

Abstract
The cellular and molecular mechanisms that govern the response of the perinatal brain to injury remain largely unexplored. We investigated the role of white matter astrocytes in a rodent model of diffuse white matter injury produced by exposing neonatal mice to chronic hypoxia—a paradigm that mimics brain injury in premature infants. We demonstrate the absence of reactive gliosis in the immature white matter following chronic hypoxia, as determined by astrocyte proliferation index and glial fibrillary acidic protein levels. Instead, Nestin expression in astrocytes is transiently increased, and the glial-specific glutamate transporters glutamate–aspartate transporter (GLAST) and glutamate transporter 1 (GLT-1) are reduced. Finally, we demonstrate that Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling—which is important in both astrocyte development and response to injury—is reduced in the white matter following hypoxia, as well as in primary astrocytes exposed to hypoxia in vitro. Hypoxia and JAK/STAT inhibition reduce glutamate transporter expression in astrocytes, but unlike hypoxia JAK/STAT inhibition downregulates GLAST expression without affecting GLT-1, as demonstrated in vitro by treatment with JAK inhibitor I and in vivo by treatment with the JAK/STAT inhibitor AG490 [(E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide]. Our findings (1) demonstrate specific changes in astrocyte function after perinatal hypoxia, which might contribute to the particular pathogenesis of perinatal white matter injury, (2) provide evidence that at least part of these changes result from a disturbance of the JAK/STAT pathway by hypoxia, and (3) identify JAK/STAT signaling as a potential therapeutic target to restore normal GLAST expression and uptake of glutamate after perinatal brain injury.
Introduction
Trauma to the adult CNS often results in reactive astrogliosis and glial scarring and the response of astrocytes to various injuries to the adult brain has been well characterized, yet the cellular and functional response of astrocytes to injury in the perinatal brain remains largely unexplored (Maxwell and Kruger, 1965; Miyake et al., 1988; Ridet et al., 1997; Sofroniew, 2005). There is substantial evidence that hypoxia is an important contributing factor to brain injury in premature infants (Claireaux, 1977; Inder et al., 2003). In the past, brain injury in premature infants commonly resulted in periventricular leukomalacia characterized by focal necrosis; however, advances in neonatal care have diminished its occurrence, and currently the most common injury observed is characterized by diffuse white matter damage (Maalouf et al., 1999; Back et al., 2001; Inder et al., 2003). One widely used rodent model of brain injury in premature infants is that of hypoxia–ischemia, which results in focal white matter and gray matter damage (Segovia et al., 2008; Scafidi et al., 2009). Yet recent studies have highlighted the importance of injury to infants by hypoxia alone, due to their immature lungs and respiratory system (Claireaux, 1977; Inder et al., 2003). In the present study, we used a well established model of diffuse white matter injury induced by chronic hypoxia in the perinatal rodent [hypoxia—postnatal day 3 (P3) to P11; 10.5 ± 0.5% O2], to study cellular and functional changes occurring in white matter astrocytes (Ment et al., 1998; Vaccarino and Ment, 2004; Bi et al., 2011). This model reproduces many major anatomical hallmarks of white matter injury observed in the brain of premature infants, including decreased white matter and gray matter volume, as well as enlargement of the lateral ventricles (Ment et al., 1998; Maalouf et al., 1999).
Studies in vitro have shown that hypoxia affects the expression of the two Na+-dependent glial-specific glutamate transporters: glutamate–aspartate transporter (GLAST) (EAAT1) and glutamate transporter 1 (GLT-1) (EAAT2) (Dallas et al., 2007). GLAST and GLT-1 are primarily expressed in astrocytes and are affected in a number of CNS pathologies (Rothstein et al., 1995; Sattler and Rothstein, 2006).
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway is active in astrocytes and is important in astrocyte differentiation. This pathway is thought to regulate the transition from immature Nestin-expressing to mature GFAP-expressing astrocytes (Sriram et al., 2004; Gautron et al., 2006). Furthermore, JAK/STAT signaling is also involved in the process of astrogliosis and scar formation in different CNS pathologies (Justicia et al., 2000; Herrmann et al., 2008).
In the present study, we examined the response of astrocytes to injury of the developing white matter using a model of chronic hypoxia in the perinatal rodent. We investigated whether chronic hypoxia affected astrocyte reactivity and function, and we examined whether JAK/STAT signaling was altered by hypoxia in astrocytes. We investigated the effects of hypoxia on astrocytes both in vivo and in vitro, and we revealed significant changes in astrocyte function in the absence of reactive gliosis. We also demonstrate a role for JAK/STAT signaling in the functional changes induced by hypoxia in astrocytes, indicating that this pathway plays a role in astrocyte pathology also in the immature brain.
Materials and Methods
Materials.
Anti-GFAP, GLT-1, and GLAST antibodies were purchased from Abcam. Antibodies against STAT3, pSTAT3 (Tyr 705), pJAK1 (Tyr 1022), JAK2, pJAK2 (Tyr 1007/1008), and actin were purchased from Santa Cruz Biotechnology. JAK1 antibody was purchased from BD Biosciences. The BrdU antibody was purchased from Covance. Anti-Nestin was purchased from Millipore. JAK inhibitor I was purchased from EMD Chemicals. Dihydrokainic acid (DHK) and (E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide (AG490) were purchased from Sigma-Aldrich. RIPA lysis buffer was purchased from Santa Cruz Biotechnology. Medium for cell culture was purchased from Invitrogen. d-[2,3-3H]Aspartic acid was purchased from PerkinElmer.
