

Abstract
The discovery of complementary methods for enantioselective transition-metal-catalyzed cyclization with silyloxyenynes has been accomplished using chiral phosphine ligands. Under palladium catalysis, 1,6-silyloxyenynes bearing a terminal alkyne led to the desired 5-membered ring with high enantioselectivities (up to 91% ee). As for reactions under cationic gold catalysis, 1,6- and 1,5-silyloxyenynes bearing an internal alkyne furnished the chiral cyclopentane derivatives with excellent enantiomeric excess (up to 94% ee). Modification of the substrate by incorporating an α,β-unsaturation led to the discovery of a tandem cyclization. Remarkably, using silyloxy-1,3-dien-7-ynes under gold catalysis conditions provided the bicyclic derivatives with excellent diastereo- and enantioselectivities (up to >20:1 dr and 99% ee).
INTRODUCTION
Enantioselective α-functionalization of enolates and enolate derivatives serves as one of the most important methods for the construction of enantioenriched carbonyl containing compounds.1 A wide range of electrophiles can be reacted with enolate derivatives, including activated carbon-carbon π-bonds.2 In this context, alkynes are potentially interesting electrophiles as the product of the addition reaction is an alkene that can be further elaborated. Therefore, a number of addition reactions of silyl enol ethers to alkynes have been reported.3,4 From these reports, two major reactivity paradigms have emerged. The first, which follows from Conia’s seminal report3a of mercury(II)-promoted addition of silyl enol ethers to alkynes, involves nucleophilic addition to the triple bond that is activated by a π-acidic transition metal complex or Lewis acid. The second more recent approach proceeds through nucleophilic addition of the enol ether to an electrophilic transition metal vinylidene generated from a terminal alkyne.5 Despite these recent developments, enantioselective variants of this class of addition reactions remain scarce. This paucity can perhaps be traced to that fact that the majority of catalysts reported for this reaction are either simple metal salts or transition metal carbonyl compounds and therefore lack a readily tunable ancillary ligand.
The past decade has witnessed the development of cationic late transition metal complexes as catalysts for addition to alkynes.6 These complexes demonstrate the ability to catalyze the addition of nucleophiles to alkynes, even when ligated with Lewis basic phosphine ligands. For example, we reported that cationic triphenylphosphinegold(I) efficiently promoted the addition of β-ketoesters and silyl enol ethers to alkynes through a π-activation pathway.4a
Therefore, we envisioned that chiral phosphine analogues could catalyze such cyclization reactions in an enantioselective manner. In support of this hypothesis, Echavarren reported a gold-catalyzed enantioselective version of a 5-exo-dig cyclization using 1,6-enynes in presence of metha-nol.7 Moreover, related enantioselective cycloisomerization reactions have been described by Mikami8 and Genet9 using catalysts based on cationic phosphinepalladium(II) and platinum(II) complexes, respectively. More recently, we10, Michelet11 and Sanz12 showed that bisphosphinegold(I) complexes also effectively catalyzed enantioselective polycyclization and cycloisomerization reactions.13,14 On the basis of this work, we aimed to develop a set of catalysts that would allow for enantioselective both 5-endo-dig and 5-exo-dig addition reactions of enol ether nucleophiles. In this context, we focused our attention on cyclizations of 1,6- and 1,5-silyloxyenynes (eq 1 and 2). catalyzed by cationic (phosphine)platinum, palladium and gold complexes as catalysts.15
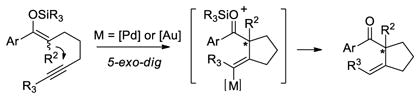 |
(1) |
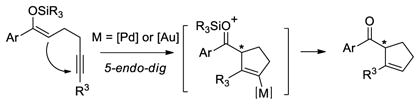 |
(2) |
Herein, we present a full account of our studies employing chiral gold and palladium catalysts to promote asymmetric 5-exo-dig and 5-endo-dig cyclizations. This work resulted in the discoveries of complimentary palladium(II) and gold(I)-catalyzed process highly enantioselective silyloxyenyne cycloisomerization reactions to yield synthetically useful exo-methylencylopentane or cyclopentene derivatives. Furthermore, we also demonstrate that silyloxy-1,3-dien-7-ynes are suitable substrate for diastereo- and enantioselective cyclization reactions to form polysubstituted bicyclo[3.3.0]octane derivatives.
