

Abstract
Background
Schizophrenia, bipolar disorder, and major depression are devastating mental diseases, each with distinctive yet overlapping epidemiologic characteristics. Microarray and proteomics data have revealed genes which expressed abnormally in patients. Several single nucleotide polymorphisms (SNPs) and mutations are associated with one or more of the three diseases. Nevertheless, there are few studies on the interactions among the disease-associated genes and proteins.
Results
This study, for the first time, incorporated microarray and protein-protein interaction (PPI) databases to construct the PPI network of abnormally expressed genes in postmortem brain samples of schizophrenia, bipolar disorder, and major depression patients. The samples were collected from Brodmann area (BA) 10 of the prefrontal cortex. Abnormally expressed disease genes were selected by t-tests comparing the disease and control samples. These genes were involved in housekeeping functions (e.g. translation, transcription, energy conversion, and metabolism), in brain specific functions (e.g. signal transduction, neuron cell differentiation, and cytoskeleton), or in stress responses (e.g. heat shocks and biotic stress).
The diseases were interconnected through several “switchboard”-like nodes in the PPI network or shared abnormally expressed genes. A “core” functional module which consisted of a tightly knitted sub-network of clique-5 and -4s was also observed. These cliques were formed by 12 genes highly expressed in both disease and control samples.
Conclusions
Several previously unidentified disease marker genes and drug targets, such as SBNO2 (schizophrenia), SEC24C (bipolar disorder), and SRRT (major depression), were identified based on statistical and topological analyses of the PPI network. The shared or interconnecting marker genes may explain the shared symptoms of the studied diseases. Furthermore, the “switchboard” genes, such as APP, UBC, and YWHAZ, are proposed as potential targets for developing new treatments due to their functional and topological significance.
Background
Schizophrenia, bipolar disorder, and major depression are suffered by approximately 1%, 5% or 20%, respectively, of human during their life time. Improving the diagnoses and treatments of these devastating diseases is an important task. However, few studies of mental diseases have used post-mortem brain samples [1]. Researchers did not have convenient access to brain samples of psychiatric patients until 1994 when the Stanley Brain Collection started.
These three diseases each have distinct characteristics, but they also share a few symptoms. All diseases above may show signs of psychosis, and in which both bipolar disorder and major depression have depressive symptoms. The shared symptoms suggest related disease mechanisms. These diseases have always been affiliated with neuron and dopamine abnormalities [2-9]. Abnormalities in the glia, GABA, and other neurotransmitter systems were revealed in more recent studies using patients’ brain samples from the Stanley Brain Collections [10-15]. The genetics of these diseases are overlapped [16,17]. Related single nucleotide polymorphisms (SNPs) and mutations, such as coding variants in the lipid transporter ABCA13, are often associated with more than one of the three diseases [1,2]. Microarrays of frontal, prefrontal, cingulate, and cerebellar cortex samples show disruption of mRNA or protein expression in intracellular signalling, synaptic neurotransmission, oligodendrocyte, stress responses, cytoskeleton, ATP biosynthesis, and translation [18-35].
The data of human protein-protein interactions (PPIs) brought insights to the network biology of diseases and explained the interrelationships among disease-related genes and proteins. Recently, the schizophrenia markers, NRG1 and CACNG2, which were considered functionally un-related, were found to be connected via the ERGG and DRL protein families in PPI network [36]. Furthermore, in our pilot study, a potential schizophrenia marker, EXOC4, was identified by analyzing the PPI network constructed using four published schizophrenic marker genes [37].
This study constructed PPI networks for schizophrenia, bipolar disorder, and major depression using abnormally expressed genes in Brodmann area (BA) 10 of prefrontal cortex. The “core” functional module of BA10 was also constructed by the most highly expressed genes in disease and control samples. Potential disease marker genes and drug targets were also identified.
Methods
This study constructed PPI networks for post-mortem prefrontal cortex of schizophrenia, bipolar disorder, and major depression patients. It focuses only on direct (physical) interactions among proteins. Genetic interactions were not investigated. The PPI networks were constructed based on the hypotheses that (1) the abundance of proteins and mRNAs were positively correlated in brains; (2) proteins were more likely to interact with proteins which had similar expression patterns or were more abundant; and (3) more abundant proteins participated in more active biological processes.
The research methodology is summarized in Figure 1. Microarray data series was used to identify genes abnormally expressed in patients’ BA10 of prefrontal cortex. These genes, together with the brain specific genes, were used to construct a PPI network for topological analyses. This network was compared with the PPI networks constructed by disease genes mentioned by in published literatures. The most abundant protein interactions in BA10 were revealed by the most highly expressed genes in the brain samples to outline the framework of prefrontal cortex biochemistry.
Figure 1.

Research methodology
Sources of microarray data
The raw data (CEL files) of microarray data series, GSE12654, were downloaded from Gene Expression Omnibus (GEO) and normalized by mas5 [38]. GSE12654 was first published by Iwamoto et al of RIKEN, Japan [39]. GSE12654 is the microarray data of post-mortem human brain sampled from the BA10 of four groups of people, including 13 schizophrenia patients, 11 bipolar disorder patients, 11 major depression patients, and 14 healthy controls [23]. Each group of the three diseases and the control samples was termed as a “sample group” in this study.
Selection of the most highly expressed genes
The z-scores of genes were calculated within each disease or control sample group. Each gene had four z-scores—three for each disease sample groups and one for the control sample group. The genes with z-scores ≥ 1.96 in ≥ 49% samples of a given sample group (e.g. in ≥ 7/13 schizophrenia samples) were defined as the most highly expressed genes of the sample types. These genes were likely to encode the most abundant proteins in BA10 of patients or healthy people.
Selection of tissue-specific “essential” genes for the healthy BA10 samples
GSE1133 (Human U133A/GNF1H) [40] was downloaded from the Novartis Research Foundation Gene Expression Database (GNF). The gcrma-normalized expression value of a gene in the prefrontal cortex was compared with the mean expression value of the same gene in all tissues examined in GSE1133. The genes were defined as prefrontal cortex-specific genes, if their expression values in the prefrontal cortex were > 4 fold higher than the mean values in all tissues. The genes which were both specific to the prefrontal cortex, as well as highly expressed (z-score ≥ 1.96 in ≥ 49% samples) in the control sample group of GSE12654, were defined as the tissue-specific “essential” genes of healthy BA10.
Selection of abnormally expressed genes in disease samples
Considering the diverse conditions of post-mortem brain samples (e.g. pH values), the profiles of subjects (e.g. age, gender, and use of medication), and the complexity of disease mechanisms, the microarrays in GSE12654 were analyzed by 2-tailed t-test. Each disease sample group was paired with the control sample group in the t-tests. The genes which expressed abnormally in disease samples would be detected by the corresponding probes with significant changes (P values < 0.01) of signal intensities in the disease sample groups as comparing to the control.
Construction of PPI networks for mental diseases
The most highly expressed genes and abnormally expressed genes were located on the human PPI network by an updated version of POINeT [37] to construct a query-query PPI (QQPPI) or level 1 PPI (L1PPI) networks. The updated version of POINeT contains the databases listed in Table 1 and has not been published. QQPPI networks only used the query marker genes as the nodes and revealed direct interactions among these queries. L1PPIs also showed the other non-query nodes directly connecting to the queries. L1PPI network allowed analysis of an extended network and could reveal indirect interactions (via common level 1 interactors as mediators) among query genes.
Table 1.
PPI databases used for constructing PPI networks in this study
| Database | Version or downloading date | Number of recorded PPIs |
|---|---|---|
| HPRD | Release 9 | 39194 |
| BioGRID | 3.1.70 | 356818 |
| IntACT | 2010-12-06 | 146227 |
| NCBI gene interactions | 2010-12-06 | 585982 |
Topology analysis of PPI networks
To analyse the QQPPI and L1PPI networks, cliques and centralities were calculated. A clique is a set of genes (nodes) in which every two genes (nodes) are connected by a protein interaction (edge). Cliques have been used successfully to identify protein functional units in PPI networks [41]. Nodes within cliques are more likely to form complexes [42]. In this study, cliques with 4 nodes (clique-4) or above were identified from PPI networks. They were searched against CORUM [43] and HPRD [44] for potential protein complexes.
Centrality analyses can assist in identifying significant proteins (nodes) which have relatively more PPIs (edges) in a network. The centrality of nodes in the L1PPI network which consisted of the abnormally expressed and “essential” genes was calculated by CentiBiN [45,46]. The centrality scores were ranked. The ranks from different centrality algorithms were fused by summing the ranking numbers [47].