Animals and hypoxia protocol.
The generation of the GFAP-GFP mouse used in this study has been described previously (Zhuo et al., 1997), and CD1 mice were obtained from Charles River Labs. All mouse colonies were maintained in the animal facility of Children's National Medical Center, and all animal procedures complied with the guidelines of the National Institutes of Health, and with the Children's Research Institute Institutional Animal Care and Use Committee guidelines. Male mice were placed in a chamber containing 10.5 ± 0.5% O2 from P3 to P11 as previously described (Ment et al., 1998; Fagel et al., 2006). Strain-matched and age-matched animals reared in normal oxygen levels were used as controls (normoxia). For studies examining proliferation, BrdU (Sigma-Aldrich; 50 μg per gram body weight) was administered 2 h before killing. Mice were killed at the given time point after hypoxia and perfused transcardially with PBS followed by 4% paraformaldehyde (PFA) and postfixed overnight in PFA followed by 20% glycerol and stored at 4°C. Treatment of mice with the JAK/STAT inhibitor AG490 has been previously described (Xu et al., 2011; Zhao et al., 2011). Briefly, CD1 mice were treated with AG490 (10 mg/kg, i.p.) or DMSO (control) twice daily (injections were ∼8–10 h apart) from P6 to P11. At P11, the white matter was carefully dissected out and lysed as described below, followed by Western blot analysis.
Primary astrocyte cultures.
Purified astrocyte cultures were obtained from 2- to 3-d-old CD1 mice. Animals were killed and cortices were dissected and mechanically dissociated with a fire-polished Pasteur pipette. Cells were then plated on poly-l-lysine-treated 75 cm2 flasks in DMEM containing 2 mm glutamine and 10% fetal bovine serum. Approximately 16 h after plating, the media was replaced. Once 80–90% confluent, cells were passaged two to three times and were cultured for no more than 21 d with media changes every 48 h. Cultures contained >95% GFAP+ cells. To expose the primary astrocytes to hypoxia, we cultured them in at 37°C in an incubator whose O2 levels were maintained at 5 ± 0.5%.
Immunohistochemistry in tissue sections and cell counting.
Floating brain sections from GFAP-GFP mouse were immunostained with antibodies against BrdU (1:200), GFAP (1:500), and Nestin (1:100). Sections were incubated in primary antibodies diluted in PBS containing 0.1% Triton X-100 (v/v) and 5% normal goat serum overnight at 4°C. Sections were incubated with secondary antibody for 1 h at room temperature. Sections were treated with DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich) to determine total cell number. Cells were visualized by confocal microscopy. All sections of subcortical white matter analyzed contained the corpus callosum, cingulum, and external capsule, and were rostral of the hippocampus.
Western blot analysis.
For Western blot analysis of white matter lysates, the subcortical white matter was dissected from 400-μm-thick sections prepared from CD1 mice that had been reared in hypoxia or normoxia. Briefly, brains were sliced coronally and only slices rostral of the hippocampus were used. Using Roboz—a fine-straight and fine-angled microdissecting forceps under a dissecting microscope—the cortex was dissected away leaving the underlying subcortical white matter attached to the striatum. The white matter was then easily pushed away from the striatum, leaving a thin ribbon of primarily white matter tissue. The dissected white matter was rinsed with ice-cold PBS, and then lysed on ice in 200–300 μl of RIPA lysis buffer (50 mm Tris-HCl, pH 7.5, 140 mm NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1 mm sodium orthovanadate, 1 mm NaF, 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin, and 1 μm microcystin-LR). For in vitro experiments, cells were cultured in six-well plates to ∼80–90% confluency and 1 μm JAK inhibitor I was added to the cultures for 24 h or they were cultured in hypoxic conditions for the indicated time period. The cells were washed twice with ice-cold PBS, and then lysed with 250 μl of RIPA lysis buffer for 30 min on ice. Protein concentrations were determined by using the Bradford protein assay kit (Bio-Rad). Western blot analysis was performed on 10–40 μg of total cell lysates. Proteins were resolved on 4–20% Tris glycine gels (Nusep) and transferred to Immobilon PVDF membranes by tank blotting in transfer buffer [25 mm Tris, 192 mm glycine, 20% (v/v) methanol, pH 8.3] for 16 h at 4°C. The membranes were then washed in Tris-buffered saline with 0.1% Tween 20 (TBST) (25 mm Tris-HCl, 140 mm NaCl, 0.1% Tween 20, pH 7.5), incubated for 1 h in TBST containing 5% bovine serum albumin (BSA), and then incubated for 16 h at 4°C with primary antibodies diluted in TBST-BSA. The membranes were then washed in TBST three times for 10 min at room temperature followed by the addition of either horseradish peroxidase-conjugated goat polyclonal anti-rabbit IgG (Santa Cruz) for polyclonal primary antibodies, or horseradish peroxidase-conjugated goat anti-mouse (Santa Cruz) for mouse monoclonal primary antibodies diluted in TBST-BSA. The chemiluminescent signals were detected using Pierce ECL Western blotting substrate. X-ray films were scanned using an Agfa T1200 scanner and densitometric measurements were obtained using ImageJ software.