RESULTS AND DISCUSSION
Palladium-catalyzed enantioselective cyclization of silyloxy-1,6-enynes
We began our investigation with the tetrasubstituted silyl enol ether 1 depicted in Table 1. We found that palladium complex (R)-DTBM-SEGPHOSPd(OTf)2 [L3Pd(OTf)2] that was successful in our previously reported enantioselective Conia-ene cyclization gave the best results in terms of enantiomeric excess (ee).16 Indeed, (Z)-isomer 1 was treated with catalyst L3Pd(OTf)2 and cyclic product 7 was isolated in 80% yield and 78% ee (entry 1). We were pleased to observe that (E)-isomer 2 led to the desired aryl ketone with an increase in enantioselectivity (91% ee, entry 2).
TABLE 1.
Pd-Cyclizations of Silyloxy-1,6-enynesa
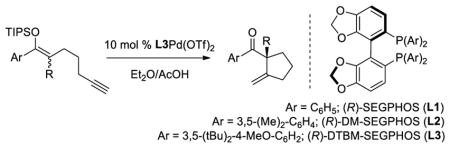 | ||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%)b | ee (%)c |
| 1 |
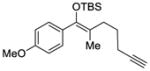 1 |
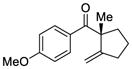 7 |
80 | 78 |
| 2 |
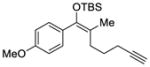 2 |
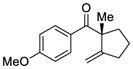 7 |
93 | 91 |
| 3 |
 3 |
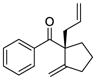 8 |
92 | 88 |
| 4 |
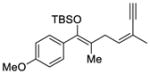 4 |
 9 |
70 | 73 |
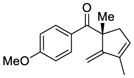
| 5 |
 5 |
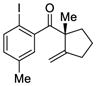 10 |
96 | 95 |
| 6d |
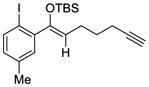 6 |
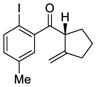 11 |
86 | 85 |
Reactions performed at 0.02 M in Et2O/AcOH (100/1) using 1 equiv of substrate and 10 mol % of L3Pd(OTf)2 for 16 h.
Isolated yields.
Determined by chiral HPLC.
At 0°C.
The scope of this reaction was studied and we found that the methyl substituent could also be modified. For example, allyl-substituted ketone 8 was formed with high selectivity (entry 3, 88% ee). We also observed that the 1,4-dien-6-yne 4 led to the desired cyclic ketone 9 with 73% ee (entry 4). The (Z)-1,6-silyloxyenyne 5 was synthesized and treated under the optimal palladium conditions to generate the desired ketone 10 with 96% yield and 95% ee (entry 5). The utility of this reaction was exemplified by the transformation of 10 into naturally occurring dimeric sesquiterpene (-)-laurebiphenyl (eq 3).15 In addition, trisubstituted silyloxyenyne 6 was reacted with catalyst L3Pd(OTf)2 at 0 °C to furnish the cyclic ketone 11 having a tertiary stereo-center with 85% enantioselectivity (entry 6).
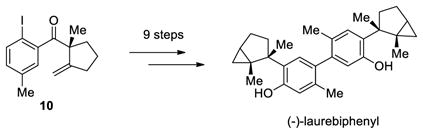 |
(3) |
The high enantioselectivity obtained with 1 and 2 suggests a transition state where the chiral complex selects the face based on placing the large (aryl/silyloxy portion) and the small (methyl) groups of the silyl enol ether in the appropriate quadrants of the chiral environment (Scheme 2). This hypothesis is supported by the low impact on the enantioselectivity obtained regardless of the olefin geometry (Table 1, entries 1 and 2).
Scheme 2.

Postulated Transition States
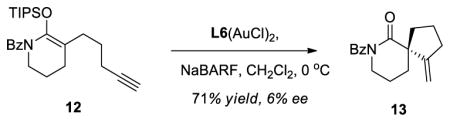
We decided to extend this methodology to the preparation of enantioenriched spirocyclic amides.17 However, L3Pd(OTf)2 with substrate 12 proved to be problematic as lower enantioselectivity was observed (Table 2, entry 1). A ligand screen was undertaken and we found that the SEGPHOS (L1) ligand gave higher enantioselectivity with moderate conversion since the hydrolysis product (corresponding lactam) was also obtained in significant amount (entry 3). A number of bisphosphine ligands having the biaryl atropisomeric backbone were tested with moderate success. We discovered that binaphane ligand (L4) was a highly effective ligand with substrate 12 (entry 6). The reaction times were generally shorter and catalyst loading could be decreased substantially compared with the conditions with L3Pd(OTf)2. Under the optimized conditions, the desired spirocyclic compound 13 was isolated with 80% yield and 98% ee.