Gene annotation
The highly expressed genes were classified into groups according to the Gene Ontology (GO) terms [48], using the Functional Classification tool in DAVID 6.7 [49]. Suitable parameters for classification stringency were chosen for the Gene Functional Classification tool as followed - a Kappa similarity term overlap of 3, a Kappa similar threshold of 0.3, an initial group membership of 3, a final group membership of 3, and a multiple linkage threshold of 0.5.
The functions of abnormally expressed disease genes (P value < 0.01) were summarized using the FatiGO+ module of Babelomics 4.2 [50,51]. Functional enrichments were performed for terms in REACTOME, GO cellular localisation, GO biological process, or GO molecular function with default parameters. The functions of genes were annotated by the Gene List Report in DAVID 6.7 [49].
Comparison with the study of Iwamoto [39]
Different analytical methods of microarray data often reveal similar but not identical results. The original contributors of microarray GSE12654 were Iwamoto et al who focused on gender- and age-related genes in their analysis. The disease genes identified from GSE12654 by Iwamoto et al[39] were constructed into QQPPI and L1PPI networks and compared with ours.
Comparison with data from Phenopedia, GWAS, HGMD, and GeneRIF
Microarray studies identify changes in gene expression patterns, whereas gene association studies focus on the identification of disease-related SNPs and mutations. The disease markers revealed by these different approaches were compared with our study.
Phenopedia collect genes which associate with human diseases by retrieving curated records from PubMed on a weekly basis since 2001 [52]. The genes which were listed in Phenopedia as being associated with schizophrenia, bipolar disorder, and major depression were constructed into QQPPI and L1PPI networks, and compared with the network constructed by the abnormally expressed disease genes. The disease-associated SNPs and mutations which were listed in A Catalog of Published Genome-Wide Association Studies (GWAS) [53] or The Human Genome Mutation Database (HGMD) [54] were also compared with our findings.
In addition, dopamine, GABA, and glutamate are three of the most significant chemicals known to affect the symptoms of the studied mental diseases. The association of abnormally expressed and tissue-specific “essential” genes with dopamine, GABA, and glutamate were searched in GeneRIF.
Results
BA10 of the prefrontal cortex is believed to be responsible for cognition, which is a function disrupted often in patients of psychiatric diseases [55,56]. By analyzing the microarrays of human BA10 samples, this study identified potential disease marker genes and drug targets of schizophrenia, bipolar disorder, and major depression. Additionally, a “core” functional module was constructed using the genes which were not only highly expressed in both disease and control samples, but also topologically significant in the PPI networks.
Abnormal gene expression in the prefrontal cortex of schizophrenia, bipolar disorder, and major depression patients
The differential expression of genes in the BA10 samples was investigated by t-tests comparing the signal intensities of corresponding probes in microarrays for the disease and control samples. The genes of which the corresponding probes had P values < 0.01 were defined as abnormally expressed and proposed as disease markers. These disease markers, together with the tissue-specific “essential” genes, were used as queries to reveal the QQPPI network shown in Figure 2. The functions of query genes are listed in Additional file 1 (titled „Abnormally expressed disease genes and functions'). The genes and P values of corresponding probes are listed in Additional file 2, 3, and 4 (titled „Abnormally expressed genes and corresponding probe IDs and P values in t-tests’ for „schizophrenia’, „bipolar disorder’, and „major depression’, respectively).
Figure 2.

QQPPI constructed by abnormally expressed disease genes and tissue-specific “essential” genes
The abnormally expressed genes could be classified into 13 groups according to their functional annotations as detailed in Additional file 5 (titled „GO functional classification for abnormally expressed genes'). They participated in transcription, translation, cytoskeleton, neuron differentiation, ATP-binding, cellular transportation, and many other biological processes. Using the functional enrichment tool FATIGO+, a few functional terms were found to be more abundant in our abnormally expressed proteins of BA10 than in the entire proteome encoded by human genome. Table 2 lists the GO biological processes which were enriched in our abnormally expressed genes. The results of functional enrichment analyses for GO cellular compartment, GO molecular function, and REACTOME are detailed in Additional file 6 (titled „FatiGO term enrichment with P values smaller than 0.01). Enriched functions can be classified into five groups—(1) neuron and signal transduction-related, such as neuron projection, transmission of nerve impulse, synaptic transmission, synaptogenesis, and signalling by NGF; (2) cytoskeleton; (3) gene expression, such as translation and ribosomes; (4) metabolisms of lipids, lipoproteins, proteins, polyamines, and sugars (diabetes); and (5) stresses, such as influenza infection. The enrichment of neuron and signal transduction-related functions was expected. Abnormality in translation-related genes has been observed in previous studies [39,57]. Abnormality in cytoskeleton genes could lead to disrupted cellular mobility of Golgi apparatus, which has a place in neuron signal transduction [58-61]. Previous studies have also shown abnormal expression of ATP-related or mitochondrial genes [20,22,62-64]. The enriched metabolism functions can be further evidences of abnormal energy conversion in patients' prefrontal cortex. The relationship to influenza infection can be explained as biotic stress; and stresses have often been reported as inducers of mental diseases [65-67].
Table 2.
Functional enrichment analysis for abnormally expressed disease markers
| GO: biological process | Go term ID | Genes with this term |
|---|---|---|
| Translation | GO:0006412 | DHX29, EIF4G1, FAU, GSPT1, KRT7, MAZ, RPL10A, RPL17, RPL 18, RPL21, RPL21P119, RPL21P16, RPL21P39, RPL21P80, RPL21P93, RPL21P97, RPL22, RPS15A, RPS5, VARS, MRPS12, PABPC4, RPS27A, TOP3A |
| Transmission of nerve impulse | GO:0019226 | CPNE6, GABARAP, MUSK, POU3F2, VGF, ADORA2A, GRM8, NF1, NMU, PARK2, DLGAP2, GNAI2, HRAS, NOS1, RPS27A, STX1A, UBB, UBC |
| Synaptic transmission | GO:0007268 | CPNE6, GABARAP, MUSK, VGF, ADORA2A, GRM8, NMU, PARK2, DLGAP2, GNAI2, HRAS, NOS1, RPS27A, STX1A, UBB, UBC |
| Negative regulation of catalytic activity | GO:0043086 | GNAT1, TNNI3, ADORA2A, GRM8, NF1, GNAI2, OXA1L, RPS27A, SH3BP5, TSC2, UBB, UBC |
| Regulation of synaptic transmission | GO:0050804 | VGF, ADORA2A, GRM8, NMU, PARK2, GNAI2, HRAS, RPS27A, STX1A, UBB, UBC |
| Translational elongation | GO:0006414 | FAU, RPL10A, RPL17, RPL18, RPL21, RPL21P119, RPL21P16, RPL21P39, RPL21P80, RPL21P93, RPL21P97, RPL22, RPS15A, RPS5, VARS, RPS27A |
| Regulation of synaptogenesis | GO:0051963 | MUSK, RPS27A, UBB, UBC |
| Polyamine metabolic process | GO:0006595 | ODC1, SLC6A11, OAZ2, SAT1 |
The ranks of centrality were calculated by various algorithms and are listed in Additional file 7 (titled „Centrality analysis of abnormally expressed genes in QQPPI network'). The top ranked nodes are summarised in Table 3. Proteins which rank higher in centrality analyses of PPI networks usually have more crucial biological functions [68]. The top ranked nodes (e.g. UBC, UBB, and ACTB) were therefore proposed as having critical roles in disease mechanisms. Only three clique-3s were found (data not shown). Clique-4 or above was not identified as a result of the looser network formed by the abnormally expressed genes (Figure 2) in comparison with the tightly knitted network formed by the highly expressed genes (Figure 3).
Table 3.