Gliosome/synaptosome
d-[3H]aspartate uptake assay and d-[3H]aspartate uptake assay in primary astrocytes. The gliosome/synaptosome uptake assays were performed using a modified method of Weller et al. (2008). Brains were removed at the given time point after hypoxic or normoxic rearing and the white matter was carefully dissected out. The tissue was then homogenized on ice using a mechanical homogenizer in tissue buffer (50 mm Tris, 0.3 m sucrose, pH 7.3) and centrifuged at 14,000 × g for 10 min. The pellet was resuspended in 250 μl of sodium containing Krebs' buffer (1200 mm NaCl, 47 mm KCl, 12 mm KH2PO4, 12 mm MgSO4, 100 mm HEPES) or sodium-free Krebs' buffer (1200 mm choline chloride instead of 1200 mm NaCl). When using primary astrocytes, 25,000 cells/cm2 were cultured on poly-l-lysine-coated 24-well plates. Cells were treated with 1 μm JAK inhibitor I or DMSO for 24 h, and then washed twice with warm Krebs' buffer followed by addition of 250 μl of Krebs' buffer. For both the gliosome/synaptosome and primary astrocyte uptake experiments, the GLT-1 inhibitor dihydrokainic acid (100 μm) was added where indicated and incubated for 10 min at 37°C before the addition of d-[3H]aspartate. d-[3H]aspartate (50 nm) was added and incubated for 10 min at 37°C, followed by three rinses with ice-cold sodium-free Krebs' buffer to halt uptake. The preparations were treated with 400 μl of 1N NaOH, and the radioactivity of 200 μl of lysate was determined by scintillation counting. Determination of protein concentration in each sample was performed using the Bradford protein assay. Data are presented as uptake velocity (picomoles per microgram of protein per minute).
Results
Perinatal hypoxia does not affect cell number or proliferation of GFAP- or Nestin-expressing cells in the white matter, but modifies GFAP and Nestin expression
To examine the cellular effects of hypoxic injury in the white matter of the immature brain, we used the GFAP-GFP transgenic mouse in which GFP expression is limited to GFAP-expressing cells. It is well established that, in response to adult brain injury, astrocytes become activated and convert to a reactive phenotype, which is characterized by increased GFAP expression, and changes in cell morphology and proliferation rate (Miyake et al., 1988; Pekny and Nilsson, 2005). To determine the effect of hypoxia on astrocyte cell number, we quantified the number of GFP+GFAP+ and GFP+GFAP+Nestin+ cells in the white matter. At P11 (immediately after hypoxia), there was no change in the number of GFP+GFAP+ or GFP+GFAP+Nestin+ cells (Fig. 1A–C). To assess the effect of hypoxia on astrocyte proliferation, we injected BrdU 2 h before killing and then quantified the number of GFP+GFAP+ and GFP+GFAP+BrdU+ cells in the white matter after hypoxia (Fig. 1A). At P11, there was no change in the number of GFP+GFAP+BrdU+ cells or in the percentage of GFP+GFAP+ cells that were BrdU+ (Fig. 1C). The percentage of GFP+GFAP+ over the total number of cells in the white matter was not significantly modified (data not shown). We also performed analysis at P5 (2 d in hypoxia; acute hypoxia), P18 (1 week after hypoxia), and P45 (5 weeks after hypoxia), and there was no difference in the number of GFP+GFAP+Nestin+, GFP+ GFAP+, GFP+GFAP+BrdU+ cells (Fig. 1A–C). We also noted no difference in astrocyte morphology or GFAP or Nestin distribution, as determined by GFAP and Nestin immunostaining, although GFAP intensity was decreased in the hypoxic white matter and Nestin intensity increased at P11 (Fig. 1A,B). Western blot analysis in the white matter of P11 mice revealed a significant decrease in GFAP protein expression and an increase in the expression of Nestin, a marker of immature astrocytes, in hypoxic animals compared with age-matched normoxic controls (Fig. 1D,E). Analysis of Nestin and GFAP protein expression at P5, P18, and P45 showed no changes compared with normoxic controls (Fig. 1D,E). Together, these results demonstrate that hypoxia does not cause reactive gliosis in the immature early postnatal brain yet is suggestive of a delay in astrocyte maturation.
Figure 1.
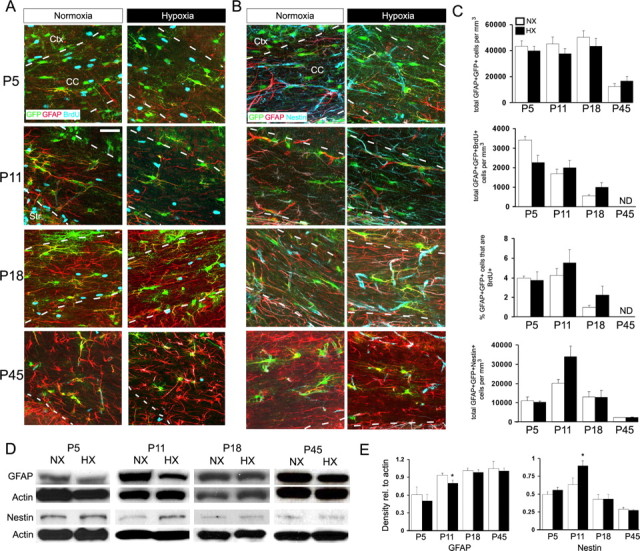
Hypoxia does not affect white matter astrocyte cell number but decreases GFAP expression and increases Nestin expression. A–C, GFAP-GFP mice were subjected to hypoxia (10.5 ± 0.5% O2) from P3 to P11 followed by an injection of BrdU 2 h before killing. Brain slices were stained with anti-GFAP (red) and anti-BrdU (blue) (A) or anti-GFAP (red) and anti-Nestin (blue) (B) antibodies. C, Quantification of percentage or number of GFAP+GFP+ cells immunostained with anti-BrdU or anti-Nestin. Scale bar, 50 μm. Cell counts are shown ± SEM. D, E, Western blot analysis of GFAP and Nestin expression in the white matter after hypoxia. Protein levels are expressed relative to actin loading control. Densitometric analysis is shown ± SD (n = 4–6 animals; *p < 0.05, unpaired Student's t test). Ctx, Cortex; CC, corpus callosum; NX, normoxia; HX, hypoxia.
