

Abstract
Mouse Xin-alpha (mXin-alpha) encodes a Xin repeat-containing, actin-binding protein localized to the intercalated disc (ICD). Ablation of mXin-alpha progressively leads to disrupted ICD structure, cardiac hypertrophy and cardiomyopathy with conduction defects during adulthood. Such conduction defects could be due to ICD structural defects and/or cell electrophysiological property changes. Here, we showed that despite the normal ICD structure, juvenile mXina-null cardiomyocytes (from 3~4-week-old mice) exhibited a significant reduction in the transient outward K+ current (ITO), similar to adult mutant cells. Juvenile but not adult mutant cardiomyocytes also had a significant reduction in the delayed rectifier K+ current. In contrast, the mutant adult ventricular myocytes had a significant reduction in the inward rectifier K+ current (IK1) on hyperpolarization. These together could account for the prolongation of action potential duration (APD) and the ease of developing early afterdepolarization observed in juvenile mXin-alpha-null cells. Interestingly, juvenile mXin-alpha-null cardiomyocytes had a notable decrease in the amplitude of intracellular Ca2+ transient and no change in the L-type Ca2+ current, suggesting that the prolonged APD did not promote an increase in intracellular Ca2+ for cardiac hypertrophy. Juvenile mXin-alpha-null ventricles had reduced levels of membrane-associated Kv channel interacting protein 2, an auxiliary subunit of ITO, and filamin, an actin cross-linking protein. We further showed that mXin-alpha interacted with both proteins, providing a novel mechanism for ITO surface expression
Keywords: Developmental Changes, Electrophysiology, KChIP2, transient outward K+ current, L-type Ca2+ current
2. INTRODUCTION
Striated muscle-specific Xin repeat-containing proteins are downstream targets of transcription factors Nkx2.5 and Mef2, localize to the intercalated disc (ICD) of the heart, and are important in cardiac morphogenesis and function (1–7). In mammals, two families of Xin repeat-containing proteins, Xinα and Xinβ, exist (8). Through alternative splicing of messages, each family of Xin gene encodes at least 2 Xin repeat-containing protein variants (major, mXinα or mXinβ, as well as minor, mXinα-a or mXinβ-a in the mouse) (5, 7, 9). In addition, a third variant (called Xin C) from Xinα gene has been detected in mouse and human heart, however, this variant, if it exists in vivo, did not contain Xin repeats (9). Loss of both mXinα and mXinα-a in the mouse (referred to mXinα-null mouse) heart results in progressive hypertrophy and cardiomyopathy with conduction defects (5). The ICD structural defects in mXinα-null hearts occur between 1 and 3 months of age and progressively worsen with aging (5, 10). Similar ICD alterations were also observed in another mXinα knockout line, which lacked all 3 mXinα variants (referred to XinABC−/− mouse) (11).
The Xin repeats are 16 amino acids repeating units, responsible for binding and bundling actin filaments (9, 12). The mXinα and mXinβ proteins contain 15 and 28 Xin repeats, respectively. mXinα directly interacts with β-catenin (12), and the β-catenin binding domain is mapped to the region overlapping with the Xin repeats of mXinα (8, 12). This interaction provides novel insight into the in vivo ICD localization (1, 4, 12) and function of Xin-containing protein. In addition, Xinα is also capable of binding to Ena/VASP and filamin c (regulatory proteins for actin dynamics) (9), and interacting with p120-catenin and filamin b (12). Therefore, Xin repeat-containing proteins play important roles in linking the N-cadherin-mediated adhesion to the underlying actin cytoskeleton for cardiac structural and functional remodeling.
Previously, we detected a slower conduction velocity and many areas of conduction block associated with both left atrial-pulmonary vein (LA-PV) (13) and ventricle (14) preparations from adult mXinα-null hearts. Interestingly, the induction of atrial fibrillation is consistently hindered in mXinα-null LA-PV preparations even under conditions that enhance its induction in wild-type preparations (13). However, the automatic and triggered rhythms are not suppressed in mXinα-null preparations (13), since their underlying mechanisms do not depend on conduction. Therefore, detailed electrophysiological characterizations on ventricular myocytes could provide information as to whether mXinα-null mice are prone to triggered arrhythmia. Using whole-cell patch-clamp techniques, we have previously detected significant reductions in transient outward K+ currents (ITO) and L-type Ca2+ currents (ICa,L) in mXinα-null ventricular myocytes isolated from 10~20-week-old mice, as compared to age-matched wild-types cells (15). Also, the maximal diastolic potential (MDP) elicited in adult mXinα-null myocytes was significantly reduced, which may render mice prone to developing early afterdepolarization (EAD) and ventricular fibrillation (15). Since mXinα-null hearts at this age already had ICD structural alteration and cardiomyopathy (5), the observed channel remodeling could be either due to the loss of mXinα or due to the secondary effects of abnormal ICD structure and/or cardiomyopathy. Thus, in the present study, we have compared electrophysiological properties of ventricular myocytes prepared from juvenile (3~4-week-old) mXinα-null and wild-type mice. At one-month-old, mXinα-null hearts have shown neither ICD structural defect nor cardiomyopathy (10). We found that both ITO and delayed rectifier K+ currents (IK) but not ICa,L are significantly depressed in juvenile mXinα-null cells. Moreover, the reduction in ITO, but not IK, was continuously detected in adult (10~20-week-old) mXinα-null cells. Using a yeast two hybrid assay, we showed that mXinα was able to bind to Kv channel interacting protein 2 (KChIP2), an auxiliary subunit of ITO channel, and filamin (12), an actin-crosslinking protein. We further found a reduced amount of KChIP2 associated with the membrane fraction of juvenile mXinα-null hearts. These results identify a novel role for mXinα in regulating ITO channel activity through its interaction with KChIP2 and filamin.
3. MATERIALS AND METHODS
3.1. Animals
All animal procedures were approved and performed in accordance with institutional guidelines. The mXinα-null line was generated as previously described and maintained in C57BL/6J. Male mice of each genotype at 1 month-old were processed for electron microscopy as described previously (5). The juvenile (3~4-week-old) and adult (10~20-week-old) wild-type and mXinα-null littermates were used for ventricular myocyte isolation and subsequent electrophysiological characterization. The adult LA-PV cardiomyocytes were obtained from wild-type and mXinα-null mice as described previously (13) and used for intracellular Ca2+ transient study.
3.2. Isolation of cardiomyocytes
The wild-type and mXinα-null littermates were sacrificed under anesthesia with sodium pentobarbital (50 mg/kg, i.p.). LA-PV or ventricular myocytes were isolated enzymatically by means of Langendorff’s preparation perfused with oxygenated Ca2+-free Tyrode solution (120 mM NaCl, 5.4 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 10 mM glucose, 6 mM HEPES, 10 mM taurine, adjusted to pH 7.4 with 1 N NaOH) containing collagenase (Sigma, type I, 1 mg/ml) and protease (Sigma, type XIV, 0.01 mg/ml), as described previously (16). Only cardiomyocytes that had clear striations and could be electrically stimulated were used.
3.3. Cellular electrophysiological study
Electrophysiological properties of single ventricular myocyte were studied with whole-cell patch-clamp techniques by means of an Axopatch 1D amplifier as described previously in detail (16). In brief, the myocytes were placed in a cell bath perfused with Tyrode solution at 35±1 °C containing 137 mM NaCl, 0.5 mM MgCl2, 1.8 mM CaCl2, 5.4 mM KCl, 10 mM glucose, 10 mM HEPES, adjusted to pH 7.4 with 1 N NaOH. Action potentials were detected with the amplifier in current-clamp mode and the pipette was filled with a solution containing 20 mM KCl, 110 mM K aspartate, 1 mM MgCl2, 0.5 mM EGTA, 5 mM Mg2ATP, 5 mM Na2phosphocreatine, 0.1 mM LiGTP, 10 mM HEPES, adjusted to pH 7.2 with 1 N KOH.