Centrality analysis of PPI network
| Gene symbol | Disease(s) or essential gene* | Gene function(s) | Sum of ranks** |
|---|---|---|---|
| UBC | S | • ubiquitin C | 7 |
| ACTB | S | • actin, beta | 15 |
| UBB | S | • ubiquitin B | 29 |
| APP | E | • amyloid beta (A4) precursor protein | 32 |
| FOS | S | • v-fos FBJ murine osteosarcoma viral oncogene homolog | 51 |
| HSPA4 | S | • heat shock 70kDa protein 4 | 54 |
| PARK2 | SD | • Parkinson disease (autosomal recessive, juvenile) 2, parkin | 67 |
| SYK | B | • spleen tyrosine kinase | 97 |
| TUBB2A | E | • tubulin, beta 2A | 117 |
| TSC2 | S | • tuberous sclerosis 2 | 127 |
| UCHL1 | E | • ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase) | 217 |
| MARK3 | S | • MAP/microtubule affinity-regulating kinase 3 | 236 |
| RPS27A | S | • ribosomal protein S27a pseudogene 12; ribosomal | 355 |
| DNM1 | E | • dynamin 1 | 476 |
| IRS2 | E | • insulin receptor substrate 2 | 497 |
| GNAI2 | S | • guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 2 | 519 |
| SMARCC2 | B | • SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily c, member 2 | 525 |
| HRAS | S | • v-Ha-ras Harvey rat sarcoma viral oncogene homolog | 527 |
| SAT1 | S | • spermidine/spermine N1-acetyltransferase 1 | 529 |
| CRKL | B | • v-crk sarcoma virus CT10 oncogene homolog (avian)-like | 531 |
The L1PPI network (not shown) was constructed by abnormally expressed disease markers, tissue-specific “essential” genes
*The listed genes were found abnormally expressed in samples of schizophrenia (S), bipolar disorder (B) or major depression (D), or were tissue-specific “essential” (E) genes.
**The sum of centrality ranks calculated using various centrality algorithms for the disease and “essential” genes shown in Figure 2.
Figure 3.

a) QQPPI constructed by highly expressed genes in disease and control samples; b) The “core” functional module
The disease marker genes identified through different analytical approaches were connected in PPI networks
The microarray data series used in this study has been analysed by the original contributors, Iwamoto et al[39]. In the studied mental diseases, Iwamoto et al revealed down-regulation of receptor-, transporter-, and channel-encoding genes; and up-regulation of transcription-, translation-, stress- and molecular chaperon-related genes [39]. Although the genes identified in our studies were not identical to the findings of Iwamoto et al, they fall into similar functional categories. Although few of our disease markers had direct protein interactions with the ones described in Iwamoto et al[39], many had indirect interactions through mediator proteins (level 1 interactors) in between. Taking bipolar disorder as an example, there was only one gene listed in both Iwamoto et al and this study as a disease marker; only two of our abnormally expressed genes for bipolar dirorder had direct interactions (PPIs) with two bipolar disorder markers of Iwamoto et al. However, our markers of bipolar disorder shared 120 level 1 interactors with the bipolar disorder markers of Iwamoto et al[39].
Similar results were observed in the data of Phenopedia as summarized in Table 4. For schizophrenia, bipolar disorder, and major depression, there were only 11, 5, and 5 genes, respectively, that were listed both as disease genes in Phenopedia and as our disease markers. Interestingly, there were many PPIs formed between the Phenopedia disease genes and our abnormally expressed genes for schizophrenia, bipolar disorder, and major depression as illustrated in Additional file 8, 9, and 10 (titled „Interrelationship between Phenopedia and abnormally expressed genes' in „schizophrenia', „bipolar disorder', and „major depression', respectively). The names of these disease genes are listed in Additional file 11 (titled „Comparison of disease genes listed in Phenopedia, HGMD, and GWAS'). The diseases associated SNPs and mutations which are listed in GWAS and HGMD were also compared with the abnormally expressed genes identified in this study and listed also in Additional file 8.
Table 4.
Comparison to the disease genes listed in Phenopedia
| Record type | Schizophrenia | Bipolar disorder | Major depression |
|---|---|---|---|
| Disease genes in Phenopedia (P) | 1001 | 722 | 226 |
| Abnormally expressed genes (T)* | 110 | 100 | 44 |
| Both P and T (shared nodes) | 11 | 5 | 1 |
| PPIs between P and T | 252 | 73 | 11 |
* Genes with P values < 0.01 in t-tests
Furthermore, dopamine, GABA, and glutamate are critical chemicals affecting the symptoms of schizophrenia, bipolar disorder, and major depression. Genes which were described in GeneRIF as involving in the biochemistry of these chemicals are listed in Table 5. This data was considered as the indirect evidence to the association of genes to the studied mental diseases.
Table 5.
Genes which participated in the biochemistry of GABA, glutamate, or dopamine
| Gene Symbol | GABA | Glutamate | Dopamine |
|---|---|---|---|
| APP | + | + | + |
| GABBR2 | + | + | |
| GABBR1 | + | + | |
| SLC12A5 | + | ||
| GABARAP | + | ||
| SLC1A3 | + | ||
| NOS1 | + | ||
| GRM8 | + | ||
| GLUL | + | ||
| UCHL1 | + | ||
| SYNGR3 | + | ||
| STXBP1 | + | ||
| STX1A | + | ||
| SNCB | + | ||
| PRKAR1B | + | ||
| PARK2 | + | ||
| IGF1 | + | ||
| GPR143 | + | ||
| EPB41L1 | + | ||
| CRYAA | + | ||
| ADORA2A | + |
The highly expressed genes and the most abundant protein interactions in the prefrontal cortex
In order to identify the most abundant protein interactions in BA10, the PPI network of highly expressed genes were constructed for schizophrenia, bipolar disorder, major depression, and healthy control samples as illustrated in Figure 3a. Many PPIs were shared by two or more of the diseases. Disease-associated SNPs, mutations, and GO functional classes were identified for each gene and labelled as shown in Figure 3a.
In this PPI network, there were one clique-5 and several clique-4s that tightly knitted into a sub-network containing 12 genes as shown in Figure 3b. These 12 genes in the BA10 cliques encode four heat shock proteins (HSPA8, HSP90AA1, HSPD1, HSP90AB1), the tubulin protein (TUBA1A), the ubiquitin protein (UBC), the actin-binding cytoskeleton protein (SPTAN1), beta-actin (ACTB), the energy generating glyceraldehyde-3-phosphate dehydrogenase (GAPDH), translation elongation factor (EEF1A1), signal transduction protein (YWHAZ), and amyloid beta (A4) precursor protein (APP). This sub-network of 12 highly expressed and topologically significant genes was proposed as the “core” functional module of BA10 for both patients and healthy people.
Discussion
This study combined data of quantitative transcriptomics and PPIs for statistical and topological analyses. New disease marker genes and potential drug targets were revealed. Shared disease mechanisms, although hypothetical at the present, were proposed based on common marker genes and interconnecting PPIs to explain the shared symptoms among diseases. A “core” functional module of BA10 was also proposed.
Disease markers and potential drug targets
The genes which had P values < 0.01 in our t-tests were defined as abnormally expressed in disease samples. These genes were proposed as disease marker genes and constructed into a PPI network as illustrated in Figure 2.
Previous studies have shown that the probes (and the genes which they represented) with lower P values appeared as more effective clustering features for separating the disease samples from the controls in hierarchical clusters (unpublished data). The observation suggested that genes with lower P values are more important disease markers.
The genes with the lowest P values in our t-tests were not well-studied genes. SBNO2 was a gene of which the probe had the lowest P value in the t-test comparing schizophrenia and control samples. SBNO2 has a strawberry notch homolog in fruit fly. The gene is involved in the anti-inflammatory signalling pathway [69]. It has also been associated with type 2 diabetes mellitus, of which shared many disease genes with mental diseases [52,70]. SEC24C was a gene of which the probe had the lowest P value in the t-test comparing bipolar disorder and control samples. SEC24C encodes a protein which may be involved in ER to Golgi vesicular transportation [71]. It has been associated another mental disease, Alzheimer's disease [52]. SRRT was a gene of which the probe had the lowest P value in the t-test comparing major depression and control samples. SRRT is possibly involved in transcriptional regulation and RNA metabolism as it is a homolog to an Arabidopsis serrate RNA effector [72]. Apart from this study, SBNO2, SEC24C and SRRT have never been associated with schizophrenia, bipolar disorder, or major depression; these genes did not form PPI with any of our abnormally expressed marker genes, either.
There appeared a weak negative correlation between the P values of a gene (probe) and its centrality ranks in this study. More “essential” proteins in PPI networks have also been shown to rank higher than less important proteins in centrality analyses [68,73]. Interestingly, the nodes which ranked highest in the centrality studies were mostly schizophrenia markers as listed in Table 3. The top ranked genes in centrality analysis, UBC, ACTB, and UBB, were all abnormally expressed in schizophrenia samples. UBC and UBB encode the polyubiquitin precursors. ACTB encodes the beta-actin protein. The roles of these proteins in mental disease mechanisms are not clear. None of these three genes have been associated with any mental disease; but UBC has been associated also with type 2 diabetes mellitus [70].