TABLE 2.
Selected Optimization Experiments With Silyloxy-1,6-Enynes 12a
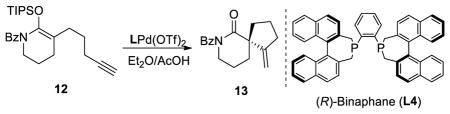 | |||
|---|---|---|---|
| Entry | Ligand | Conv (%)b | ee (%)c |
| 1 | (R)-DTBM-SEGPHOS (L3) | 15 | 48 |
| 2 | (R)-DTBM-SEGPHOS (L3) | >95 | 47 |
| 3 | (R)-SEGPHOS (L1) | 50 | 88 |
| 4 | (R,S)-SynPhos | 50 | 84 |
| 5 | (R,S)-CyJosiPhos | >95 | 5 |
| 6 | (R)-Binaphane (L4) | >95 (80)d | 98 |
Reactions performed at 0.02 M in Et2O/AcOH (100/1) using 1 equiv of substrate 12 and 10 mol % of LPd(OTf)2 for 2 h
Determined by 1H NMR
Determined by chiral HPLC.
Isolated yield.
Next, we examined the scope of this cyclization with other O-silylketene aminals. As shown in Table 3, 2-silyloxy indole 14 afforded the desired spiro-oxindole product 18 with 83% yield and 91% ee (entry 1). The acyclic O-silylketene aminal 15 was also reacted under similar conditions to obtain the amide 19 with satisfactory enantioselectivity (entry 2). The efficiency of this catalyst is particularly noteworthy as treatment of silyl enol ethers 16 and 17 with L4Pd(OTf)2 gave the corresponding cyclopentane adducts with high enantioselectivity (entries 3 and 4), while attempts at the L3Pd(OTf)2-catalyzed cyclization of these substrates provided trace amounts of the desired products.18
TABLE 3.
Pd-Cyclizations of Silyloxy-1,6-enynesa
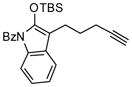
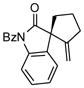
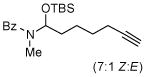
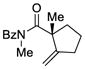
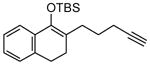
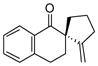
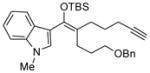
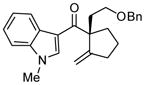
Reactions performed at 0.02 M in CH2Cl2/AcOH (100/1) using 1 equiv of substrate and 5 mol % of L4Pd(OTf)2 for 120 min.
Isolated yields.
Determined by chiral HPLC.
Gold-catalyzed enantioselective cyclization of silyloxy-1,6-enynes
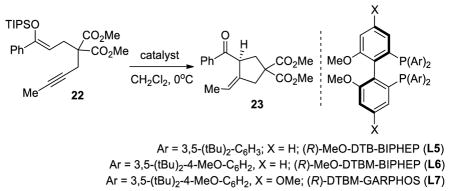
During the course of our study of this enantioselective palladium-catalyzed 5-exo-dig cyclization reaction, we found that substrates bearing an internal alkyne such as 22 were not reactive with catalyst L3Pd(OTf)2 (Table 4, entry 1). With the more reactive binaphane ligand (L4), the ketone derived from the hydrolysis of the silyl enol ether was obtained as the major product (entry 2). We also tested substrate 22 under platinum catalysis with L3Pt(OTf)2 and no reaction was observed (entry 3). Despite little success with chiral cationic gold complexes and substrates bearing a terminal alkyne,19 we decided to explore the reactivity of 22 with a different set of ligands under gold catalysis. An initial screen revealed that L3(AuCl)2 could promote the desired 5-exo-dig cylization to give 23, albeit with low yield and enantioselectivity (entry 4).20 We subjected 22 to previously reported conditions for the 6-exo-dig cyclization10b (L5(AuCl)2 and AgSbF6) and moderate yields (42%) of desired cyclized product 23 were obtained with a slight improvement in the enantioselectivity (entry 6). The low conversions were associated with the hydrolysis of the enolsilane, probably due to trace amounts of acid formed under the reaction conditions with silver salts.21 Performing the reaction at low temperature and using sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBARF) as a chloride scavenger in combination with L5(AuCl)2 led to the desired cyclized ketone 23 in 67% yield and 52% ee (entry 7). Then, lowering the temperature and switching to L6(AuCl)2 was promising in terms of enantioselectivity (entries 8 and 9). Attempts to improve the reaction parameters were performed and the desired product was isolated in 84% yield and 93% ee when the reaction was performed at −30 °C using dichloroethane as the solvent (entry 10).