For measurement of transient outward K+ currents (ITO) and delayed rectifier outward currents (IK), perfusate containing 200 μM CdCl2 was used. The ITO currents were elicited in two steps from a holding potential of −80 to −40 mV for 30 ms to inactivate Na+ currents (INa) and then to test potentials up to +60 mV in 10 mV increments for 300 ms at a frequency of 0.1 Hz. The IK currents were measured from a holding potential of −40 mV to test potentials ranging from −40 to +60 mV in 10 mV steps for 1,000 ms. The inward rectifier K+ currents (IK1) were determined in the presence of 200 μM Cd2+ and induced on test potentials ranging from −20 to −120 mV from a holding potential of −40 mV. The presence of IK1 current was verified by addition of 1 mM Ba2+, a potent IK1 inhibitor (17) in the bathing solution. The L-type Ca2+ inward currents (ICa,L) were determined on depolarization from a holding potential of −50 mV to test potentials ranging from −40 to +60 mV in 10 mV increments for 300 ms in Na+-free and K+-free perfusate (replaced by TEA and Cs+, respectively) (16).
3.4. Measurement of intracellular calcium concentration
The intracellular Ca2+ concentration was recorded using a fluorimetric ratio technique with Indo-1 fluorescence in cardiomyocytes. The fluorescent indicator Indo-1 was loaded by incubating the ventricular myocytes at room temperature for 20~30 min with 10 μM of Indo-1/AM (Sigma Chemical, St Louis, MO, USA) (18). The cardiomyocytes were then perfused with the Tyrode solution at 35±1 °C for at least 20 min to wash out the extracellular indicator and to allow for the intracellular de-esterification of the Indo-1/AM. The background and cell autofluorescence were cancelled out by zeroing the output of the photomultiplier tubes using cells without Indo-1 loading. The experiments were performed at 35±1 °C. An UV light of 350nm with a monochromator was used for the excitation of the Indo-1 from a xenon arc lamp controlled by the microfluorimetry system (OSP100-CA, Olympus, Tokyo, Japan) and the excitation light beam was directed into an inverted microscope IX-70 (Olympus). The emitted fluorescence signals from the Indo-1/AM loaded myocytes were digitized at 200 Hz. The ratio of the fluorescence emission at 410 and 485nm (R410/485) was used as the index of the intracellular Ca2+ concentration (18). This approach avoided uncertainties from the calibration of the fluorescent Ca2+ indicators. The intracellular Ca2+ transient, peak systolic and diastolic intracellular Ca2+ concentrations were measured during a 2 Hz field-stimulation with 10-ms twice-threshold strength square-wave pulses. The intracellular Ca2+ transient was calculated from the difference of the peak systolic and diastolic intracellular concentrations. The fluorescence ratio data were processed and stored in a computer using software OSP-SFCA (Olympus).
As previously reported (19), LA cardiomyocytes and PV cardiomyocytes without spontaneous activity of adult rabbit hearts have indistinguishable intracellular Ca2+ sparks/transients. Thus, we prepared LA-PV cardiomyocytes from adult wild-type and mXinα-null mice and used for Ca2+ transients with a laser scanning confocal microscope (Zeiss LSM 510) as described previously (19). Briefly, cells were loaded with 10 μM of Fluo-3/AM for 30 minutes at room temperature. The excess extracellular dye was removed by changing the bathing solution and allowing for the intracellular hydrolysis of the Fluo-3/AM after 30 minutes. The Fluo-3 fluorescence was excited with a 488nm line of an argon ion laser. The emission was recorded at >515nm. Cells were stimulated with a 2 Hz field-stimulation with 10-ms twice-threshold strength square-wave pulses and repetitively scanned at 3-ms intervals for a total duration of 6 s. The intracellular Ca2+ concentration was presented as background-subtracted normalized fluorescence (F/F0) where F was the fluorescence intensity and F0 the resting fluorescence recorded under steady-state conditions.
3.5. Northern blot analysis and quantitative RT-PCR (qRT-PCR)
Total RNAs were isolated from freshly dissected hearts of 3 sets of adult wild-type and mXinα-deficient littermates using TRI Reagent RNA isolation kit (Molecular Research Center, Inc., Cincinnati, OH, USA) as described previously (1). Ten μg of total RNA from each sample were used in Northern blot analysis. DNA probes including KChIP2 cDNA (20), mXinα cDNA (1), and GAPDH cDNA were labeled with α-32P-dATP using a random primed DNA labeling kit (Boehringer Mannheim, Indianapolis, IN). Northern blot experiments were then performed as described previously (1).
The qRT-PCR was carried out using ABI Prism 7000 sequence detection system and SYBR Green reagents from Applied Biosystems (Foster City, CA). Primer pairs were designed using Primer Express version 2.0 (Applied Biosystems): CTCCAGGGCCCAGTAAAAAG and GCACGGCAGCAGCTTGA for KChIP2 and TCAAGAAGGTGGTGAAGCAG and ACCACCCTGTTGCTGTAGCC for GAPDH. The RT reaction mixture containing 5 μg of total RNA (DNA-free), 0.5 μg Oligo (dT), 1 unit RNasin, 50 mM Tris-HCl pH8.3, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 0.5 mM dNTPs, and 200 units of Superscript II reverse transcriptase in 20 μl was incubated at 42 °C for 50 min to convert into cDNA and then inactivated by heating at 70 °C for 15 min. The cDNA mixture was further diluted 5–10 times with H2O. For amplification in PCR, each reaction in 20 μl (1 μl each of diluted cDNA template, 5 pmoles each of forward and reverse primers and 1X SYBR Green master mix) was performed in triplicate in clear 96-well plates at 95 °C, 10 min; then 95 °C, 15 seconds, and 60 °C, 1 min, for 40 cycles. The internal control for qRT-PCR was GAPDH from each ventricle sample. The 2−ΔΔCt method was used to calculate relative changes in gene expression determined from qRT-PCR experiments (21–23).
3.6. Membrane preparation and Western blot analysis
Total membranes were prepared from ventricles of mouse littermates at 1 month of age by using a protocol of Barry et al (24). All procedures were performed at 4 °C, and all solutions contained complete protease inhibitor cocktail (cat#11836145001, Roche Applied Sciences). Briefly, dissected ventricles were minced, diluted in 10 volume of TE (20 mM Tris-HCl and 1 mM EDTA, pH 7.4), and homogenized with a Pro200 homogenizer with Multi-Gen 7 generators (Pro Scientific Inc). After centrifugation at 1,000×g for 10 min, the pellet was washed once with TE. The supernatants from both low-speed spins were pooled and further centrifuged at 40,000×g for 10 min. The pellet was resuspended in TE containing 0.6 M KI and incubated on ice for 10 min. After centrifugation at 40,000×g for 10 min, the supernatant was labeled as “cytosolic” fraction, and the resulting pellet was washed twice in TE to ensure completely removing KI. The final pellet was then labeled as “membrane” fraction. Aliquots of 1,000×g supernatants and pellets as well as cytosol and membrane fractions were solubilized in SDS-PAGE sample buffer and analyzed by Western blot.
Western blot analysis was performed as described previously with ECL detection system (5). The primary antibodies included rabbit polyclonal anti-mXin proteins (U1013), and mouse monoclonal anti-KChIP2 (UC Davis/NIH NeuroMab Facility), anti-Kv4.2 (NeuroMab), anti-Kv4.3 (NeuroMab), anti-filamin (Zymed), anti-N-cadherin (Zymed) and anti-β-tubulin (DM1B, Sigma).
3.7. Yeast two hybrid interaction assay
The interaction between mXinα and KChIP2 was tested by the Matchmaker yeast two-hybrid assay (Clontech, Palo Alto, CA) as described previously (12). The generation of the bait plasmid containing full-length mXinα cDNA was previously described (12). The full-length cDNAs for KChIP2 (20), p120 catenin (12), and α-catenin (a generous gift from Dr. W. J. Nelson) were separately subcloned into the pGADT7 plasmids and used as preys. The α-catenin and p120 catenin prey plasmids were used as negative and positive controls, respectively. After transformation with both bait and prey plasmids, yeast AH109 (trp1-901, leu2-3, his3-200, ura3-52, GAL1UAS-HIS3, GAL2UAS-ADE2, and URA3::MEL1UAS-lacZ) cells were selected on Trp/Leu double dropout (DDO) plate to verify the presence of both plasmids. To test for the interaction, cells were subsequently replicated and selected on Trp/Leu/His triple dropout (TDO), Trp/Leu/His/Ade quadruple dropout (QDO) and QDO plus X-gal plates. If there is no interaction between bait and prey, cells will show no growth on TDO and QDO selective plates and no expression of β-galactosidase on X-gal plate.