Schizophrenia samples had the largest number of abnormally expressed genes, and major depression had the fewest. The above observations aligned with the most severe and complex symptoms of schizophrenia in comparison to the other two diseases; major depression encompassed the least of symptoms. All three disorders may show signs of psychosis, but symptoms such as hallucinations and poverty of speech are usually only seen with schizophrenia. The delusions associated with bipolar disorder and major depression is usually the milder “mood-congruent” delusion contributing to the overly positive or negative self perceptions of manic or depressive patients. Whereas the delusions occur with schizophrenia can be completely implausible and “bizarre”. The additional symptoms of schizophrenia may be caused by additional aberrations of genes. It must be noted, however, that few evidences are available to support or clarify this assumption.
Shared disease mechanisms
The three studied mental diseases are genetically related and share disease genes [16,17]. Each disease may have malfunctions in multiple biological pathways, and the same malfunctions may occur in multiple diseases [74]. There are also increasing numbers of evidences which indicate correlations in genetic risks for schizophrenia and bipolar disorder [75-79].
A few genes were found to be abnormally expressed in more than one disease in this study. As shown in Figure 2, MUSK, which encodes a protein responsible for the assembly of receptors in post-synaptic neuromuscular junctions, was abnormally expressed in samples of bipolar disorder and major depression [80,81]. PARK2, a gene of unknown function associated with related phenotypes such as Parkinson disease, nerve degeneration, cognition disorders, visual perception, attention, and memory, was abnormally expressed in schizophrenia and major depression [52,82-84]. AP4M1, which encodes a subunit of AP-4 complex responsible for transportation of proteins from Golgi, was abnormally expressed in schizophrenia and bipolar disorder [52,85]. Many of the genes were also found to be highly expressed in more than one disease as shown in Figure 3a.
One might hypothesize that the shared genes are the reasons for shared symptoms— the abnormal expression of MUSK may cause the depression of bipolar disorder and major depression; PARK2 may cause the psychosis symptoms such as delusions, avolition, blunted effect, asociality, and cognitive dysfunction, which are commonly seen with varying severities in both schizophrenia and major depression; AP4M1 may cause the psychosis symptoms such as the less “bizarre” forms of delusions and restlessness sometimes observed in both schizophrenia and bipolar disorder. These hypotheses are far from being conclusive. Further researches such as mutant studies in animal models, genetic association studies, and gene and protein expression analyses are required to better explain the observed phenomenon of overlapping PPI network of the mental diseases. Unfortunately, as symptoms such as hallucinations and delusions are uniquely human, the exact roles of disease-associated genes play in mental disease mechanisms are difficult to be studied using mutant model animals [86].
The centrality ranks of MUSK, PARK2, and AP4M1 might indicate the divergence or similarity of the studied mental diseases, if phenotypical similarities were positively correlated with similarities in disease mechanisms. In the PPI network constructed using 3550 human disease genes retrieved from OMIM Morbid Map, shared genes were more central than disease-specific genes, and the genes shared by phenotypically similar diseases are less central than the ones shared by phenotypically divergent diseases [73]. AP4M1 ranked the lowest in centrality among the three genes as shown in Fig. 2; PARK2 ranked the highest. We might deduce that the disease mechanisms of schizophrenia and bipolar disorder are most similar among the three studied diseases; and the disease mechanisms of schizophrenia and major depression are most diverged.
“Switchboards” in PPI sub-networks of psychiatric diseases
Apart from shared marker genes, marker genes of multiple diseases sometimes interact with the same critical nodes. These critical nodes were designated as “switchboard” nodes to describe their place in interconnecting PPIs of different diseases. The “switchboards” usually ranked higher in centrality analyses, suggesting they are more “essential” and effect diverged ranges of phenotypes. The abnormalities of different protein interactions with the same gene may explain the relevant but different symptoms of the studied diseases.
In the schizophrenia PPI network shown in figure 2, the “switchboard” APP interacted with the abnormally expressed ACTB and FOS. APP was a tissue-specific “essential” gene, which encodes a highly expressed beta-amyloid precursor protein. ACTB encodes the beta-actin, which is responsible for cellular structure and (neuron signal) mobility [87-89]. FOS is likely to associate with cell differentiation, apoptotic cell death, and depression-related diseases such as bulimia and anorexia [52,90]. In bipolar disorder, APP interacted with the abnormally expressed F12. F12 encodes a coagulation factor which circulates in blood as zymogen. Mutations in F12 may prolong whole-blood clotting time; and the gene has been associated with type 1 and 2 diabetes mellitus [52,91-93]. In major depression, APP interacted with the abnormally expressed NF1. NF1 encodes a neurofibromin responsible for signal transduction, and has been associated with mental retardation and autism [52,94,95]. APP has been listed in the HGMD as being associated with schizophrenia [96]. However, the transcript of APP was consistently highly abundant in all disease and control samples. The mutation of APP in patients might not have affected its transcription.
A “switchboard” can also be a disease gene. For example, the ubiquitous protein, UBC, was abnormally expressed in schizophrenia samples and interacted with the maker genes of schizophrenia (i.e. TSC2, SCNN1A and USP2), bipolar disorder (i.e. MUSK), and major depression (i.e. MUSK and FLT3).
The same “switchboard” mechanism was observed in the network constructed by the most highly expressed genes. One such example was YWHAZ, which encodes a signal transduction protein. YWHAZ interacted with 43 nodes as shown in Figure 3a and was highly expressed in disease and control samples. The abnormal PPIs (i.e. PPIs of highly expressed genes that did not occur in the controls) between YWHAZ and many other proteins were observed in disease samples. The interaction with NCL was abnormal in schizophrenia, bipolar disorder, and major depression. The other YWHAZ abnormalities were the interactions with RNPS1 and LGALS1 in schizophrenia; the interactions with MYCBP2, PRDX1 or TP1l in bipolar disorder; the interactions with RPLP2 and VIM in major depression; and the interactions with RPLP0 in both schizophrenia and major depression.
The “core” functional module
We proposed that the 12 genes in the clique-5 and -4s in PPI constructed by the highly expressed genes were central to the functioning of BA10 (Figure 3a and b). These nodes were the ones with the highest ranks in the centrality analysis of PPI network of highly expressed gene. A few genes, such as UBC and ACTB, were also highly ranked in the centrality analysis of the abnormally expressed PPI network as listed in Table 3. Many of these genes encode members of important protein complexes as summarised in Table 6. They were mostly tissue-specific “essential” genes and highly expressed in all three studied mental diseases as well as healthy control. The nodes in cliques were sometimes involved in biological processes which were disrupted in schizophrenia, bipolar disorder, or major depression.
Table 6.
Cliques in the PPI network constructed by highly expressed genes in disease and control samples
| List of clique-4 | Protein complex | |||
|---|---|---|---|---|
| HSPA8 | HSP90AA1 | APP | YWHAZ | • Nil |
| HSPA8 | HSP90AA1 | APP | ACTB | • Amyloid beta protein oligomer (PMID:15834427) |
| HSPA8 | HSP90AA1 | ACTB | YWHAZ | • APP-FE65-LRP complex (PMID:15115822) |
| HSPA8 | APP | ACTB | YWHAZ | • APP-TIMM23 complex (PMID:16943564) |
| HSP90AA1 | APP | ACTB | YWHAZ | • APP-TOMM40 complex (PMID:16943564) |
| HSP90AA1 | ACTB | YWHAZ | TUBA1A | • Emerin architectural complex (PMID:17620012) |
| GAPDH | HSP90AB1* | ACTB | YWHAZ | • Emerin complex 25 (PMID:17620012) |
| GAPDH | APP | ACTB | YWHAZ | • Emerin-actin-NMI- (PMID:alphaII) spectrin complex (PMID:17620012) |
| HSPA8 | ACTB | UBC* | YWHAZ | • Kinase maturation complex 1 (PMID:14743216) |
| HSP90AB1* | ACTB | SPTAN1 | YWHAZ | • P2X7 receptor signaling complex (PMID:11707406) |
| HSP90AA1 | HSPD1 | ACTB | YWHAZ | • Profilin 2 complex (PMID:9463375) |
| EEF1A1* | ACTB | UBC* | YWHAZ | • Profilin 2 complex (PMID:9463375) |
| APP | ACTB | SPTAN1 | YWHAZ | • Nil |
The protein complexes which may contain nodes within these cliques-4s are listed.
* This protein was not documented as a member of the suggested protein complex.