Hypoxia reduces expression of GLAST and GLT-1, and decreases d-[3H]aspartate transport in the white matter
Previous in vitro studies demonstrated that exposing primary astrocyte cultures to hypoxia decreases GLAST and GLT-1 protein levels (Dallas et al., 2007). To test whether chronic hypoxia in the perinatal rodent decreased GLAST and GLT-1 expression in the subcortical white matter in vivo, we performed Western blot analysis on white matter lysates. At P11, GLAST and GLT-1 levels were significantly decreased, compared with normoxic controls, but at P5, P18, and P45 no difference was detected (Fig. 2A,B). To test whether hypoxia alters glutamate transport activity in the white matter, we measured uptake of d-[3H]aspartate in white matter membrane gliosome/synaptosome fractions. At P11, total d-[3H]aspartate uptake was significantly decreased after hypoxia (Fig. 3A). To determine the contribution of GLT-1 to total uptake, we pretreated the gliosome/synaptosome preparation with the GLT-1 inhibitor DHK. Hypoxia decreased both GLT-1-specific and nonspecific (DHK-treated) uptake at P11 but, consistent with Western blot results, had no effect at P18 (Fig. 3A). To confirm that this uptake was Na+-dependent, we performed uptake assays in the absence of Na+, which resulted in uptake that was <1% of the total uptake measured in the presence of Na+ (Fig. 3B). Together, these data demonstrate that hypoxia transiently reduces glutamate transporter function in astrocytes by decreasing GLAST and GLT-1 protein expression.
Figure 2.

Hypoxia decreases GLAST and GLT-1 expression in the white matter. A, Western blot analysis of GLAST and GLT-1 expression in the white matter after hypoxia. B, Protein levels are expressed relative to actin loading control. Densitometric analysis is shown ± SD (n = 4–6 animals; *p < 0.05, unpaired Student's t test).
Figure 3.

Hypoxia decreases d-[3H]aspartate uptake in the white matter. A, Na+-dependent uptake velocity of d-[3H]aspartate (50 nm) in white matter gliosome/synaptosome membrane fraction. Dihydrokainate (100 μm) was added 10 min before assay to inhibit GLT-1-specific uptake. B, d-[3H]aspartate uptake measured under the same conditions as in A, but in the absence of Na+. Uptake velocities are shown ± SEM (n = 4–6 animals; *p < 0.05, unpaired Student's t test).
Hypoxia reduces JAK/STAT signaling in the white matter
It has been previously shown that the JAK/STAT pathway is important both in astrocyte maturation—as onset of GFAP expression is dependent on a STAT3 mechanism—and in astrocyte response to pathological insults (Sriram et al., 2004; Gautron et al., 2006; Herrmann et al., 2008). Since we observed an immature astrocyte phenotype in the white matter after perinatal hypoxia, we wanted to determine whether changes in the JAK/STAT signaling pathway also occurred. At P11, Western blot analysis revealed a decrease in pSTAT3, pJAK1, and pJAK2 in the hypoxic white matter, compared with normoxic controls (Fig. 4A,B). Levels of total STAT3, JAK1, and JAK2 were similar in the hypoxic and normoxic groups (Fig. 4A,B). At P5, P18, and P45, levels of pSTAT3, pJAK1 and pJAK2 were not modified (Fig. 4A,B). These results demonstrate that hypoxia transiently reduces JAK/STAT signaling in white matter with a time course similar to the reduction in glutamate transporter expression and function.
Figure 4.

Hypoxia alters JAK/STAT signaling in the white matter. A, Western blot analysis of components of the JAK/STAT signaling pathway in the white matter of hypoxic and normoxic mice at P5, P11, P18, and P45. B, Protein levels are expressed relative to actin loading control. Densitometric analysis is shown ± SD (n = 4–6 animals; *p < 0.05, unpaired Student's t test).
Hypoxia reduces expression of GFAP, GLAST, GLT-1, and pSTAT3, increases Nestin expression, and decreases d-[3H]aspartate transport in primary astrocytes
We exposed primary astrocyte cultures to hypoxia (5% O2) for 24, 48, and 72 h. Consistent with our findings in vivo (Figs. 1, 4), we observed a decrease in GFAP protein expression, as well as in pSTAT3, pJAK1, and pJAK2 levels at 48 h after hypoxia, and an increase in Nestin expression suggestive of an immature phenotype (Fig. 5A,B). Furthermore, as previously shown by Dallas et al. (2007), a decrease in both GLAST and GLT-1 expression was also observed (Fig. 5A,B). Therefore, exposure of astrocytes to hypoxia in culture reproduces the effects of hypoxia on astrocytes in vivo.
Figure 5.