TABLE 4.
Selected Optimization Experiments With Silyloxy-1,6-Enynesa
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Yield (%)b | ee (%)c |
| 1 | L3Pd(OTf)2 | none | NR | ND |
| 2 | L4Pd(OTf)2 | none | traces | ND |
| 3 | L3Pt(OTf)2 | none | NR | ND |
| 4 | L3(AuCl)2 | AgOTf | 39d | 17 |
| 5 | L3(AuCl)2 | AgSbF6 | 37d | 32 |
| 6 | L5(AuCl)2 | AgSbF6 | 42d | 35 |
| 7 | L5(AuCl)2 | NaBARF | 67 | 52 |
| 8 | L6(AuCl)2 | NaBARF | 81 | 72 |
| 9e | L6 (AuCl)2 | NaBARF | 74 | 85 |
| 10e,f | L6(AuCl)2 | NaBARF | 84 | 93 |
| 11e,f | L7(AuCl)2 | NaBARF | 88 | 87 |
Reactions performed at 0.1 M using 1 equiv of 22, 5 mol % of catalyst and 10 mol % of additive for 16 h. NR and ND mean no reaction and not determined, respectively
Isolated yields.
Determined by chiral HPLC.
Determined 1H NMR versus using an internal standard (diethyl phthalate).
Reaction was performed at −30 °C.
Using 1,2-dichloroethane (DCE) as a solvent.
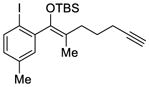
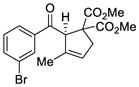
We explored the optimized conditions with substrate having different substituents on the aryl moiety. As shown in Table 5, the cyclization proceeded cleanly with substrates bearing methyl substitution (24, 25 and 26) to give the desired products with high enantiomeric excess (86% to 91%). Having a chlorine substituent such as in 28 was deleterious to reactivity, and a higher reaction temperature was required (entry 5). Finally, modifying the position of the gem-diester as in substrate 29 had a significant impact on the enantioselectivity and the cyclic product 35 was obtained with 50% ee (entry 6).
TABLE 5.
Enantioselective Au(I)-Cyclizations with Silyloxy-1,6-Enynesa
| Entry | Substrate | Product | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 |
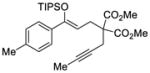 24 |
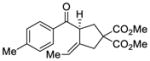 30 |
83 | 86 |
| 2 |
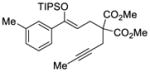 25 |
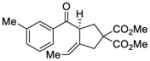 31 |
86 | 90 |
| 3 |
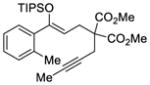 26 |
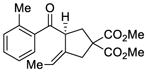 32 |
88 | 91 |
| 4 |
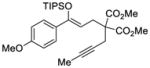 27 |
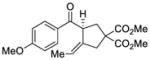 33 |
81 | 79 |
| 5d |
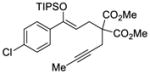 28 |
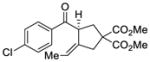 34 |
73 | 71 |
| 6e |
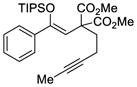 29 |
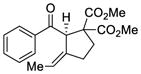 35 |
79f | 50 |
Reactions performed at 0.1 M using 1 equiv of substrate, 5 mol % (R)-MeO-DTBM-BIPHEP(AuCl)2 [L5(AuCl)2] and 10 mol % of NaBARF for 16 h in DCE at −30 °C.
Isolated yields.
Determined by chiral HPLC.
Reaction was performed at −10 °C.
Using dichloromethane as a solvent.
Isolated as an inseparable, 4:1 mixture of cyclic products.