3.8 Data analysis
Data were presented as mean±SEM. ANOVA test, Student’s t test or Mann-Whitney Rank Sum test was used to test the significance between wild type (mXinα+/+) and homozygous (mXinα−/−) cardiomyocytes for statistical analysis. A p value ≦ 0.05 was considered statistically significant.
4. RESULTS
4.1. Prolonged action potential duration (APD) and higher incidence of early afterdepolarization (EAD) were associated with both juvenile and adult mXinα-null ventricular myocytes
At 1 month of age, the intercalated disc (Figure 1B) and sarcomere (Figure 1D) organizations of the mXinα-null heart did not appear to be significantly different from that of age-matched wild type heart (Figure 1A and C), except less electron-density was detected around the mutant disc. In contrast, we have previously shown that a separation of the membrane faces of the intercalated disc and myofibril disarray in the region surrounding the disc were readily detected in mXinα-null heart by 3 months of age (5). These defects were even greater by 6 months of age as observed in both heterozygous and mXinα-null hearts, leading to hypertrophy and cardiomyopathy (5). At 1 month of age, the mXinα-null cardiomyocytes appeared to have the same cell size, as measured by membrane capacitance (Table 1), as the age-matched wild type cardiomyocytes. Therefore, at the juvenile age, mXinα-null hearts appeared to have neither structural defects nor hypertrophy.
Figure 1.

Transmission electron microscopy. Comparisons of wild-type (A, C) and mXinα-null mouse hearts (B, D) at 1 month of age demonstrate no significant difference in the intercalated disc (A, B) and sarcomere (C, D) organizations, except less electron-density detected in mXinα-null ICD. Bar = 0.5 μm for A and B; 1 μm for C and D.
Table 1.
Action potential characteristics of juvenile and adult wild type and mXinα−/− ventricular myocytes
| Juvenile (3~4-week-old) | Adult (10~20-week-old) | |||
|---|---|---|---|---|
| mXinα+/+ | mXinα−/− | mXinα+/+ | mXinα−/− | |
| Cm, pF | 75.5±2.7 (n=47) | 72.6±3.5 (n=58) | 102.2±4.6 (n=113) | 134.3±5.6* (n=104) |
| MDP, −mV | 70.5±1.4 (n=15) | 70.2±1.1 (n=17) | 77.0±1.4 (n=17) | 60.2±2.5* (n=10) |
| APA, mV | 85.9±4.9 (n=15) | 81.7±1.1 (n=17) | 91.9±2.9 (n=17) | 83.3±3.0 (n=10) |
| APD20, ms | 3.1±0.1 (n=15) | 4.1±1.0* (n=17) | 5.9±0.8 (n=17) | 9.2±1.6# (n=10) |
| APD50, ms | 9.0±0.6 (n=15) | 11.6±2.9* (n=17) | 20.0±4.2 (n=17) | 34.5±7.8 (n=10) |
| APD90, ms | 37.1±2.7 (n=15) | 46.8±3.5* (n=17) | 167.4±8.4 (n=17) | 213.6±31.0 (n=10) |
The cardiomyocytes were driven electrically at 1 Hz. Cm, membrane capacitance; MDP, maximum diastolic potential; APA, action potential amplitude; APD20, APD50 and APD90, action potential duration at 20%, 50% and 90% repolarization levels, respectively. Values are mean±SEM. Number in parenthesis, number of ventricular myocytes tested;
p<0.05, significantly different from wild type control values by Student’s t test or
p<0.05 by Mann-Whitney Rank Sum test.
Action potentials of ventricular myocytes prepared from juvenile wild-type and mXinα-null hearts were elicited by supra-threshold electrical stimulation at a frequency of 1 Hz. As illustrated in Figure 2 and Table 1, action potentials of juvenile mXinα−/− cardiomyocytes had a longer APD at 20, 50 and 90% repolarization levels as compared to those obtained from juvenile mXinα+/+ controls. The prolonged APDs at 20% repolarization, and a trend of increased APDs at 50 and 90% repolarization levels, were also observed in adult mXinα-null ventricular myocytes (Table 1). However, the longer APD was not accompanied by a larger membrane capacitance (Cm) in juvenile mXinα−/− cardiomyocytes (Table 1). In contrast, adult mXinα-null cardiomyocytes exhibited larger membrane capacitance (Table 1), suggesting that cardiac hypertrophy is an adult late-onset process.
Figure 2.

Action potential configurations of the typical juvenile wild-type (mXinα+/+) and mXinα−/− mouse ventricular myocytes. Panels A and B illustrate typical tracings of action potentials in mXinα+/+ and mXinα−/− ventricular myocytes, respectively, driven electrically at 1 Hz. Panel C shows the superimposed action potentials of panels A and B. Panel D illustrates another mXinα−/− ventricular myocyte developing a series of early afterdepolarizations (EAD) after a driven action potential. Upward arrows underneath the first driven action potential in each panel indicate time of electrical stimulation.
As shown in Figure 2B and 2C, there was a hindrance of phase 3 repolarization associated with APD90 prolongation in mXinα−/− ventricular myocytes. This delay in repolarization could lead to the development of EAD and the subsequent spontaneous rhythms (Figure 2D) observed in some mXinα−/− myocytes. Similar EAD was observed in 5 of 17 (29%) juvenile mXinα−/− ventricular myocytes, but less frequently in juvenile mXinα+/+ ventricular myocytes (in 2 of 15, 13%). Previously, a relatively high frequency in the development of EAD was also observed in adult mXinα-null ventricular myocytes (15). Although the maximum diastolic potential (MDP) of juvenile mXinα-null ventricular myocytes was not different from age-matched wild-type cells, the MDP of adult mXinα-null ventricular myocytes was significantly reduced (Table 1), which may further contribute to the observed high incidence of the EAD development (15).
4.2. Depressed transient outward currents (ITO) were detected in both juvenile and adult mXinα-null ventricular myocytes
The ITO currents were recorded on depolarization of ventricular myocytes from a holding potential of −80 mV and a prepulse to −40 mV for 300 ms then to +60 mV in 10 mV increments. As shown in Figure 3(I)A and 3(I)B, the ITO currents were significantly reduced in size in juvenile mXinα−/− ventricular myocytes as compared to the ITO of age-matched wild-type ventricular myocytes. Figure 3(I)C shows the voltage-current density plot from recording 12 juvenile wild-type ventricular myocytes and 16 juvenile mXinα-null ventricular myocytes. Clearly, the ITO current densities at various pulsed voltages were significantly depressed in juvenile mXinα-null cardiomyocytes. At this juvenile stage, the mXinα-null ventricular myocytes showed neither hypertrophy (Table 1) nor ICD structural defect (Figure 1). Therefore, the reduction in ITO current density is a direct result of the loss of mXinα in ventricular myocytes and independent of structural defects or hypertrophy. The depressed ITO currents were also detected in adult mXinα−/− ventricular myocytes, as compared to age-matched control cells (Figure 3(II)). This is consistent with our previous observation of the reduced ITO in adult mXinα-null ventricular myocytes (15).
Figure 3.

The ITO current densities of both juvenile and adult mXinα-null ventricular myocytes are significantly depressed as compared to those of wild-type counterparts. Membrane currents were elicited on depolarization from a holding potential of −80 mV to a test potential from −40 to +60 mV. Examples of transient outward currents (ITO) recorded in the (I) juvenile and (II) adult mXinα+/+ (Panel A) and mXinα−/− (Panel B) ventricular myocytes. Inset, various clamp protocols. Panel C summarizes current density-voltage (mean±SEM) relationships of ITO. n, Number of ventricular myocytes tested. * p<0.05, significant difference between wild-type and mXinα-null ventricular myocytes.