Teams of disease marker genes
The abnormally expressed genes identified in this study were compared with published disease associated genes from previous analysis of the same data series (GSE12654), Phenopedia, GWAS, and HGMD. Although few genes were consistently identified in diseases by different groups of researchers, many of these genes were found to form QQPPIs or share the same level 1 PPI interactors as shown in Table 4 and Additional file 8, 9, and 10.
The observations above suggested that, in brains of patients, disease genes can become defected due to various abnormalities and lead to the same symptoms. Each research approach may only detect a sub-set of abnormalities, such as mutation, significant changes in gene expression, or smaller changes in gene expression. Besides, only a snapshot of transcript abundance can be detected in post-mortem brain samples. The proteins were likely to work in teams—the failure of any team members at any given time may similarly disrupt the participating biological process and cause similar phenotypes. A protein team may be a set of proteins which have direct physical interactions, such as QQPPIs, or via a common protein, such as in the more extended L1PPIs. A more stringent definition of a protein team can be a set of proteins which form cliques. The teams may also be defined by genetic interactions, although it may not be applicable for human samples which cannot have synthetic lethal experiments; nonetheless, redundancy or overlapping of gene functions may be speculated with sequence homology.
Network medicine
Psychiatric drugs may be developed based on the concepts of network medicine. Network analysis of disease genes have been shown to significantly accelerate the trials for new treatments [97].
Combining drugs to target the largest number of disease genes in a PPI network, while avoiding non-disease genes to avoid side effects, has been shown to create effective new treatments for complex diseases [98]. Besides, topologically significant non-markers can also be potential drug targets so that the “switchboard” gene APP and its neighbouring nodes were proposed as potential drug targets [99]. APP is a cell surface receptor and trans-membrane precursor which can be cleaved to from peptides. The exact function of APP was not clear, but its roles in cellular signalling neuronal adhesion and positioning in cortical layers have been observed in mice models [100,101]. APP has also been reported to participate in the biochemistry of GABA, dopamine, and glutamate (Table 5), which are all known to have significant places in the symptoms of the studied mental diseases. Abnormal accumulation of APP protein has long been associated with Alzheimer's disease [102]. The polymorphism of APP has also been associated with schizophrenia as listed in HGMD, although its gene expression was not significantly different in disease samples of this study. The gene's association with cognition, dementia, and type 2 diabetes mellitus have also been mentioned [52]. The other “switchboards”, including UBC and YWHAZ, and their neighbouring nodes can also be potential targets for developing new treatments.
Limitations of the research methods
This study analysed the disease mechanisms by considering the interactions of the proteins and employing topological analyses. However, the extensiveness of PPI subnetworks was limited by the availability of PPI recorded in the PPI databases listed in Table 1. Markers whose PPI data were not recorded in databases would be excluded in the proposed PPI networks. The incomplete human PPI network could lead to incomplete PPI network for the disease samples; it could also bias the topological analyses. The significance of disease markers was therefore evaluated by both t-tests and centrality analyses. Besides, abnormalities such as deformed protein structures were not observable in this study. This study only proposed disease markers which can be detected by examining the transient abundances of mRNAs. The functions of many disease marker genes were speculated by homologous genes in other model organisms and required confirmation.
Conclusions
By identifying abnormally expressed genes in post-mortem brain samples of mental disease patients, several disease marker genes were proposed for schizophrenia, bipolar disorder, and major depression. The disease markers were constructed into PPI networks and analysed by topological theories.
A few genes were shared among the studied diseases, such as MUSK, PARK2, and AP4M1. They are evidences of shared disease mechanisms. The studied diseases also shared disease genes with the other mental diseases, such as Alzheimer's and Parkinson, and metabolic diseases, such as type 1 and 2 diabetes mellitus. The research methodology of this study may be applied to expand our investigations to related diseases. Genes with higher P values, ranked lower in centrality analyses and not shared among diseases are proposed as more effective disease marker genes; the abnormal expression of these genes are more likely to be unique to a specific disease.
Nevertheless, disease markers which ranked higher in centrality analyses, interacted with the “switchboards”, or were members of the “core” functional module, were considered to have more “essential” roles in the disease mechanisms. These genes included SBNO2, CEACAM5, AKAP1, UBC, ACTB, UBB, and FOS for schizophrenia; SEC24C, PGLYRP1, ARHGAP4, RPL22, SLC6A11, and SYK for bipolar disorder; and SRRT, PARK2, LILRA4, STK17A, IGFBP2, and NF1 for major depression. These markers, together with the “switchboards” such as APP, UBC, and YWHAZ, were proposed as targets for drug development.
The three studied mental diseases showed aberrations in common biological processes. Most of the disease markers fall into the functional categories which have been previously proposed as being related to the three studied mental diseases. Based on published data and the results of this study, it might be said that the stress responses, cellular signal transduction, neuron cell differentiation and aging, energy metabolism, and translation were dysfunctional in patients suffering schizophrenia, bipolar disorder, or major depression. However, we are still far from drawing a clear picture of the molecular biology of patients’ brains. Extensive studies are still required for establishing the disease mechanisms.
List of abbreviations used
BA: Brodmann Area; Bi: bipolar disorder; Co: control; De: major depression; GEO: Gene Expression Omnibus; GO: Gene Ontology; GOA: Gene Ontology Annotation; GWAS: A Catalog of Published Genome-Wide Association Studies; HGMD: The Human Genome Mutation Database; L1PPI: level 1 protein-protein interaction; LDL: low-density lipoprotein; PPI: protein-protein interaction; QQPPI: query-query protein-protein interaction; Sc: schizophrenia; single SNP: nucleotide polymorphism.
Authors' contributions
SAL conceived the study, programmed the bioinformatics analysis tools, carried out the data analysis and assisted in drafting the manuscript. TTHT conceived the study, interpreted the results, drafted the manuscript, surveyed the microarray data, and contributed to the design of the bioinformatics analysis tools. KCY assisted in the interpretation of results, drafting of the manuscripts, and contributed to the design of the bioinformatics analysis tools. LH programmed the bioinformatics analysis tools and carried out the data analysis. YLK and CHH assisted in data analyses. WKL assisted in the interpretation of results. KCH contributed to drafting the manuscript, interpretation of the data, and project management. CYK conceived the study and participate in coordination and management of the research project.
Conflict of interest
The authors confirm that they have no conflict of interest.