Gold-catalyzed enantioselective cyclization of silyloxy-1,5-enynes
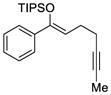
Decreasing the tether length by one carbon allowed us to examine the 5-endo-dig process in further detail. We suspected that silyloxy-1,5-enynes such as 36 would be suitable substrates and could favor only the product derived from the 5-endo attack. We initially subjected 36 to palladium and platinum catalysis and reactions with d8 metal complexes gave none of the desired product (Table 6, entries 1 and 2). Treatment of the same substrate with gold catalyst L6(AuCl)2 and NaBARF gave the desired product 37 but with low enantioselectivity (entry 3). The catalyst derived from (R)-SEGPHOS(AuCl)2 [L1(AuCl)2] and NaBARF was also tested and furnished the desired product with no significant enantiomeric excess (entry 4). However, increasing the size of the substituents on the phosphorus aryl moieties proved to be beneficial for enantioselectivity and led to an encouraging 73% ee (entry 5). Moreover, performing the cyclization using L3(AuCl)2 and NaBARF at lower temperature (−50 °C) greatly improved the selectivity, as 37 was isolated with 94% ee and 75% yield (entry 9).
TABLE 6.
Selected Optimization Experiments With Silyloxy-1,5-Enynesa
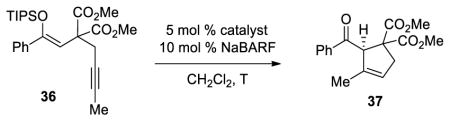 | ||||
|---|---|---|---|---|
| Entry | Catalyst | T (°C) | Yield (%)b | ee (%)c |
| 1d | L3Pd(OTf)2 | r.t. | NR | ND |
| 2d | L3Pt(OTf)2 | r.t | NR | ND |
| 3 | L6(AuCl)2 | r.t. | 76 | 24 |
| 4 | L1(AuCl)2 | r.t. | 69 | 4 |
| 5 | L2(AuCl)2 | r.t. | 72 | 28 |
| 6 | L3(AuCl)2 | r.t. | 74 | 73 |
| 7 | L3(AuCl)2 | −10 | 79 | 82 |
| 8 | L3(AuCl)2 | −30 | 81 | 88 |
| 9 | L3(AuCl)2 | −50 | 75 | 94 |
Reactions performed at 0.1 M using 1 equiv of 36, 5 mol % catalyst and 10 mol % of additive for 16 h.
Isolated yields.
Determined by chiral HPLC.
No NaBARF was added during this reaction.
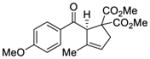
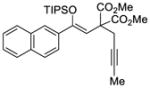
With these results in hand, we next sought to evaluate the substrate scope of our optimized conditions (Table 7). Various substituted phenyl derived silyl enol ethers (38–42) furnished the cyclized products (45–49) in good yields (67–92%) and high enantiomeric excess (91–94% ee) (entries 1–5). The reactions with substrates bearing an electron-poor aryl substituent showed lower reaction rates but still provided the desired product in high enantiomeric purity. Similarly, 2-naphthyl (43) and 2-thiophenyl (44) substituted substrates reacted under these conditions to generate the desired ketones in high yields and enantioselectivities (entries 6 and 7, 89 and 90 % ee respectively). The absolute stereochemistry of the cyclopentene derivatives was assigned by analogy to an x-ray structure obtained after re-crystallization of aryl ketone 47.22
TABLE 7.
Enantioselective Au(I)-Cyclization with Silyloxy-1,5-Enynesa
| Entry | Substrate | Product | Yield (%)b | ee (%)c |
|---|---|---|---|---|
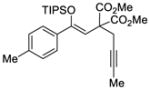
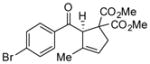
| 1 |
 38 |
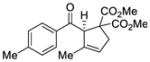 45 |
81 | 91 |
| 2 |
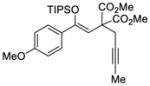 39 |
 46 |
92 | 94 |
| 3e |
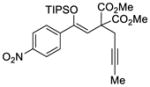 40 |
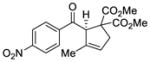 47 |
71 | 94 |
| 4 |
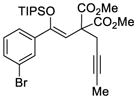
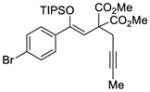 41 |
 48 |
67 | 92 |
| 5 |
 42 |
 49 |
72 | 93 |
| 6 |
 43 |
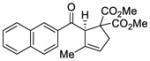 50 |
82 | 89 |
| 7 |
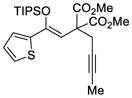 44 |
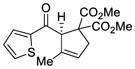 51 |
94 | 90 |
Reactions performed at 0.1 M using 1 equiv of substrate, 5 mol % (R)-DTBM-SEGPHOS(AuCl)2 [L3(AuCl)2] and 10 mol % of NaBARF for 16 h at −30 °C.