4.3. A small but significant reduction in the delayed rectifier outward K+ currents (IK) was observed in juvenile but not adult mXinα-null ventricular myocytes
Another K+ current, IK, near the end of depolarizing pulse (indicated by arrowheads in Figure 4) can also affect the duration of repolarization of an action potential, leading to the prolongation of APD. The IK current density of juvenile mXinα−/− ventricular myocytes was modestly reduced, which was statistically significant (Figure 4(I)C). On the other hand, the IK of adult mXinα-null ventricular myocytes was not different from that of age-matched control ventricular myocytes (Figure 4(II)).
Figure 4.

The delayed rectifier outward K+ current densities of juvenile but not adult mXinα-null ventricular myocytes are significantly depressed as compared to those of wild-type counterparts. Membrane currents were elicited on depolarization from a holding potential of −40 mV to a test potential from −40 to +60 mV. Examples of delayed rectifier K+ currents (indicated by downward solid triangles, IK) recorded in the (I) juvenile and (II) adult mXinα+/+ (Panel A) and mXinα−/− (Panel B) ventricular myocytes. Inset, various clamp protocols. Panel C summarizes current density-voltage (mean±SEM) relationships of IK. n, Number of ventricular myocytes tested. * p<0.05, significant difference between juvenile wild-type and mXinα-null ventricular myocytes.
4.4. No changes in the inward rectifier K+ currents (IK1) and the L-type Ca2+ currents (ICa,L) were detected in juvenile mXinα-null ventricular myocytes, but both current densities were consistently reduced in adult mutant myocytes
In addition to voltage-gated K+ (Kv) currents ITO and IK, the background inward rectifier K+ current (IK1 or IKir) that setup a negative resting membrane potential can also contribute to myocyte action potential repolarization (17). Therefore, we analyzed and compared the Ba2+-sensitive IK1 currents in wild type and mutant cardiomyocytes. As illustrated in Figure 5(I), there was no significant change in the IKI from cardiomyocytes of juvenile wild-type and mXinα-null mice in the potential range from −20 mV to −120 mV. However, the current density of Ba2+-sensitive IK1 was consistently reduced in the adult mXinα-null cardiomyocytes at potential range from −90 to −120 mV (p<0.05) (Figure 5(II)C). Note the consistent reduction in IK1 was observed only in adult ventricular myocytes, in agreement with change in MDP of action potential shown in Table 1.
Figure 5.

Ba2+-sensitive inward rectifier K+ current (IK1) in juvenile and adult ventricular myocytes. Top panel (I) illustrates examples of a series of IK1 currents elicited on hyperpolarization in a wild-type (mXinα+/+) (A) and a mXinα-null myocyte (B). The clamp protocol is shown below the calibration bar. The differences in current densities before and after Ba2+ (mean ± SEM) were then plotted against the test potentials to obtain current-voltage relationship curves. No change in the IK1 was detected in juvenile mXinα-null ventricular myocytes (I) as compared to juvenile wild-type myocytes (C). In contrast, the IK1 currents of adult mXinα-null ventricular myocytes (II) were significantly depressed at test potentials more negative than −90 mV as compared to the wild-type counterparts (II). * p<0.05, significant difference between wild-type and mXinα-null ventricular myocytes.
The phase 2 of cardiac action potential is a much slower rate of repolarization. This not only ensures adequate time for Ca2+ entry via ICa,L channels into the myocyte for optimum excitation-contraction coupling, but also makes cardiac muscle refractory to premature excitation. The important components of the phase 2 repolarization are the slow delayed rectifier K+ currents (portion of IK) and the inward ICa,L currents (25). To study whether the ICa,L currents changed in mXinα-null ventricular myocytes, we performed whole-cell patch-clamp experiments in the presence of TEA and Cs+ to block both outward and inward K+ currents. Under this condition, the depolarizing steps (from −40 to +60 mV in 10 mV increments) induced only voltage-dependent L-type inward Ca2+ currents (ICa,L). As shown in Figure 6(I)C, both juvenile wild-type and mXinα-null ventricular myocytes had very similar voltage-current density curves. The peaks of ICa,L current density for both ventricular myocytes occurred at the depolarizing step of +10 mV. In contrast, the ICa,L of adult mXinα-null ventricular myocytes was significantly smaller (~40% reduction at the peak) than that of adult wild-type cells (Figure 6(II)). This reduction in ICa,L of adult mXinα-null cardiomyocytes may be a consequence of ICD structural defects and/or cell hypertrophy.
Figure 6.

The ICa,L current densities of juvenile mXinα-null ventricular myocytes are not significantly different from that of wild type counterparts, whereas the ICa,L current densities of adult mXinα-null ventricular myocytes are significantly reduced. Membrane currents were elicited on depolarization from a holding potential of −50 mV to a test potential from −40 to +60 mV. Examples of L-type Ca2+ current (ICa,L) recorded in the (I) juvenile and (II) adult mXinα+/+ (Panel A) and mXinα−/− (Panel B) ventricular myocytes. Inset, various clamp protocols. Panel C summarizes current density-voltage (mean±SEM) relationships of ICa,L. n, Number of ventricular myocytes tested. * p<0.05, significant difference between wild-type and mXinα-null ventricular myocytes. Note the current density-voltage curves of ICa,L originated from both mutant and wild type myocytes are peaked near +10 mV.
4.5. Intracellular Ca2+ concentration was significantly decreased in both juvenile and adult mXinα-null cardiomyocytes
We had previously imaged the Ca2+ transients as an index of the intracellular Ca2+ concentration by the confocal laser scanning microscopy (26) in adult LA-PV cardiomyocytes loaded with Fluo-3 fluorescence. Examples of the fluorescence (F/F0) tracings of the Ca2+ transients are shown in Figure 7A for a mXinα+/+ and a mXinα−/− LA-PV cardiomyocyte. The amplitude in adult mXinα-null LA-PV cells appeared to be smaller than that of wild type counterparts. The average amplitudes (F/F0: 0.5±0.1) and decay time (τ: 85.1±13.3 ms) of the Ca2+ transients obtained from 7 adult LA-PV mXinα-null cardiomyocytes were significantly reduced, as compared to wild type counterparts (F/F0: 1.6±0.5 and τ: 220.6±56.4 ms). Similar findings were also observed in juvenile ventricular myocytes loaded with Indo-1 fluorescence and analyzed by ratio fluorimetric technique. Figure 7B and 7C show the tracings of the intracellular Ca2+ transients in juvenile and adult, respectively, wild type and mXinα-null ventricular myocytes. The amplitude of the Ca2+ transient measured as Indo-1 fluorescence ratio at 410nm and 485nm (R410/485) was used as an index of intracellular Ca2+ concentration (18). Results from analyzing 7~13 ventricular myocytes again revealed a significant reduction of Ca2+ transient in juvenile mXinα-null ventricular myocytes (Figure 7D) and adult myocytes (Figure 7E). Thus, two different methods of Ca2+ transient measurements gave similar results, suggesting that the mXinα-null cardiomyocytes had a significant decrease in the intracellular Ca2+ concentration.
Figure 7.
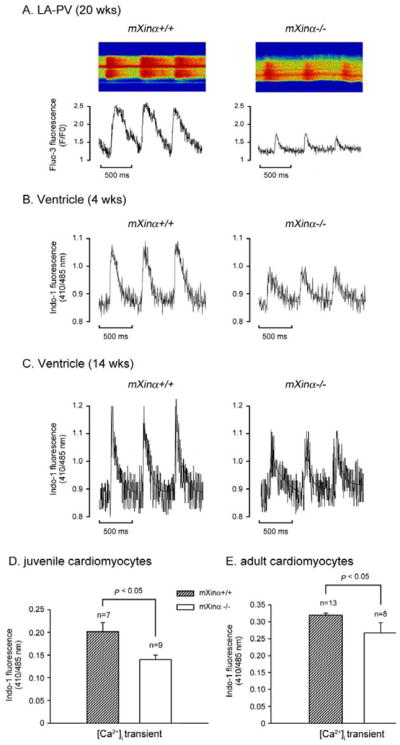
Intracellular Ca2+ concentration was decreased in both juvenile and adult mXinα-null cardiomyocytes. Panel A illustrates typical examples of Ca2+ transient in a wild type and a mXinα-null LA-PV cardiomyocytes (20-wks-old) as detected by Fluo-3 fluorescence (F/F0). Panels B and C illustrate examples of Ca2+ transient in juvenile (4 wks-old) and adult (14-wks-old) ventricular myocytes of wild type and mXinα-null mice, as detected by the Indo-1 fluorescence ratio (R410/485). Panels D and E summarize the average amplitudes of Ca2+ transients (R410/485) in juvenile (n = 7~9) and adult ventricular myocytes (n = 8~13). *p<0.05, significant difference in Ca2+ transient between wild-type and mXinα-null cells.