Hypoxia decreases JAK/STAT signaling and expression of GFAP, GLAST, and GLT-1 in primary astrocytes. A, Western blot analysis of glial-specific glutamate transporter GLT-1 and GLAST, and GFAP in primary astrocytes after hypoxia (5% O2). B, Western blot data are expressed ± SD (n = 3 individual experiments; *p < 0.05, **p < 0.01, unpaired Student's t test). Treatment with JAK inhibitor I decreases GLAST and GFAP expression in primary astrocytes. C, Primary astrocytes were treated with JAK inhibitor I (1 μm) for 24 h, and then assessed for protein expression by Western blot analysis. D, Western blot data are expressed ± SD (n = 3 individual experiments; *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student's t test). E, Treatment with JAK Inhibitor decreases total and non-GLT-1-specific uptake of d-[3H]aspartate (50 nm) in primary astrocyte cultures. Dihydrokainic acid (100 μm) was added to inhibit GLT-1-specific uptake. Uptake data are expressed ± SEM (n = 3 individual experiments; **p < 0.01, unpaired Student's t test).
Disruption of JAK/STAT signaling in primary astrocyte cultures reduces GFAP and GLAST expression, increases Nestin expression, and decreases d-[3H]aspartate transport
To determine whether JAK/STAT signaling could be responsible for the decreases in GLAST and GLT-1 expression observed after hypoxia, we treated primary astrocyte cultures with the JAK/STAT inhibitor JAK inhibitor I (1 μm). As expected, after 24 h of treatment with JAK inhibitor I, pJAK1, pJAK2, and pSTAT3 levels were decreased (Fig. 5C, D). We also analyzed expression of GFAP, Nestin, GLAST, and GLT-1. Treatment with JAK inhibitor I decreased levels of GFAP and GLAST, and increased levels of Nestin, yet GLT-1 levels were similar to untreated cultures (Fig. 5C, D). To determine whether glutamate uptake was also affected by JAK inhibitor I treatment, we performed a d-[3H]aspartate uptake assay on JAK inhibitor I-treated astrocytes. JAK inhibitor I decreased total uptake as well as non-GLT-1 (DHK-treated) uptake, but GLT-1-specific uptake was unaffected (Fig. 5E). These experiments were also performed in the absence of Na+ to determine the contribution of non-Na+-dependent uptake to the total uptake measured and this accounted for <1% of the total uptake (data not shown). Our results indicate that disruption of JAK/STAT signaling in primary astrocytes is causally linked to a decrease in glutamate transporter function in these cells.
Pharmacological inhibition of JAK/STAT signaling in vivo decreases GLAST expression in the white matter
To determine whether inhibition of JAK/STAT signaling in vivo also decreases GLAST expression, we treated perinatal mice that have not been exposed to hypoxia with the JAK/STAT inhibitor AG490 from P6 to P11. It has been previously demonstrated that administration of AG490 affects JAK/STAT signaling in the brain (Xu et al., 2011; Zhao et al., 2011). After AG490 administration, levels of pJAK1, pJAK2, pSTAT3 were significantly decreased in P11 white matter lysates—compared with untreated animals—confirming that the pharmacological treatment inhibited JAK/STAT signaling in vivo (Fig. 6A,B). Both GFAP and GLAST expression were also proportionally reduced (Fig. 6A,B). Conversely, levels of JAK1, JAK2, STAT3, and GLT-1 were not affected (Fig. 6A,B). We also noted that Nestin levels were not modified (Fig. 6A,B), as observed both in white matter of mice exposed to hypoxia (Fig. 1) and in primary astrocyte cultures treated with JAK inhibitor I (Fig. 5). These in vivo results support our observation that GLAST expression is decreased in primary astrocyte cultures exposed to JAK inhibitor I, and while regulation of GLAST and GLT-1 is complex, our data indicate that JAK/STAT signaling plays a role in GLAST expression.
Figure 6.
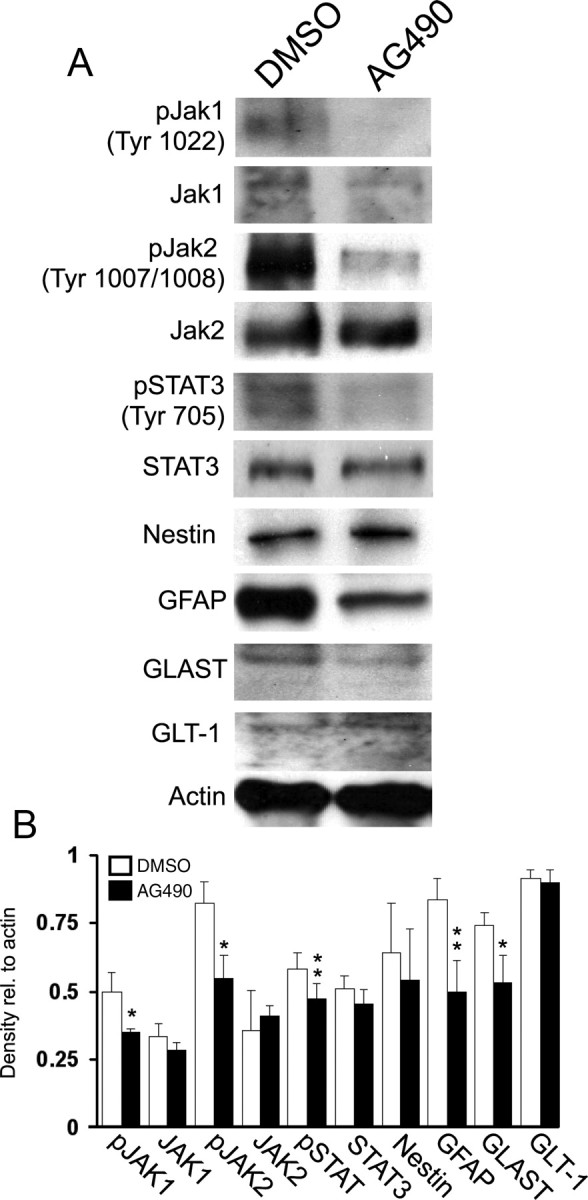
Treatment of early postnatal mice with the JAK/STAT inhibitor AG490 decreases expression of GFAP and GLAST in the white matter. Mice were treated with AG490 (10 mg/kg, i.p.) from P6 to P11. Western blot analysis was performed on P11 lysates from AG490-treated and control mice. A, Treatment with AG490 decreases GFAP and GLAST expression in the white matter. B, Protein levels are expressed relative to actin loading control. Data are shown ± SD (n = 4–6 animals; *p < 0.05, **p < 0.01 unpaired Student's t test).