Isolated yields.
Determined by chiral HPLC.
Using 1,2-dichloroethane as a solvent.
As depicted in Table 8, decreasing the size of the substituents on the silyl group (52, TES and 53, TBDMS) was slightly detrimental in terms of yields but still led to good enantioselectivities (87% and 86% ee, respectively). Next, we observed that substrate 54 having no substitution on the backbone gave lower enantioselectivity (entry 4, 55% ee). Treatment of malononitrile derivative 55 under the optimal conditions gave 58 with an increase in enantioselectivity (entry 5, 71% ee). Finally, we were pleased to find that the gold-catalyzed cyclization of 56 led to the desired dime-thyl-substituted product 59 in 81% yield and 90% ee (entry 6).
TABLE 8.
Enantioselective Au(I)-Cyclization with Silyloxy-1,5-Enynesa
| Entry | Substrate | Product | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 |
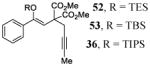
|
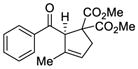 37 |
71 | 86 |
| 2 | 67 | 87 | ||
| 3 | 75 | 94 | ||
| 4 |
 54 |
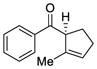 57 |
77 | 55 |
| 5e |
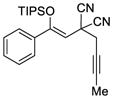 55 |
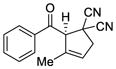 58 |
58 | 71 |
| 6 |
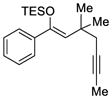 56 |
 59 |
81 | 90 |
Reactions performed at 0.1 M using 1 equiv of substrate, 5 mol % (R)-DTBM-SEGPHOS(AuCl)2 [L3(AuCl)2] and 10 mol % of NaBARF for 16 h at −30 °C.
Isolated yields.
Determined by chiral HPLC.
Using 1,2-dichloroethane as a solvent.
Gold-catalyzed enantioselective tandem cyclization of silyloxy-1,3-dien-7-yne
Subsequently, as depicted in Scheme 3, we explored the reactivity of the analogous 3-siloxy-1,3-diene-7-yne towards gold(I) catalysis, anticipating the formation of bicyclo[3.3.0]octane derivatives. For this scenario to be successful, the vinyl gold intermediate B obtained after the cyclization would perform a conjugate addition on the activated unsaturated ketone to form an additional carbon-carbon bond.23 Under this proposed mechanism, the carbene intermediate24 would undergo subsequent 1,2-hydrogen migration to give the desired bicylic diene.25
Scheme 3.

Proposed intermediates for gold-catalyzed tandem cyclization of silyloxy-1,3-dien-7-yne.
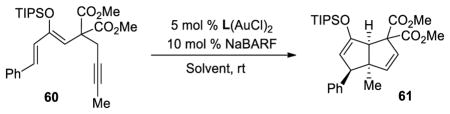
To our delight, the reaction performed with substrate 60 and L3(AuCl)2 gave the desired product 61 with excellent diastereoselectivity and enantioselectivity (Table 9, entry 1). In this case, the reaction was performed at room temperature and dry molecular sieves were added to the reaction mixture in order to avoid the formation of a complex mixture of ketones. The optimized conditions using dichloroethane as a solvent (entry 2) gave 61 in a very impressive 99% ee, with excellent yield (91%) and diastereoselectivity (>20:1). We noted that both the electron density and steric hindrance associated with bulky ligand L3 were essential to obtain excellent yields and enantioselectivities (entries 3 and 4).
TABLE 9.
Selected Optimization for Enantioselective Au(I)-Cyclization with Silyloxy-1,3-dien-7-ynesa
 | |||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | drb | Yield (%)c | ee (%)d |
| 1 | L3 | CH2Cl2 | >20:1 | 76 | 98 |
| 2 | L3 | DCE | >20:1 | 91 | 99 |
| 3 | L1 | CH2Cl2 | >20:1 | 36 | 41 |
| 4 | L2 | CH2Cl2 | >20:1 | 72 | 56 |
Reactions performed at 0.1 M using 1 equiv of 60, 5 mol % L3(AuCl)2 and 10 mol % of NaBARF for 16 h.