4.6. The KChIP2 message was significantly decreased in adult mXinα-deficient hearts
In addition to conduction defects, as detected in adult mXinα-null hearts by ECG recordings (5) and by optical mapping on ventricle and LA-PV preparations with a MED64 multi-electrode system (13, 14), juvenile mXinα-null ventricular myocytes already exhibited significantly reduced ITO and IK current densities (Figure 3 and 4). Therefore, it is possible that the loss of mXinα would result in altered ion channel expression in cardiomyocytes. Toward this end, we first carried out qRT-PCR for analyzing the changes in expression of many known channel components. The results showed 23% and 41% decreases in relative expression of KChIP2, an auxiliary subunit of ITO, in heterozygous and mXinα-null hearts, respectively, as compared to the wild type hearts (p<0.05). The Northern blot analysis further confirmed that mXinα-null ventricles expressed significantly reduced levels of KChIP2 messages (Figure 8).
Figure 8.
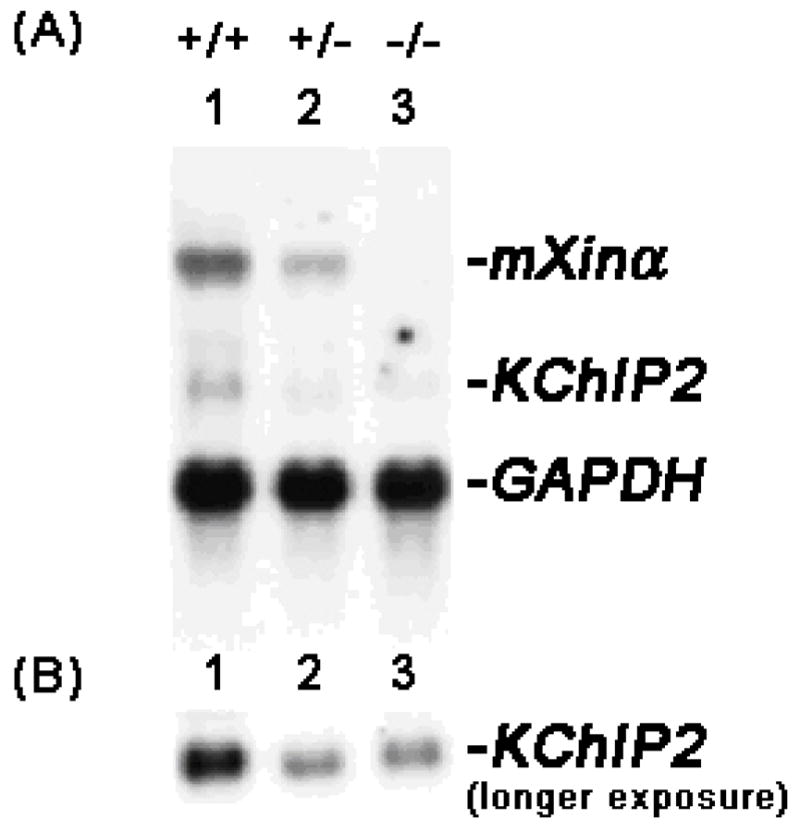
Down-regulation of KChIP2 in mXinα-deficient mouse hearts. (A) Total RNAs (10 μg each lane) prepared from wild-type (+/+, lane 1), heterozygous (+/−, lane 2) and homozygous (−/−, lane 3) mXinα mouse hearts were blotted and hybridized with a mixture of 32P-labeled mXinα cDNA and KChIP2 cDNA probes. For the RNA loading control, the same blot was further hybridized with GAPDH cDNA probe. (B) A longer exposure of the same Northern blot was shown to reveal the KChIP2 message band in mXinα-deficient hearts.
4.7. The membrane-associated KChIP2 and filamin proteins were reduced in juvenile mXinα-null hearts
Nkx2.5 heterozygous mice (27) with depressed ITO are susceptible to the induction of cardiac tachycardia. Similar tachycardia phenotype is also observed in KChIP2-null mice with complete loss of ITO (20). The studies of KChIP2-deficient mice further reveal that the KChIP2 expression level can quantitatively determine the ITO current density in the ventricles. Since juvenile mXinα-null cardiomyocytes without ICD structural defects already expressed reduced ITO current density (Figure 3), we then asked whether these mutant cells would express a decreased amount of KChIP2 protein and/or Kv4.2/4.3 α-subunit of ITO channel in their membrane fraction. Using previously established method of subcellular fractionation (24), we prepared membrane and cytosolic fractions from juvenile hearts and characterized them by Western blot analysis. As shown in Figure 9, both KChIP2 and filamin (indicated by arrows) associated with the membrane fraction of juvenile mXinα-null heart were significantly reduced, as compared to that from age-matched wild-type and heterozygous hearts. In contrast, both Kv4.2 and Kv4.3 in this membrane fraction (40,000×g pellet after KI extraction) of mXinα-null hearts showed no reduction. Similarly, there were no significant changes in N-cadherin and β-tubulin detected in the membrane of juvenile mXinα-null heart (Figure 9). Therefore, the complete loss of mXinα results in the reductions of membrane-associated KChIP2 and filamin, which likely accounts for the observed decrease in ITO current density in juvenile mXinα-null hearts.
Figure 9.

Juvenile mXinα-null ventricles exhibited significant decreases in both membrane-associated KChIP2 and filamin proteins (indicated by arrows). Total ventricular extracts were prepared as described under Materials and Methods in a low salt buffer, and spinned at 1,000×g centrifugation. The 1,000×g pellet contained nuclei, debris and low salt-resistant protein aggregates. The membrane vesicles, myofibrils and organelles in the 1,000×g supernatant (sup’t) were collected by 40,000×g centrifugation. After extracted with 6 M KI to remove myofibrils, the 40,000×g pellet and sup’t were termed “membrane” and “cytosolic” fractions, respectively. Western blot analysis was performed on these fractions prepared from ventricles of wild type (+/+, lane 1), heterozygous (+/−, lane 2) and homozygous (−/−, lane 3) littermates at 1 month of age. An equivalent volume from each fraction was loaded onto the SDS-PAGE gel, except 10 times concentrated sample of membrane fraction was used, and analyzed by Western blot with antibodies including U1013, anti-KChIP2, anti-Kv4.2, anti-Kv4.3, anti-filamin, anti-N-cadherin and anti-β-tubulin. The majority of Kv4.2 and Kv4.3, α-subunits, of ITO channel together with most of N-cadherin were detected in the membrane fraction, whereas most of β-tubulin was found in the cytosolic fraction.
After low speed centrifugation, the low salt (TE) buffer effectively extracted about 90% of β-tubulin into the 1,000×g supernatant (sup’t), whereas only <10~20% of N-cadherin and filamin could be extracted (Figure 9). In addition to N-cadherin and filamin, the 1,000×g pellet fraction contained about 50% of mXinα and mXinα-a, as well as the majority of mXinβ (Figure 9). After KI extraction and 40,000×g centrifugation, the membrane pellet contains ITO channel components as well as mXinα and mXinα-a (Figure 9).