Supplementary Material
Abnormally expressed disease genes and functions
Abnormally expressed genes and corresponding probe IDs and P values in t-tests for schizophrenia
Abnormally expressed genes and corresponding probe IDs and P values in t-tests for bipolar disorder
Abnormally expressed genes and corresponding probe IDs and P values in t-tests for major depression
GO functional classification for abnormally expressed genes
FATIGO term enrichment with P values smaller than 0.01
Centrality analysis of abnormally expressed genes in QQPPI network
Interrelationship between Phenopedia and abnormally expressed genes in schizophrenia
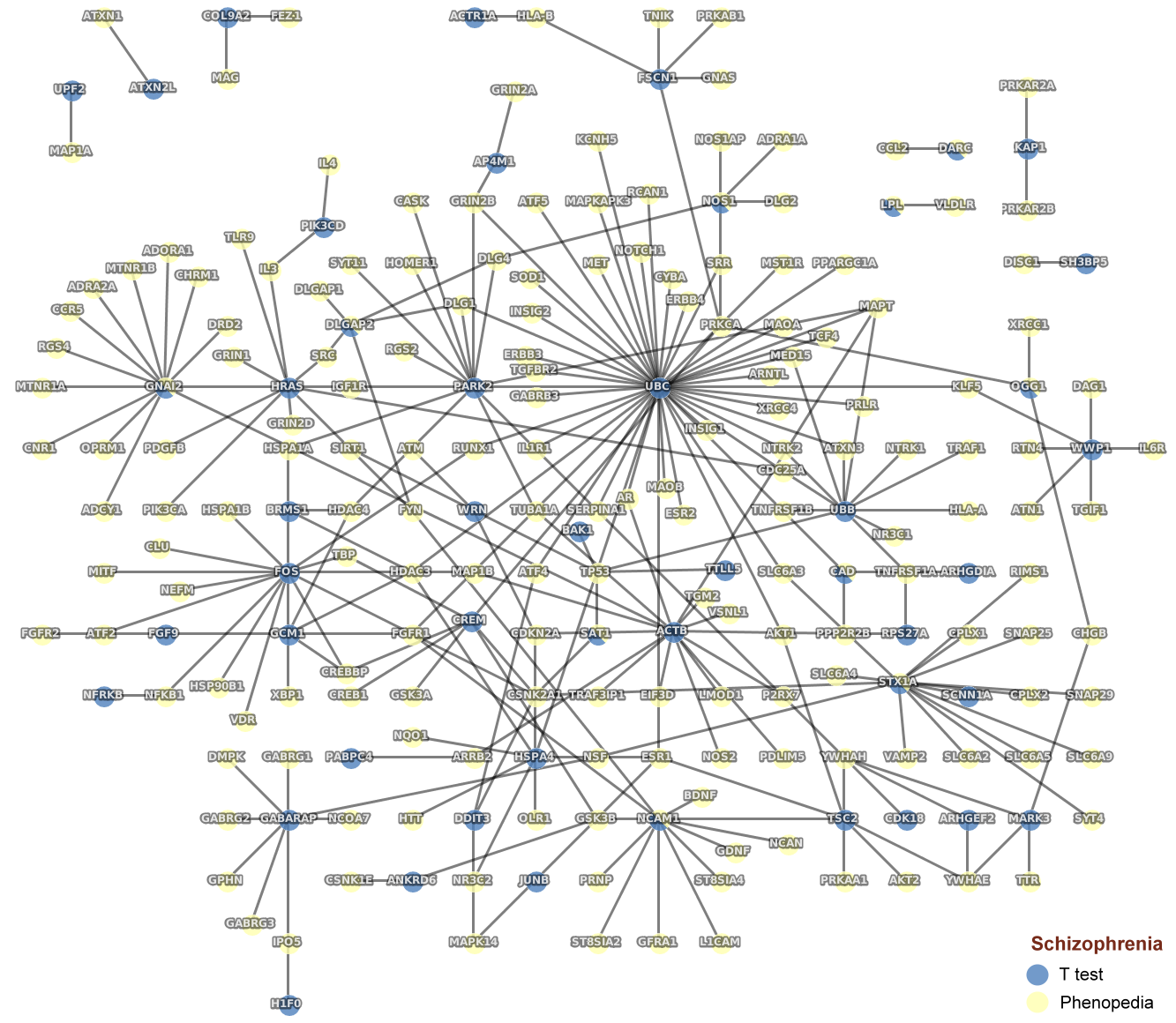{kind=link}
Interrelationship between Phenopedia and abnormally expressed genes in bipolar disorder
{kind=link}
Interrelationship between Phenopedia and abnormally expressed genes in major depression
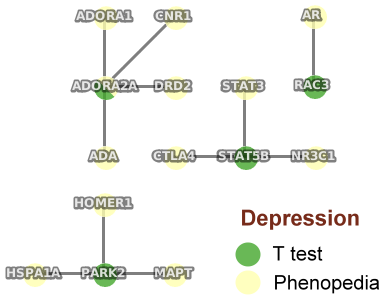{kind=link}
Comparison of disease genes listed in Phenopedia, HGMD, and GWAS
Contributor Information
Sheng-An Lee, Email: shengan@mail.knu.edu.tw.
Theresa Tsun-Hui Tsao, Email: postergrey@gmail.com.
Ko-Chun Yang, Email: f97945020@ntu.edu.tw.
Han Lin, Email: hotdogee@gmail.com.
Yu-Lun Kuo, Email: sscc6991@gmail.com.
Chien-Hsiang Hsu, Email: r98945020@ntu.edu.tw.
Wen-Kuei Lee, Email: akwei1@yahoo.com.tw.
Kuo-Chuan Huang, Email: d94922009@ntu.edu.tw.
Cheng-Yan Kao, Email: cykao@csie.ntu.edu.tw.
Acknowledgements
We wish to thank National Science Council, Taiwan, for funding this study and National Taiwan University for providing space and facilities. We were grateful that Computer and Information Networking Center, National Taiwan University provided high-performance computing facilities. This research project was funded by NSC grant (Project ID: NSC98-2221-E-002-141-MY2). TTHT was also funded by the NSC grants (Project ID: 098-2811-E-002-106 and 099-2811-E-002-021). This study was also supported by Armed Forces Beitou Hospital. We also thanked RIKEN for generously making their precious microarray data series, GSE12654, available on-line. This paper was kindly edited and proofread by Yan Ge.
This article has been published as part of BMC Bioinformatics Volume 12 Supplement 13, 2011: Tenth International Conference on Bioinformatics – First ISCB Asia Joint Conference 2011 (InCoB/ISCB-Asia 2011): Bioinformatics. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/12?issue=S13.
References
- Harrison PJ. Using our brains: the findings, flaws, and future of postmortem studies of psychiatric disorders. Biol Psychiatry. 2011;69:102–103. doi: 10.1016/j.biopsych.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Egan MF, el-Mallakh RS, Suddath RL, Lohr JB, Bracha HS, Wyatt RJ. Cerebrospinal fluid and serum levels of neuron-specific enolase in patients with schizophrenia. Psychiatry Res. 1992;43:187–195. doi: 10.1016/0165-1781(92)90133-N. [DOI] [PubMed] [Google Scholar]
- Heckers S, Heinsen H, Geiger B, Beckmann H. Hippocampal neuron number in schizophrenia. A stereological study. Arch Gen Psychiatry. 1991;48:1002–1008. doi: 10.1001/archpsyc.1991.01810350042006. [DOI] [PubMed] [Google Scholar]
- Goode DJ, Manning AA. Specific imbalance of right and left sided motor neuron excitability in schizophrenia. J Neurol Neurosurg Psychiatry. 1988;51:626–629. doi: 10.1136/jnnp.51.5.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KA, Holcomb LA, Yazdani U, Hicks PB, German DC. Elevated neuron number in the limbic thalamus in major depression. Am J Psychiatry. 2004;161:1270–1277. doi: 10.1176/appi.ajp.161.7.1270. [DOI] [PubMed] [Google Scholar]
- Manaye KF, Lei DL, Tizabi Y, Davila-Garcia MI, Mouton PR, Kelly PH. Selective neuron loss in the paraventricular nucleus of hypothalamus in patients suffering from major depression and bipolar disorder. J Neuropathol Exp Neurol. 2005;64:224–229. doi: 10.1093/jnen/64.3.224. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Nolte-Zenker B, Patzer J, Bauer M, Schmidt LG, Heinz A. Psychopathological correlates of reduced dopamine receptor sensitivity in depression, schizophrenia, and opiate and alcohol dependence. Pharmacopsychiatry. 2001;34:66–72. doi: 10.1055/s-2001-15184. [DOI] [PubMed] [Google Scholar]
- Benkert O, Grunder G, Wetzel H. Dopamine autoreceptor agonists in the treatment of schizophrenia and major depression. Pharmacopsychiatry. 1992;25:254–260. doi: 10.1055/s-2007-1014417. [DOI] [PubMed] [Google Scholar]
- Zhan L, Kerr JR, Lafuente MJ, Maclean A, Chibalina MV, Liu B, Burke B, Bevan S, Nasir J. Altered expression and coregulation of dopamine signalling genes in schizophrenia and bipolar disorder. Neuropathol Appl Neurobiol. 2011;37:206–219. doi: 10.1111/j.1365-2990.2010.01128.x. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex. 2002;12:386–394. doi: 10.1093/cercor/12.4.386. [DOI] [PubMed] [Google Scholar]
- Cotter DR, Pariante CM, Everall IP. Glial cell abnormalities in major psychiatric disorders: the evidence and implications. Brain Res Bull. 2001;55:585–595. doi: 10.1016/S0361-9230(01)00527-5. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- Ongur D, Jensen JE, Prescot AP, Stork C, Lundy M, Cohen BM, Renshaw PF. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry. 2008;64:718–726. doi: 10.1016/j.biopsych.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowley MP, Drevets WC, Ongur D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry. 2002;52:404–412. doi: 10.1016/S0006-3223(02)01404-X. [DOI] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier W, Hofgen B, Zobel A, Rietschel M. Genetic models of schizophrenia and bipolar disorder: overlapping inheritance or discrete genotypes? Eur Arch Psychiatry Clin Neurosci. 2005;255:159–166. doi: 10.1007/s00406-005-0583-9. [DOI] [PubMed] [Google Scholar]
- Berrettini W. Bipolar disorder and schizophrenia: not so distant relatives? World Psychiatry. 2003;2:68–72. [PMC free article] [PubMed] [Google Scholar]
- Harris LW, Lockstone HE, Khaitovich P, Weickert CS, Webster MJ, Bahn S. Gene expression in the prefrontal cortex during adolescence: implications for the onset of schizophrenia. BMC Med Genomics. 2009;2:28. doi: 10.1186/1755-8794-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LW, Wayland M, Lan M, Ryan M, Giger T, Lockstone H, Wuethrich I, Mimmack M, Wang L, Kotter M. et al. The cerebral microvasculature in schizophrenia: a laser capture microdissection study. PLoS One. 2008;3:e3964. doi: 10.1371/journal.pone.0003964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins B, Martin MV, Sequeira PA, Moon EA, Morgan LZ, Watson SJ, Schatzberg A, Akil H, Myers RM, Jones EG. et al. Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS One. 2009;4:e4913. doi: 10.1371/journal.pone.0004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Menke A, Binder EB. Gene expression studies in major depression. Curr Psychiatry Rep. 2010;12:135–144. doi: 10.1007/s11920-010-0100-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet. 2005;14:241–253. doi: 10.1093/hmg/ddi022. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Bundo M, Washizuka S, Kakiuchi C, Kato T. Expression of HSPF1 and LIM in the lymphoblastoid cells derived from patients with bipolar disorder and schizophrenia. J Hum Genet. 2004;49:227–231. doi: 10.1007/s10038-004-0136-5. [DOI] [PubMed] [Google Scholar]
- Choi KH, Zepp ME, Higgs BW, Weickert CS, Webster MJ. Expression profiles of schizophrenia susceptibility genes during human prefrontal cortical development. J Psychiatry Neurosci. 2009;34:450–458. [PMC free article] [PubMed] [Google Scholar]
- Kim S, Webster MJ. Correlation analysis between genome-wide expression profiles and cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Mol Psychiatry. 2010;15:326–336. doi: 10.1038/mp.2008.99. [DOI] [PubMed] [Google Scholar]