Discussion
The cellular responses to hypoxia-induced diffuse white matter injury are still largely unknown. Animal models of this pathology will help elucidate fundamental cellular mechanisms of injury and define physiological changes triggered by hypoxia in distinct cell populations. In the present study, we used a well established model of chronic hypoxia in the perinatal rodent, which displays many of the same histopathological hallmarks seen in infants born premature (Ment et al., 1998; Maalouf et al., 1999). Our study demonstrates that, in the immature white matter, astrocyte response to diffuse injury is developmentally regulated, being evident after 1 week of hypoxia, but not at later time points. This astrocytic response is different from what is observed in hypoxia–ischemia [i.e., in another well established model of brain injury in premature infants (Sizonenko et al., 2008)], although a recent study by Schmitz et al. (2011) reported similar observations in a model of hyperoxia-induced brain injury, which also results in diffuse white matter injury. Hypoxia–ischemia causes focal necrotic lesioning and astrocyte activation, which leads to long-term changes in their cellular properties (Miyake et al., 1988; Pekny and Nilsson, 2005; Sofroniew, 2005; Segovia et al., 2008; Scafidi et al., 2009). Conversely, our study demonstrates that chronic hypoxia causes a decrease in GFAP and an increase in Nestin expression, as well as attenuation of JAK/STAT signaling, which is suggestive of an immature astrocytic phenotype. The decrease in GFAP expression is similar to what is observed in hyperoxia-induced perinatal white matter injury (Schmitz et al., 2011).
Our results show transient changes in the expression of the glial-specific glutamate transporters GLAST and GLT-1 after hypoxia. Changes in expression and function of glial-specific glutamate transporters have been demonstrated in a variety of brain insults and CNS pathologies (Cimarosti et al., 2005; Chao et al., 2010). In rodent models of injury that result in reactive gliosis and scar formation, including focal cerebral ischemia and demyelination, reactive astrocytes found in and around the glial scar area in the subcortical white matter display increased expression of the glial-specific glutamate transporters GLAST and GLT-1 (Arranz et al., 2010; Chao et al., 2010). The effect of injury on glutamate transporter expression is most likely region specific, because a different study demonstrated that, after hypoxic–ischemic injury, GLT-1 levels are increased in cortex, but decreased in striatum (Cimarosti et al., 2005). In some brain pathologies, as seen in patients with schizophrenia, levels of GLAST and GLT-1 mRNA, and levels of GLT-1 mRNA were increased in the thalamus and prefrontal cortex, respectively (Smith et al., 2001; Matute et al., 2005). Similar to what we observed in rodent white matter after hypoxia, other brain pathologies also result in decreased glutamate transporter expression and function (Rothstein et al., 1995; Boycott et al., 2008). For example, decreased GLT-1 and decreased glutamate uptake were demonstrated in CNS tissue obtained from ALS patients (Rothstein et al., 1995). Hyperoxia-induced white matter injury in the perinatal rodent results in a similar transient decrease in expression of GLAST and GLT-1 (Schmitz et al., 2011). Although the molecular pathways that regulate GLAST expression after hypoxic injury in vivo are still undefined, it is well established that differential mechanisms regulate hypoxia-induced changes in GLAST and GLT-1 transcription in vitro, and that reduction of GLT-1 expression is selectively mediated by NF-κB and its associated pathway (Boycott et al., 2008).
The JAK/STAT pathway is important in astrocyte maturation and in their cellular response to injury. Previous studies demonstrated that GFAP transcription is regulated by a STAT3-dependent mechanism and cellular characterization of astrocytes in the developing rodent cortex during the first 2 postnatal weeks demonstrated that both immature Nestin-expressing astrocytes from P0 to P3 and GFAP-expressing astrocytes around P10 express STAT3 and pSTAT3 (Sriram et al., 2004; Gautron et al., 2006). Since we induced hypoxic injury during this same developmental time window, our findings that JAK/STAT signaling and expression of Nestin and GFAP are affected by hypoxia in white matter strongly suggest that this insult inhibits astrocyte maturation through the STAT3 pathway. This hypothesis is confirmed by the finding that astrocyte proliferation was not affected. Furthermore, Sarafian et al. (2010) recently reported that disruption of STAT3 signaling in primary astrocyte cultures increases oxidative stress, indicating a strong link between oxidative injury and JAK/STAT signaling in astrocytes.