4.8. mXinα interacts with KChIP2 and filamin
KChIP2, the only KChIP family of proteins expressed in the heart, directly interacts with both Kv4.2 and Kv4.3 and regulates the expression of ITO currents (20, 28–30). It has also been shown that the C-terminus of Kv4.2 directly interacts with actin-binding protein, filamin, and that this interaction is essential for the generation of appropriate ITO current density (31). We (12) and others (9) have previously demonstrated that mXinα can also interact with both filamin and actin filaments. As described above, the juvenile mXinα-null hearts expressed reduced amounts of both membrane-associated filamin and KChIP2 proteins and reduced ITO current density. Therefore, mXinα likely plays an important role in the assembly and/or surface expression of ITO channel, possibly through its interaction with KChIP2 and filamin. Using a yeast two hybrid assay (Figure 10), we showed that the interaction between full-length mXinα bait and full-length KChIP2 prey rendered the yeast cells to be able to grow on TDO and QDO selective plates and to express β-galactosidase activity (blue color) on QDO with X-gal plate. Under the same condition, the α-catenin prey as a negative control did not interact with mXinα and no growth was observed on TDO and QDO selective plates. On the other hand, the positive control prey, p120-catenin, readily interacted with mXinα, and supported cell growth on selective plates as well as the expression of β-galactosidase, as previously demonstrated (12).
Figure 10.
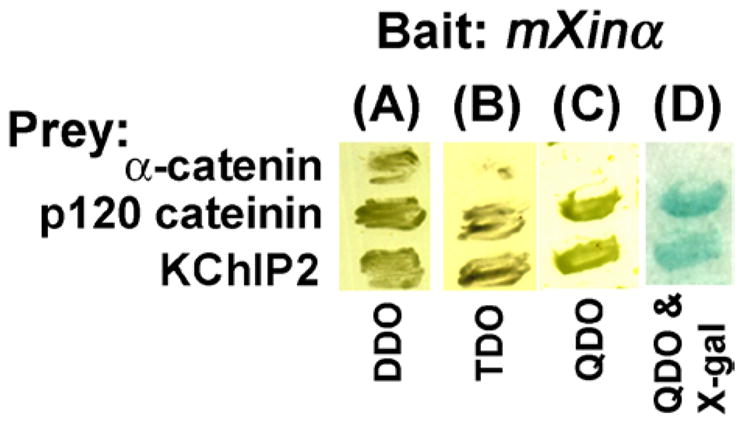
The yeast two hybrid assays showed the interaction of mXinα with KChIP2. The α-catenin and p120 catenin preys were used in the same assay condition as a negative and a positive control, respectively. Yeast cells after transformation with both bait and prey plasmids were grown on double dropout (DDO) plate. Only the interaction occurring between bait and prey would support the cell growth on triple (TDO) and quadruple (QDO) dropout plates as well as the expression of β-galactosidase (blue color) on QDO & X-gal plate.
5. DISCUSSION
5.1. Reductions in ITO and IK of juvenile mXinα-null cardiomyocytes were primarily responsible for the prolonged APD and higher incidence of EAD
In the present study, we show that the complete loss of mXinα in cardiomyocytes leads to depressed ITO current density. This ITO depression is detected in cells prepared from both juvenile and adult mXinα-null hearts, independently of ICD structural defects and cardiomyopathy. On the other hand, a depressed ICa,L current density is only associated with adult mXinα-null cardiomyocytes but not with juvenile mXinα-null cells. Since the ICD structural defect can be detected in adult mutant hearts (5, 10), the ICa,L depression may be the secondary consequences of the altered ICD structure. The IK current density of juvenile mXinα-null cardiomyocytes is also reduced but to a lesser extent. However, the IK in adult wild-type and mXinα-null cardiomyocytes show no significant difference. These results suggest that the complete loss of mXinα may slightly impede the maturation of IK channel assembly during development. The reductions in ITO and IK of juvenile mXinα-null cardiomyocytes likely slow down the repolarization process and result in prolonged APD and a higher incidence of EAD. On the other hand, no difference in ICa,L between juvenile wild-type and mXinα-null cardiomyocytes and the reduced amplitude of Ca2+ transients in juvenile mXinα-null cardiomyocytes suggests that there was no Ca2+ overload in these mutant cardiomyocytes. Thus, it is likely that juvenile mutant cells also lack the delay afterdepolarization (DAD), as we have preliminarily observed for adult LA-PV cardiomyocytes. The DAD is an indication of intracellular Ca2+ overload (32). Although the underlying cellular mechanisms are different, both DAD and EAD could lead to triggered arrhythmias (13–15, 33).
5.2. Reduced ITO and prolonged APD did not cause cardiac hypertrophy in juvenile mXinα-null hearts
In many animal models of cardiac hypertrophy and human heart failure, hypertrophied myocytes undergo K+ channel remodeling that causes a prolongation of APD (34–38). A common target of K+ channel alteration is the depression of ITO in ventricular myocytes. However, the causal relationship between reduced ITO and cardiac hypertrophy remains a controversial issue (34). Most studies with ITO α-subunit (39, 40) and KChIP2 (20) knockout mice demonstrate that a decrease in ITO does not necessarily lead to hypertrophy or cardiomyopathy. Instead, the prolonged APD caused by reduced ITO leads to conduction defects/arrhythmias. However, other studies in cultured cardiomyocytes and in transgenic mice expressing dominant-negative Kv4.2 reveal that reduced ITO causes prolonged APD, which leads to hypertrophy and cardiomyopathy (41, 42) via a Ca2+/calcineurin-dependent signaling pathway (34). In the present study, we show that juvenile mXinα-null cardiomyocytes exhibit reduced ITO and prolonged APD (Figure 2 and 3). However, these cells have neither hypertrophy nor increases in ICa,L and intracellular Ca2+ transient (Table 1 and Figure 6 and 7). Although the cardiac hypertrophy and cardiomyopathy can be detected in adult mXinα-null hearts (5, 15), the ICa,L of adult mutant ventricular myocytes (Figure 6) and the intracellular Ca2+ transient in adult mutant LA-PV myocytes (Figure 7A) remained depressed. These results strongly support that the reduced ITO and prolonged APD in mXinα-null cardiomyocytes are not responsible for cardiac hypertrophy, instead, for conduction defects and arrhythmias.
5.3. Molecular mechanisms underlying the depressed ITO in mXinα-null cardiomyocytes
In mice, the major components of ITO channel are pore-forming α-subunits (Kv4.2 and 4.3), β-subunit (Kvβ1) and an auxiliary subunit (KChIP2) (30, 43). The Kv4 α-subunit is known to mediate the α-subunit interaction (44), and the binding of filamin (31), Kvβ1 (30) and KChIP2 (28, 30). The association of KChIP2 with Kv4.2 enhances the surface expression of Kv4.2 (45), although the molecular mechanisms remain unclear. In the present study, we show down-regulation of the KChIP2 message and membrane-associated KChIP2 protein in mXinα-null hearts. Yeast two-hybrid interaction assays reveal that mXinα directly interacts with KChIP2. Previous studies also show that mXinα directly binds to filamin and actin filaments (9, 12). Together, we propose a model illustrating how mXinα regulates surface expression and activity of ITO channel (Figure 11). Through its interactions with KChIP2 and filamin, mXinα stabilizes both proteins and then enhances ITO current density. Filamin is an actin cross-linking protein, whereas mXinα is capable of binding and bundling actin filaments. Thus mXinα can affect the filamin’s cross-linking activity and/or coordinate with filamin’s activity to provide actin dynamics for surface expression of the ITO channel.
Figure 11.
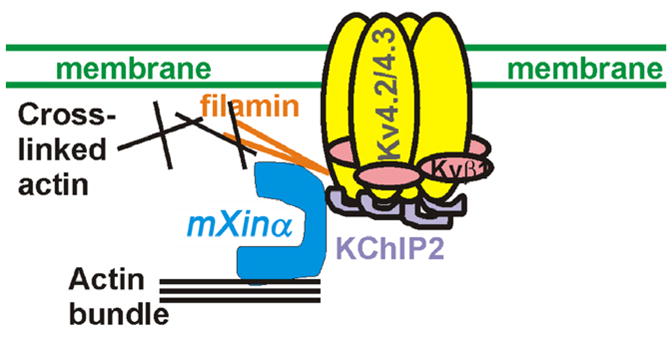
A schematic model suggesting that mXinα participates the assembly of ITO channel to the ICD of ventricular myocytes. In mice, the ITO channels reflect the assembly of Kv4.2/Kv4.3 α-subunits, Kvβ1 and KChIP2. A population of the ITO channels is known to localize at the ICD. Furthermore, it has been shown that the Kv4 pore-forming α-subunit is able to bind to filamin, and KChIP2. The associations with both proteins greatly enhance the surface expression of Kv4.2. In this study and our previous studies, we have clearly demonstrated that mXinα is capable of interacting with both KChIP2 and filamin. Through these interactions, mXinα likely influences surface expression of ITO channel.