- Klempan TA, Ernst C, Deleva V, Labonte B, Turecki G. Characterization of QKI gene expression, genetics, and epigenetics in suicide victims with major depressive disorder. Biol Psychiatry. 2009;66:824–831. doi: 10.1016/j.biopsych.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Klempan TA, Sequeira A, Canetti L, Lalovic A, Ernst C, ffrench-Mullen J, Turecki G. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol Psychiatry. 2009;14:175–189. doi: 10.1038/sj.mp.4002110. [DOI] [PubMed] [Google Scholar]
- Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, Rehal S, Klempan T, Gratton A, Benkelfat C. et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One. 2009;4:e6585. doi: 10.1371/journal.pone.0006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Elashoff M, Higgs BW, Song J, Kim S, Sabunciyan S, Diglisic S, Yolken RH, Knable MB, Torrey EF, Webster MJ. Putative psychosis genes in the prefrontal cortex: combined analysis of gene expression microarrays. BMC Psychiatry. 2008;8:87. doi: 10.1186/1471-244X-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, Schatzberg AF, Myers RM, Akil H, Watson SJ. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2010. [DOI] [PMC free article] [PubMed]
- Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS. et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004;9:684–697. doi: 10.1038/sj.mp.4001511. 643. [DOI] [PubMed] [Google Scholar]
- Kim S, Choi KH, Baykiz AF, Gershenfeld HK. Suicide candidate genes associated with bipolar disorder and schizophrenia: an exploratory gene expression profiling analysis of post-mortem prefrontal cortex. BMC Genomics. 2007;8:413. doi: 10.1186/1471-2164-8-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MJ, O'Grady J, Kleinman JE, Weickert CS. Glial fibrillary acidic protein mRNA levels in the cingulate cortex of individuals with depression, bipolar disorder and schizophrenia. Neuroscience. 2005;133:453–461. doi: 10.1016/j.neuroscience.2005.02.037. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PC, Yang UC, Shih KH, Liu CM, Liu YL, Hwu HG. A protein interaction based model for schizophrenia study. BMC Bioinformatics. 2008;9(Suppl 12):S23. doi: 10.1186/1471-2105-9-S12-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SA, Chan CH, Chen TC, Yang CY, Huang KC, Tsai CH, Lai JM, Wang FS, Kao CY, Huang CY. POINeT: protein interactome with sub-network analysis and hub prioritization. BMC Bioinformatics. 2009;10:114. doi: 10.1186/1471-2105-10-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell E, Liu WM, Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004;9:406–416. doi: 10.1038/sj.mp.4001437. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TC, Lee SA, Chan CH, Juang YL, Hong YR, Huang YH, Lai JM, Kao CY, Huang CY. Cliques in mitotic spindle network bring kinetochore-associated complexes to form dependence pathway. Proteomics. 2009;9:4048–4062. doi: 10.1002/pmic.200900231. [DOI] [PubMed] [Google Scholar]
- Spirin V, Mirny LA. Protein complexes and functional modules in molecular networks. P Natl Acad Sci USA. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, Mewes HW. CORUM: the comprehensive resource of mammalian protein complexes--2009. Nucleic Acids Res. 2010;38:D497–501. doi: 10.1093/nar/gkp914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A. et al. Human Protein Reference Database--2009 update. Nucleic Acids Res. 2009;37:D767–772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker BH, Koschutzki D, Schreiber F. Exploration of biological network centralities with CentiBiN. BMC Bioinformatics. 2006;7:219. doi: 10.1186/1471-2105-7-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- Hsu DF, Taksa I. Comparing rank and score combination methods for data fusion in information retrieval. Inform Retrieval. 2005;8:449–480. doi: 10.1007/s10791-005-6994-4. [DOI] [Google Scholar]
- Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The Gene Ontology Annotation (GOA) Database: sharing knowledge in Uniprot with Gene Ontology. Nucleic Acids Research. 2004;32:D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- Medina I, Carbonell J, Pulido L, Madeira SC, Goetz S, Conesa A, Tarraga J, Pascual-Montano A, Nogales-Cadenas R, Santoyo J. et al. Babelomics: an integrative platform for the analysis of transcriptomics, proteomics and genomic data with advanced functional profiling. Nucleic Acids Res. 2010;38:W210–213. doi: 10.1093/nar/gkq388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shahrour F, Minguez P, Tarraga J, Medina I, Alloza E, Montaner D, Dopazo J. FatiGO +: a functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucleic Acids Res. 2007;35:W91–96. doi: 10.1093/nar/gkm260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Clyne M, Khoury MJ, Gwinn M. Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics. 2010;26:145–146. doi: 10.1093/bioinformatics/btp618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Howells K, Phillips AD, Mort M, Cooper DN. The Human Gene Mutation Database: providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum Genomics. 2009;4:69–72. doi: 10.1186/1479-7364-4-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, Nopoulos P, O'Leary DS, Miller DD, Wassink T, Flaum M. Defining the phenotype of schizophrenia: cognitive dysmetria and its neural mechanisms. Biol Psychiatry. 1999;46:908–920. doi: 10.1016/S0006-3223(99)00152-3. [DOI] [PubMed] [Google Scholar]
- Koechlin E, Hyafil A. Anterior prefrontal function and the limits of human decision-making. Science. 2007;318:594–598. doi: 10.1126/science.1142995. [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Chandler SD, Bousman CA, Chana G, Lucero GR, Tatro E, May T, Lohr JB, Kremen WS, Everall IP, Tsuang MT. Alternatively spliced genes as biomarkers for schizophrenia, bipolar disorder and psychosis: a blood-based spliceome-profiling exploratory study. Curr Pharmacogenomics Person Med. 2009;7:164–188. doi: 10.2174/1875692110907030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham B. Glutamate and its role in psychiatric illness. Hum Psychopharmacol. 2001;16:139–146. doi: 10.1002/hup.279. [DOI] [PubMed] [Google Scholar]
- Manji HK, Chen G. PKC, MAP kinases and the bcl-2 family of proteins as long-term targets for mood stabilizers. Mol Psychiatry. 2002;7(Suppl 1):S46–56. doi: 10.1038/sj.mp.4001018. [DOI] [PubMed] [Google Scholar]
- Gaspar PA, Bustamante ML, Silva H, Aboitiz F. Molecular mechanisms underlying glutamatergic dysfunction in schizophrenia: therapeutic implications. J Neurochem. 2009;111:891–900. doi: 10.1111/j.1471-4159.2009.06325.x. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Verge B, Alonso Y, Valero J, Miralles C, Vilella E, Martorell L. Mitochondrial DNA (mtDNA) and schizophrenia. Eur Psychiatry. 2011;26:45–56. doi: 10.1016/j.eurpsy.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Clay HB, Sillivan S, Konradi C. Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci. 2011;29:311–324. doi: 10.1016/j.ijdevneu.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry KM, Nimgaonkar VL. Mitochondrial DNA variants in schizophrenia: association studies. Psychiatr Genet. 2000;10:27–31. doi: 10.1097/00041444-200010010-00005. [DOI] [PubMed] [Google Scholar]
- Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. 2011;29:215–223. doi: 10.1016/j.ijdevneu.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. Neuronal-immune interactions in mediating stress effects in the etiology and course of schizophrenia: role of the amygdala in developmental co-ordination. Med Hypotheses. 2011;76:54–60. doi: 10.1016/j.mehy.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Bennett AOM. Stress and anxiety in schizophrenia and depression: glucocorticoids, corticotropin-releasing hormone and synapse regression. Aust N Z J Psychiatry. 2008;42:995–1002. doi: 10.1080/00048670802512073. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Kern AD. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol Biol Evol. 2005;22:803–806. doi: 10.1093/molbev/msi072. [DOI] [PubMed] [Google Scholar]