Astrocytes contribute to the cellular and molecular mechanisms associated with white matter injury observed after chronic hypoxia; however, several of cell types—including oligodendrocytes and their progenitors—are also involved (Back et al., 2006; Bi et al., 2011). We are currently analyzing hypoxia-induced damage to the oligodendrocyte lineage in our animal model of chronic perinatal hypoxia. Our preliminary results demonstrate that oligodendrocyte death occurs after the first week of hypoxia (B. Jablonska and V. Gallo, unpublished observations). Our studies also show that more mature (CC1+) stages of the oligodendrocyte lineage are particularly vulnerable to hypoxia-induced toxicity (B. Jablonska and V. Gallo, unpublished observations). In other models of perinatal brain injury, including hypoxia–ischemia and hyperoxia-induced injury, it has been demonstrated that late oligodendrocyte progenitors (O4+ cells) are most vulnerable to injury (Back et al., 2002; Schmitz et al., 2011). Therefore, it appears that different types of insults to the developing white matter affect distinct stages of the oligodendrocyte lineage. Future experimental analysis will define the different cellular and molecular mechanisms that underlie white matter injury in chronic hypoxia-, hypoxia–ischemia-, and hyperoxia-induced injury to the developing brain.
Our finding that expression of GLAST and GLT-1 is reduced after hypoxia is suggestive that changes in the concentration of extracellular glutamate likely occur in the white matter environment. What are the physiological consequences of hypoxia-induced reduction in astrocytic glutamate uptake? In the presence of excess glutamate, oligodendrocytes and their progenitors are damaged as a consequence of overactivation of AMPA receptors and subsequent Ca2+ influx, which ultimately causes excitotoxicity (Yoshioka et al., 1996; Matute et al., 2007). It is well established that glutamate vesicles are released from unmyelinated axons in the white matter and this is a potential source of excess glutamate in the hypoxic animal (Ziskin et al., 2007). We demonstrate that in vivo at P11 (7 d after hypoxia) both GLAST and GLT-1 expression are significantly reduced in the white matter, and that d-[3H]aspartate uptake in white matter gliosomes is significantly decreased after hypoxia. These findings strongly suggest that astrocyte impaired ability to clear glutamate after hypoxia would lead to excess glutamate in the extracellular space, which in turn causes excitotoxic damage to immature oligodendrocytes and/or prevents their maturation to myelinating oligodendrocytes (Steinhauser and Gallo, 1996; Yoshioka et al., 1996). In conclusion, dysregulation of glutamate homeostasis in white matter astrocytes after hypoxia is most likely one of the contributing factors underlying oligodendrocyte pathology after hypoxic injury.
In the present study, we demonstrate that the cellular response of astrocytes to hypoxic injury in vivo involves not only a reduction in glutamate transporter expression in the developing white matter, which in turn likely affects glutamate homeostasis, but also attenuation of JAK/STAT signaling, resulting in an immature phenotype, and these two responses are likely to be causally related. Defining how the JAK/STAT pathway regulates GLAST expression will be important to develop molecular therapeutic targets to promote neuroprotection or prevention of damage to the premature brain.
Footnotes
This work was supported by NIH Grant P01 NS0626860, NIH–Intellectual and Developmental Disabilities Research Center Grant P30 HD40677 (V.G.), and NIH Grant F31 NS073355 (M.R.). We thank Drs. Flora Vaccarino, Laura Ment, and Joey Scafidi for discussion. We thank Dr. Adan Aguirre for his technical help and assistance in experimental design.
References
- Arranz AM, Gottlieb M, Pérez-Cerdá F, Matute C. Increased expression of glutamate transporters in subcortical white matter after transient focal cerebral ischemia. Neurobiol Dis. 2010;37:156–165. doi: 10.1016/j.nbd.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Craig A, Luo NL, Ren J, Akundi RS, Ribeiro I, Rivkees SA. Protective effects of caffeine on chronic hypoxia-induced white matter injury. Ann Neurol. 2006;60:696–705. doi: 10.1002/ana.21008. [DOI] [PubMed] [Google Scholar]
- Bi B, Salmaso N, Komitova M, Simonini MV, Silbereis J, Cheng E, Kim J, Luft S, Ment LR, Horvath TL, Schwartz ML, Vaccarino FM. Cortical glial fibrillary acidic protein-positive cells generate neurons after perinatal hypoxic injury. J Neurosci. 2011;31:9205–9221. doi: 10.1523/JNEUROSCI.0518-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott HE, Wilkinson JA, Boyle JP, Pearson HA, Peers C. Differential involvement of TNFα in hypoxic suppression of astrocyte glutamate transporters. Glia. 2008;56:998–1004. doi: 10.1002/glia.20673. [DOI] [PubMed] [Google Scholar]
- Chao XD, Fei F, Fei Z. The role of excitatory amino acid transporters in cerebral ischemia. Neurochem Res. 2010;35:1224–1230. doi: 10.1007/s11064-010-0178-3. [DOI] [PubMed] [Google Scholar]