Several lines of evidence support that interactions with the actin cytoskeleton can effectively determine the localization and function of many ion channels in excitable cells (31, 46–51). A new functional spectrin-rich domain, called the transitional junction, has been identified at the adherens junctions of ICDs (52). Spectrin directly interacts with actin filaments and ankyrin, and together with their associated proteins forms a membrane cytoskeleton, which plays an important role in targeting of ion channels, transporters and cell adhesion molecules to specialized compartments within the plasma membrane (53). Accumulated lines of evidence further reveal that mutations affecting functions of ankyrins result in cardiac arrhythmia (54–56). For example, the human SCN5A missense mutation results in defective binding to ankyrin-G and leads to reduction of Na+ inward current (INa) at T-tubules and ICDs and Brugada syndrome (47). Thus, ICD components and the underlying actin cytoskeleton play regulatory roles in the surface expression of INa and possibly other channels. The Kv4.2 of ITO also localizes to the ICDs and the T-tubules of cardiomyocytes (24, 57), and directly interacts with filamin (31) and KChIP2 (30). It is known that these interactions between Kv4.2 and filamin/KChIP2 influence the ITO surface expression and activity (31), although the molecular mechanisms remain unknown. The findings (i) that mXinα is able to interact with both filamin and KChIP2 and (ii) that the cardiomyocytes completely lacking mXinα express reduced ITO current density strongly identify a novel role of mXinα in targeting the ITO channel in cardiomyocytes.
Reductions in ITO and IK of juvenile mXinα-null cardiomyocytes were primarily responsible for the prolonged APD and higher incidence of EAD. However, reduction of IK1 of adult mXinα-null cardiomyocytes would result in a less negative MDP and promote the development of EAD (58), although this reduction could be due to cardiac hypertrophy and/or ICD structural defects.
Acknowledgments
The present work was supported in parts by grants NSC96-2320-B016-013 and NSC98-2320-B016-010-MY3 (to CIL) from National Science Council and a grant from Chen-Han Foundation for Education (to CHH), Taipei, Taiwan and by grant HL075015 and HL107383 (to JJCL) from National Institutes of Health, USA.
Abbreviations
- ICD
intercalated disc
- KChIP2
Kv channel interacting protein 2
- ITO
transient outward K+ current
- IK
delayed rectifier outward K+ current
- IK1
inward rectifier K+ (Kir) current
- ICa,L
L-type Ca2+ current
- APD
action potential duration
- MDP
maximal diastolic potential
- EAD
early afterdepolarization
- DAD
delayed afterdepolarization
References
- 1.Wang DZ, Reiter RS, Lin JL, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJ. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development. 1999;126(6):1281–94. doi: 10.1242/dev.126.6.1281. [DOI] [PubMed] [Google Scholar]
- 2.Lin JJ-C, Gustafson-Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang DZ, Evans S, Lin JL-C. Structure, expression, and function of a novel intercalated disc protein, Xin. J Med Sci. 2005;25(5):215–222. doi: 10.1901/jaba.2005.25-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang HT, Brand OM, Mathew M, Ignatiou C, Ewen EP, McCalmon SA, Naya FJ. Myomaxin is a novel transcriptional target of MEF2A that encodes a Xin-related alpha-actinin-interacting protein. J Biol Chem. 2006;281(51):39370–9. doi: 10.1074/jbc.M603244200. [DOI] [PubMed] [Google Scholar]
- 4.Sinn HW, Balsamo J, Lilien J, Lin JJ. Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development. Dev Dyn. 2002;225(1):1–13. doi: 10.1002/dvdy.10131. [DOI] [PubMed] [Google Scholar]
- 5.Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293(5):H2680–92. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCalmon SA, Desjardins DM, Ahmad S, Davidoff KS, Snyder CM, Sato K, Ohashi K, Kielbasa OM, Mathew M, Ewen EP, Walsh K, Gavras H, Naya FJ. Modulation of angiotensin II-mediated cardiac remodeling by the MEF2A target gene Xirp2. Circ Res. 2010;106(5):952–60. doi: 10.1161/CIRCRESAHA.109.209007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, Gustafson-Wagner EA, Scholz TD, Yang B, Lin JJ. Essential roles of an intercalated disc protein, mXinbeta, in postnatal heart growth and survival. Circ Res. 2010;106(9):1468–78. doi: 10.1161/CIRCRESAHA.109.212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grosskurth SE, Bhattacharya D, Wang Q, Lin JJ. Emergence of Xin demarcates a key innovation in heart evolution. PLoS ONE. 2008;3(8):e2857. doi: 10.1371/journal.pone.0002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Ven PFM, Ehler E, Vakeel P, Eulitz S, Schenk JA, Milting H, Micheel B, Furst DO. Unusual splicing events result in distinct Xin isoforms that associate differentially with filamin c and Mens/VASP. Exp Cell Res. 2006;312:2154–2167. doi: 10.1016/j.yexcr.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 10.Gustafson-Wagner E. Charactering the function of the novel Xin genes in the mouse heart. University of Iowa; Iowa City, IA: 2007. [Google Scholar]
- 11.Otten J, van der Ven PF, Vakeel P, Eulitz S, Kirfel G, Brandau O, Boesl M, Schrickel JW, Linhart M, Hayess K, Naya FJ, Milting H, Meyer R, Furst DO. Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs. Cardiovasc Res. 2010;85(4):739–50. doi: 10.1093/cvr/cvp345. [DOI] [PubMed] [Google Scholar]
- 12.Choi S, Gustafson-Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL, Lin JJ. The intercalated disc protein, mXinα, is capable of interacting with β-catenin and bundling actin filaments. J Biol Chem. 2007;282(49):36024–36. doi: 10.1074/jbc.M707639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai YJ, Huang EY, Yeh HI, Chen YL, Lin JJ, Lin CI. On the mechanisms of arrhythmias in the myocardium of mXinalpha-deficient murine left atrial-pulmonary veins. Life Sci. 2008;83(7–8):272–83. doi: 10.1016/j.lfs.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai YJ, Chen YY, Cheng CP, Lin JJ, Chudorodova SL, Roshchevskaya IM, Roshchevsky MP, Chen YC, Lin CI. Changes in ionic currents and reduced conduction velocity in hypertrophied ventricular myocardium of Xinα-deficient mice. Anatol J Cardiol. 2007;7(Suppl 1):90–2. [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng CP, Loh YX, Lin CI, Lai YJ, Chen YC, Sytwn HK, Gustafson-Wagner EA, Lin JJ-C. Electrophysiological characteristics of ventricular myocytes of Xinα-deficient mice. In: Kimchi A, editor. International Proceedings: Advances in heart disease. MEDIMOND; Bologna, Italy: 2005. [Google Scholar]
- 16.Chen YJ, Chen SA, Chen YC, Yeh HI, Chan P, Chang MS, Lin CI. Effects of rapid atrial pacing on the arrhythmogenic activity of single cardiomyocytes from pulmonary veins: implication in initiation of atrial fibrillation. Circulation. 2001;104(23):2849–54. doi: 10.1161/hc4801.099736. [DOI] [PubMed] [Google Scholar]
- 17.Wible BA, De Biasi M, Majumder K, Taglialatela M, Brown AM. Cloning and functional expression of an inwardly rectifying K+ channel from human atrium. Circ Res. 1995;76(3):343–50. doi: 10.1161/01.res.76.3.343. [DOI] [PubMed] [Google Scholar]
