112–123. doi: 10.1101/gad.1493207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee D, Ezhkova E, Li B, Pattenden SG, Tansey WP, Workman JL. The proteasome regulatory particle alters the SAGA coactivator to enhance its interactions with transcriptional activators. Cell. 2005;123:423–436. doi: 10.1016/j.cell.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 50.Perlmann T, Rangarajan PN, Umesono K, Evans RM. Determinants for selective RAR and TR recognition of direct repeat HREs. Genes Dev. 1993;7:1411–1422. doi: 10.1101/gad.7.7b.1411. [DOI] [PubMed] [Google Scholar]
- 51.Depoix C, Delmotte MH, Formstecher P, Lefebvre P. Control of retinoic acid receptor heterodimerization by ligand-induced structural transitions. A novel mechanism of action for retinoid antagonists. J Biol Chem. 2001;276:9452–9459. doi: 10.1074/jbc.m008004200. [DOI] [PubMed] [Google Scholar]
- 52.Zechel C, Shen XQ, Chambon P, Gronemeyer H. Dimerization interfaces formed between the DNA binding domains determine the cooperative binding of RXR/RAR and RXR/TR heterodimers to DR5 and DR4 elements. EMBO J. 1994;13:1414–1424. doi: 10.1002/j.1460-2075.1994.tb06395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Halappanavar SS, St-Germain JR, Tsang BK, Li Q. Role of Akt/protein kinase B in the activity of transcriptional coactivator p300. Cell Mol Life Sci. 2004;61:1675–1683. doi: 10.1007/s00018-004-4103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, Ghazawi FM, Bakkar W, Li Q. Valproic acid and butyrate induce apoptosis in human cancer cells through inhibition of gene expression of Akt/protein kinase B. Mol Cancer. 2006;5:71. doi: 10.1186/1476-4598-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lefebvre B, Brand C, Lefebvre P, Ozato K. Chromosomal integration of retinoic acid response elements prevents cooperative transcriptional activation by retinoic acid receptor and retinoid X receptor. Mol Cell Biol. 2002;22:1446–1459. doi: 10.1128/mcb.22.5.1446-1459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillespie RF, Gudas LJ. Retinoid regulated association of transcriptional co-regulators and the polycomb group protein SUZ12 with the retinoic acid response elements of Hoxa1, RARbeta(2) and Cyp26A1 in F9 embryonal carcinoma cells. J Mol Biol. 2007;372:298–316. doi: 10.1016/j.jmb.2007.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]