- El Kasmi KC, Smith AM, Williams L, Neale G, Panopoulos AD, Watowich SS, Hacker H, Foxwell BM, Murray PJ. Cutting edge: A transcriptional repressor and corepressor induced by the STAT3-regulated anti-inflammatory signaling pathway. J Immunol. 2007;179:7215–7219. doi: 10.4049/jimmunol.179.11.7215. [DOI] [PubMed] [Google Scholar]
- Barrenas F, Chavali S, Holme P, Mobini R, Benson M. Network properties of complex human disease genes identified through genome-wide association studies. PLoS One. 2009;4:e8090. doi: 10.1371/journal.pone.0008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamayoshi S, Noda T, Ebihara H, Goto H, Morikawa Y, Lukashevich IS, Neumann G, Feldmann H, Kawaoka Y. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe. 2008;3:168–177. doi: 10.1016/j.chom.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Wang D, Wagner R, Breyssens H, Gertsenstein M, Lobe C, Lu X, Nagy A, Burke RD, Koop BF, Howard PL. ARS2 is a conserved eukaryotic gene essential for early mammalian development. Mol Cell Biol. 2008;28:1503–1514. doi: 10.1128/MCB.01565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavali S, Barrenas F, Kanduri K, Benson M. Network properties of human disease genes with pleiotropic effects. BMC Syst Biol. 2010;4:78. doi: 10.1186/1752-0509-4-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25:528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivleva E, Thaker G, Tamminga CA. Comparing genes and phenomenology in the major psychoses: schizophrenia and bipolar 1 disorder. Schizophr Bull. 2008;34:734–742. doi: 10.1093/schbul/sbn051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E, Owen MJ, O'Donovan MC. Gene-wide analyses of genome-wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry. 2009;14:252–260. doi: 10.1038/mp.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, Craddock N, Jablensky A. The genetic deconstruction of psychosis. Schizophr Bull. 2007;33:905–911. doi: 10.1093/schbul/sbm053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazanfari N, Fernandez KJ, Murata Y, Morsch M, Ngo ST, Reddel SW, Noakes PG, Phillips WD. Muscle specific kinase: organiser of synaptic membrane domains. Int J Biochem Cell Biol. 2011;43:295–298. doi: 10.1016/j.biocel.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Maselli RA, Arredondo J, Cagney O, Ng JJ, Anderson JA, Williams C, Gerke BJ, Soliven B, Wollmann RL. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum Mol Genet. 2010;19:2370–2379. doi: 10.1093/hmg/ddq110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoritaka A, Shimo Y, Inoue Y, Yoshino H, Hattori N. Nonmotor Symptoms in Patients with PARK2 Mutations. Parkinsons Dis. 2011;2011:473640. doi: 10.4061/2011/473640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura Y. The long journey to the discovery of PARK2. Neuropathology. 2010.
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, Arends MJ. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Schot R, Dumee B, Schellekens K, Swagemakers S, Bertoli-Avella AM, Lequin MH, Dudink J, Govaert P, van Zwol AL. et al. Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet. 2009;85:40–52. doi: 10.1016/j.ajhg.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BP, Waddington JL, O'Tuathaigh CM. Advancing a functional genomics for schizophrenia: psychopathological and cognitive phenotypes in mutants with gene disruption. Brain Res Bull. 2010;83:162–176. doi: 10.1016/j.brainresbull.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Shiozawa S, Kawai K, Okada Y, Tomioka I, Maeda T, Kanda A, Shinohara H, Suemizu H, James Okano H, Sotomaru Y, Gene targeting and subsequent site-specific transgenesis at the beta-actin (ACTB) locus in common marmoset embryonic stem cells. Stem Cells Dev. 2011. [DOI] [PubMed]
- Dahlen A, Mertens F, Mandahl N, Panagopoulos I. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12) Biochem Biophys Res Commun. 2004;325:1318–1323. doi: 10.1016/j.bbrc.2004.10.172. [DOI] [PubMed] [Google Scholar]
- Dahlen A, Fletcher CD, Mertens F, Fletcher JA, Perez-Atayde AR, Hicks MJ, Debiec-Rychter M, Sciot R, Wejde J, Wedin R. et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12) Am J Pathol. 2004;164:1645–1653. doi: 10.1016/S0002-9440(10)63723-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestre DC, Gil GA, Tomasini N, Bussolino DF, Caputto BL. Growth of peripheral and central nervous system tumors is supported by cytoplasmic c-Fos in humans and mice. PLoS One. 2010;5:e9544. doi: 10.1371/journal.pone.0009544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral J, Anton AI, Quiroga T, Gonzalez-Conejero R, Pereira J, Roldan V, Vicente V, Mezzano D. Influence of the F12 -4 C>T polymorphism on hemostatic tests. Blood Coagul Fibrinolysis. 2010;21:632–639. doi: 10.1097/MBC.0b013e32833a9048. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kwon EH, Lee KO, Park IA, Kim SH. Novel deleterious mutation in the F12 gene in a Korean family with severe coagulation factor XII deficiency. Blood Coagul Fibrinolysis. 2010;21:683–686. doi: 10.1097/MBC.0b013e32833e429c. [DOI] [PubMed] [Google Scholar]
- Gary T, Hafner F, Froehlich H, Stojakovic T, Scharnagl H, Pilger E, Brodmann M. High factor VIII activity, high plasminogen activator inhibitor 1 antigen levels and low factor XII activity contribute to a thrombophilic tendency in elderly venous thromboembolism patients. Acta Haematol. 2010;124:214–217. doi: 10.1159/000321530. [DOI] [PubMed] [Google Scholar]
- Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C, Kehrer-Sawatzki H. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32:213–219. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- Harder A, Titze S, Herbst L, Harder T, Guse K, Tinschert S, Kaufmann D, Rosenbaum T, Mautner VF, Windt E. et al. Monozygotic twins with neurofibromatosis type 1 (NF1) display differences in methylation of NF1 gene promoter elements, 5' untranslated region, exon and intron 1. Twin Res Hum Genet. 2010;13:582–594. doi: 10.1375/twin.13.6.582. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Linding R. Network-based drugs and biomarkers. J Pathol. 2010;220:290–296. doi: 10.1002/path.2646. [DOI] [PubMed] [Google Scholar]
- Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, Furnari FB, White FM. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A. 2007;104:12867–12872. doi: 10.1073/pnas.0705158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, Linggi B, Kalra A, Paragas V, Bukhalid R. et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- Aydin D, Filippov MA, Tschape JA, Gretz N, Prinz M, Eils R, Brors B, Muller UC. Comparative transcriptome profiling of amyloid precursor protein family members in the adult cortex. BMC Genomics. 2011;12:160. doi: 10.1186/1471-2164-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anliker B, Muller U. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegener Dis. 2006;3:239–246. doi: 10.1159/000095262. [DOI] [PubMed] [Google Scholar]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Abnormally expressed disease genes and functions
Abnormally expressed genes and corresponding probe IDs and P values in t-tests for schizophrenia
Abnormally expressed genes and corresponding probe IDs and P values in t-tests for bipolar disorder
Abnormally expressed genes and corresponding probe IDs and P values in t-tests for major depression
GO functional classification for abnormally expressed genes
FATIGO term enrichment with P values smaller than 0.01
Centrality analysis of abnormally expressed genes in QQPPI network
Interrelationship between Phenopedia and abnormally expressed genes in schizophrenia
Interrelationship between Phenopedia and abnormally expressed genes in bipolar disorder
Interrelationship between Phenopedia and abnormally expressed genes in major depression
Comparison of disease genes listed in Phenopedia, HGMD, and GWAS