- Cimarosti H, Jones NM, O'Shea RD, Pow DV, Salbego C, Beart PM. Hypoxic preconditioning in neonatal rat brain involves regulation of excitatory amino acid transporter 2 and estrogen receptor alpha. Neurosci Lett. 2005;385:52–57. doi: 10.1016/j.neulet.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Claireaux AE. Pathology of perinatal hypoxia. J Clin Pathol Suppl (R Coll Pathol) 1977;11:142–148. doi: 10.1136/jcp.s3-11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas M, Boycott HE, Atkinson L, Miller A, Boyle JP, Pearson HA, Peers C. Hypoxia suppresses glutamate transport in astrocytes. J Neurosci. 2007;27:3946–3955. doi: 10.1523/JNEUROSCI.5030-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagel DM, Ganat Y, Silbereis J, Ebbitt T, Stewart W, Zhang H, Ment LR, Vaccarino FM. Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp Neurol. 2006;199:77–91. doi: 10.1016/j.expneurol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Gautron L, De Smedt-Peyrusse V, Layé S. Characterization of STAT3 expressing cells in the postnatal rat brain. Brain Res. 2006;1098:26–32. doi: 10.1016/j.brainres.2006.04.115. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inder TE, Wells SJ, Mogridge NB, Spencer C, Volpe JJ. Defining the nature of the cerebral abnormalities in the premature infant: a qualitative magnetic resonance imaging study. J Pediatr. 2003;143:171–179. doi: 10.1067/S0022-3476(03)00357-3. [DOI] [PubMed] [Google Scholar]
- Justicia C, Gabriel C, Planas AM. Activation of the JAK/STAT pathway following transient focal cerebral ischemia signaling through Jak1 and Stat3 in astrocytes. Glia. 2000;30:253–270. doi: 10.1002/(sici)1098-1136(200005)30:3<253::aid-glia5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Maalouf EF, Duggan PJ, Rutherford MA, Counsell SJ, Fletcher AM, Battin M, Cowan F, Edwards AD. Magnetic resonance imaging of the brain in a cohort of extremely preterm infants. J Pediatr. 1999;135:351–357. doi: 10.1016/s0022-3476(99)70133-2. [DOI] [PubMed] [Google Scholar]
- Matute C, Melone M, Vallejo-Illarramendi A, Conti F. Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia. 2005;49:451–455. doi: 10.1002/glia.20119. [DOI] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, Sánchez-Gómez MV, Pérez-Samartín A, Rodríguez-Antigüedad A, Pérez-Cerdá F. Excitotoxic damage to white matter. J Anat. 2007;210:693–702. doi: 10.1111/j.1469-7580.2007.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DS, Kruger L. The fine structure of astrocytes in the cerebral cortex and their response to focal injury produced by heavy ionizing particles. J Cell Biol. 1965;25:141–157. doi: 10.1083/jcb.25.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ment LR, Schwartz M, Makuch RW, Stewart WB. Association of chronic sublethal hypoxia with ventriculomegaly in the developing rat brain. Brain Res Dev Brain Res. 1998;111:197–203. doi: 10.1016/s0165-3806(98)00139-4. [DOI] [PubMed] [Google Scholar]
- Miyake T, Hattori T, Fukuda M, Kitamura T, Fujita S. Quantitative studies on proliferative changes of reactive astrocytes in mouse cerebral cortex. Brain Res. 1988;451:133–138. doi: 10.1016/0006-8993(88)90757-3. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Montes C, Imura T, Qi J, Coppola G, Geschwind DH, Sofroniew MV. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS One. 2010;5:e9532. doi: 10.1371/journal.pone.0009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Rothstein JD. Regulation and dysregulation of glutamate transporters. Handb Exp Pharmacol. 2006;175:277–303. doi: 10.1007/3-540-29784-7_14. [DOI] [PubMed] [Google Scholar]
- Scafidi J, Fagel DM, Ment LR, Vaccarino FM. Modeling premature brain injury and recovery. Int J Dev Neurosci. 2009;27:863–871. doi: 10.1016/j.ijdevneu.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz T, Ritter J, Mueller S, Felderhoff-Mueser U, Chew LJ, Gallo V. Cellular changes underlying hyperoxia-induced delay of white matter development. J Neurosci. 2011;31:4327–4344. doi: 10.1523/JNEUROSCI.3942-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS, Back SA. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:520–530. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizonenko SV, Camm EJ, Dayer A, Kiss JZ. Glial responses to neonatal hypoxic-ischemic injury in the rate cerebral cortex. Int J Dev Neurosci. 2008;26:37–45. doi: 10.1016/j.ijdevneu.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. Am J Psychiatry. 2001;158:1393–1399. doi: 10.1176/appi.ajp.158.9.1393. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration. J Biol Chem. 2004;279:19936–19947. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- Steinhäuser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Ment LR. Injury and repair in developing brain. Arch Dis Child Fetal Neonatal Ed. 2004;89:F190–F192. doi: 10.1136/adc.2003.043661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller ML, Stone IM, Goss A, Rau T, Rova C, Poulsen DJ. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience. 2008;155:1204–1211. doi: 10.1016/j.neuroscience.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Xue T, Zhang Z, Wang X, Xu P, Zhang J, Lei X, Li Y, Xie Y, Wang L, Fang M, Chen Y. Role of signal transducer and activator of transcription-3 in up-regulation of GFAP after epilepsy. Neurochem Res. 2011;36:2208–2215. doi: 10.1007/s11064-011-0576-1. [DOI] [PubMed] [Google Scholar]
- Yoshioka A, Bacskai B, Pleasure D. Pathophysiology of oligodendroglial excitotoxicity. J Neurosci Res. 1996;46:427–437. doi: 10.1002/(SICI)1097-4547(19961115)46:4<427::AID-JNR4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Zhao JB, Zhang Y, Li GZ, Su XF, Hang CH. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci Lett. 2011;498:147–152. doi: 10.1016/j.neulet.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Sun B, Zhang CL, Fine A, Chiu SY, Messing A. Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev Biol. 1997;187:36–42. doi: 10.1006/dbio.1997.8601. [DOI] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]