- 18.Chen YJ, Chen YC, Wongcharoen W, Lin CI, Chen SA. Effect of K201, a novel antiarrhythmic drug on calcium handling and arrhythmogenic activity of pulmonary vein cardiomyocytes. Br J Pharmacol. 2008;153(5):915–25. doi: 10.1038/sj.bjp.0707564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang SH, Chen YC, Chiang SJ, Higa S, Cheng CC, Chen YJ, Chen SA. Increased Ca(2+) sparks and sarcoplasmic reticulum Ca(2+) stores potentially determine the spontaneous activity of pulmonary vein cardiomyocytes. Life Sci. 2008;83(7–8):284–92. doi: 10.1016/j.lfs.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 20.Kuo H-C, Cheng C-F, Clark RB, Lin JJ-C, Lin JL-C, Hoshijima M, Nguyen-Tran VTB, Gu Y, Ikeda Y, Chu PH, Jr, JR, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of Ito and confers genetic susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 21.Winer J, Jung CK, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270(1):41–9. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- 22.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem. 2000;285(2):194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Barry DM, Trimmer JS, Merlie JP, Nerbonne JM. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ Res. 1995;77(2):361–9. doi: 10.1161/01.res.77.2.361. [DOI] [PubMed] [Google Scholar]
- 25.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–9. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 26.Chan FC, Lai YJ, Chen YC, Lin CI. Electrophysiological characteristics of the left-atrium-pulmonary vein (LA-PV) cardiomyocytes obtained from Xinα–knockout mouse heart. Proc 23rd Joint Ann Conf Biomed Sci; Taipei, Taiwan. 2008. [Google Scholar]
- 27.Tanaka M, Berul CI, Ishii M, Jay PY, Wakimoto H, Douglas P, Yamasaki N, Kawamoto T, Gehrmann J, Maguire CT, Schinke M, Seidman CE, Seidman JG, Kurachi Y, Izumo S. A mouse model of congenital heart disease: cardiac arrhythmias and atrial septal defect caused by haploinsufficiency of the cardiac transcription factor Csx/Nkx2.5. Cold Spring Harb Symp Quant Biol. 2002;67:317–25. doi: 10.1101/sqb.2002.67.317. [DOI] [PubMed] [Google Scholar]
- 28.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403(6769):553–6. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 29.Nerbonne JM. Studying cardiac arrhythmias in the mouse--a reasonable model for probing mechanisms? Trends Cardiovasc Med. 2004;14(3):83–93. doi: 10.1016/j.tcm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85(4):1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 31.Petrecca K, Miller DM, Shrier A. Localization and enhanced current density of the Kv4.2 potassium channel by interaction with the actin-binding protein filamin. J Neurosci. 2000;20(23):8736–44. doi: 10.1523/JNEUROSCI.20-23-08736.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vassalle M, Lin CI. Calcium overload and cardiac function. J Biomed Sci. 2004;11(5):542–65. doi: 10.1007/BF02256119. [DOI] [PubMed] [Google Scholar]
- 33.Wu SH, Chen YC, Higa S, Lin CI. Oscillatory transient inward currents in ventricular myocytes of healthy versus myopathic Syrian hamster. Clin Exp Pharmacol Physiol. 2004;31(10):668–76. doi: 10.1111/j.1440-1681.2004.04082.x. [DOI] [PubMed] [Google Scholar]
- 34.Sanguinetti MC. Reduced transient outward K+ current and cardiac hypertrophy: causal relationship or epiphenomenon? Circ Res. 2002;90(5):497–9. doi: 10.1161/01.res.0000014284.39141.8c. [DOI] [PubMed] [Google Scholar]
- 35.Furukawa T, Kurokawa J. Potassium channel remodeling in cardiac hypertrophy. J Mol Cell Cardiol. 2006;41(5):753–61. doi: 10.1016/j.yjmcc.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 36.Mitarai S, Reed TD, Yatani A. Changes in ionic currents and β-adrenergic receptor signaling in hypertrophied myocytes overexpressing G αq. Am J Physiol Heart Circ Physiol. 2000;279(1):H139–48. doi: 10.1152/ajpheart.2000.279.1.H139. [DOI] [PubMed] [Google Scholar]
- 37.Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol. 2000;525(Pt 2):483–98. doi: 10.1111/j.1469-7793.2000.t01-1-00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42(2):270–83. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 39.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res. 1998;83(5):560–7. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- 40.Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97(12):1342–50. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- 41.Wickenden AD, Lee P, Sah R, Huang Q, Fishman GI, Backx PH. Targeted expression of a dominant-negative K(v)4.2 K(+) channel subunit in the mouse heart. Circ Res. 1999;85(11):1067–76. doi: 10.1161/01.res.85.11.1067. [DOI] [PubMed] [Google Scholar]
- 42.Kassiri Z, Zobel C, Nguyen TT, Molkentin JD, Backx PH. Reduction of I(to) causes hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2002;90(5):578–85. doi: 10.1161/01.res.0000012223.86441.a1. [DOI] [PubMed] [Google Scholar]
- 43.Guo W, Li H, Aimond F, Johns DC, Rhodes KJ, Trimmer JS, Nerbonne JM. Role of heteromultimers in the generation of myocardial transient outward K+ currents. Circ Res. 2002;90(5):586–93. doi: 10.1161/01.res.0000012664.05949.e0. [DOI] [PubMed] [Google Scholar]
- 44.Choe S, Roosild T. Regulation of the K channels by cytoplasmic domains. Biopolymers. 2002;66(5):294–9. doi: 10.1002/bip.10309. [DOI] [PubMed] [Google Scholar]
- 45.Shibata R, Misonou H, Campomanes CR, Anderson AE, Schrader LA, Doliveira LC, Carroll KI, Sweatt JD, Rhodes KJ, Trimmer JS. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. J Biol Chem. 2003;278(38):36445–54. doi: 10.1074/jbc.M306142200. [DOI] [PubMed] [Google Scholar]
- 46.Maltsev VA, Undrovinas AI. Cytoskeleton modulates coupling between availability and activation of cardiac sodium channel. Am J Physiol. 1997;273(4 Pt 2):H1832–40. doi: 10.1152/ajpheart.1997.273.4.H1832. [DOI] [PubMed] [Google Scholar]
- 47.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101(50):17533–8. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Z, Eldstrom JR, Jantzi J, Moore ED, Fedida D. Increased focal Kv4.2 channel expression at the plasma membrane is the result of actin depolymerization. Am J Physiol Heart Circ Physiol. 2004;286(2):H749–59. doi: 10.1152/ajpheart.00398.2003. [DOI] [PubMed] [Google Scholar]
- 49.Maruoka ND, Steele DF, Au BP, Dan P, Zhang X, Moore ED, Fedida D. α-actinin-2 couples to cardiac Kv1.5 channels, regulating current density and channel localization in HEK cells. FEBS Lett. 2000;473(2):188–94. doi: 10.1016/s0014-5793(00)01521-0. [DOI] [PubMed] [Google Scholar]
- 50.Lader AS, Kwiatkowski DJ, Cantiello HF. Role of gelsolin in the actin filament regulation of cardiac L-type calcium channels. Am J Physiol. 1999;277(6 Pt 1):C1277–83. doi: 10.1152/ajpcell.1999.277.6.C1277. [DOI] [PubMed] [Google Scholar]
- 51.Steele DF, Eldstrom J, Fedida D. Mechanisms of cardiac potassium channel trafficking. J Physiol. 2007;582(Pt 1):17–26. doi: 10.1113/jphysiol.2007.130245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett PM, Maggs AM, Baines AJ, Pinder JC. The transitional junction: a new functional subcellular domain at the intercalated disc. Mol Biol Cell. 2006;17(4):2091–100. doi: 10.1091/mbc.E05-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81(3):1353–92. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 54.Mohler PJ, Bennett V. Ankyrin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr Opin Cardiol. 2005;20(3):189–93. doi: 10.1097/01.hco.0000160372.95116.3e. [DOI] [PubMed] [Google Scholar]
- 55.Cunha SR, Mohler PJ. Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc Res. 2006;71(1):22–9. doi: 10.1016/j.cardiores.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 56.Cunha SR, Mohler PJ. Ankyrin protein networks in membrane formation and stabilization. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeuchi S, Takagishi Y, Yasui K, Murata Y, Toyama J, Kodama I. Voltage-gated K+ Channel, Kv4.2, localizes predominantly to the transverse-axial tubular system of the rat myocyte. J Mol Cell Cardiol. 2000;32(7):1361–9. doi: 10.1006/jmcc.2000.1172. [DOI] [PubMed] [Google Scholar]
- 58.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73(2):379–85. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]