The diastereoselectivity was determined by 1H NMR of the crude reaction mixture.
Isolated yields.
Determined by chiral HPLC.
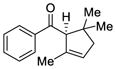
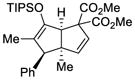
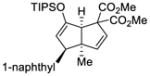
This tandem gold(I)-catalyzed process showed great compatibility with the presence of a wide range of substrates (Table 8). Tetrasubstituted diene 62 gave the desired bicyclic product 69 with high yield (76%) and enantioselectivity (95% ee, entry 1). Modification of the terminal substituent of the alkyne to an ethyl group gave the desired silyl enol ether 63 in a satisfactory yield (61%) and enantioselectivity (96% ee, entry 2). The reactions were faster with substrates 64 and 65 and resulted in the formation of the bicyclic dienes 71 and 72 with high enantioselectivities (98% and 89% ee, entries 3 and 4). However, substrate having para-methoxy substitution on the aryl ring gave a complex mixture of products. We found that the reaction was efficient with a substrate bearing the 1-naphthyl group (66); the bicyclic product 73 was obtained with high enantioselectivity (91% ee, entry 5). Finally, the reaction was found to tolerate various alkyl substituents at R1 (c-hexyl and n-butyl) and gave the bicyclic ketones 74 and 75 in good yield with slightly lower enantioselectivites (73 and 81% ee, entries 6 and 7).
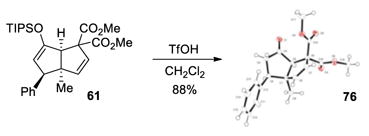
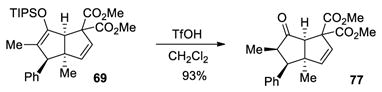
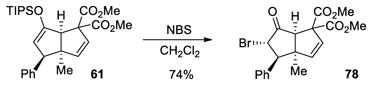
To exemplify the utility of this tandem cyclization, silyl enol ether 61 was hydrolyzed in the presence of acid to give the corresponding ketone 76 (eq 4). The absolute stereochemistry was determined by x-ray analysis of this crystalline compound and the stereochemistry of the related bicyclo[3.3.0]octane derivatives was assigned by analogy. Ketone 77 containing four contiguous stereogenic centers was obtained as a single stereoisomer by treatment of 69 under similar conditions (eq 5). Furthermore, bromination of 61 afforded the α-bromoketone 78 with high diastereoselectivity (>20:1, eq 6). The preference for formation of the anti-substituted product may be explained by the increased steric repulsion between the β-substituent and the methyl group in transition state 80 leading to the syn-substituted adduct (Figure 1).
Figure 1.

Rationale for trans Diastereoselectivity
 |
(4) |
 |
(5) |
 |
(6) |
CONCLUSION
In summary, we have described novel asymmetric metal-catalyzed 5-exo and 5-endo-dig cyclizations of syliloxyenynes using palladium and gold complexes as catalysts. These reactions showed excellent enantioselectivity and provide entry into a wide range of cyclopentanoid structures. While the palladium and gold-catalyzed cyclization reactions are mechanistically related, the two catalyst have complimentary limitations and preferences; the palladium(II) complexes were generally limited to catalysis of 5-exo-dig cyclization reactions of terminal alkynes, while the gold(I) catalysts showed preference for cyclization reactions of non-terminal alkynes. Moreover, the chiral phosphine gold complexes provide access to enantioselective 5-endo-dig cyclization reactions previously unachievable through catalysis with cationic group 10 metal complexes. Taken together, these results further highlight the great potential of electrophilic late transition metal complexes to serve as catalysts for enantioselective formation of carbon–carbon bonds by alkyne activation.
Supplementary Material
TABLE 10.
| Entry | Substrate | Product | Yield (%)c | ee (%)d |
|---|---|---|---|---|
| 1 |
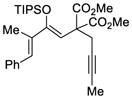 62 |
 69 |
76 | 95 |
| 2 |
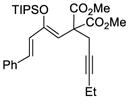 63 |
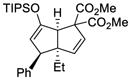 70 |
61 | 96 |
| 3 |
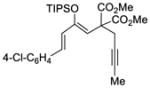 64 |
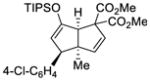 71 |
64 | 98 |
| 4 |
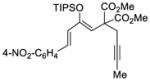 65 |
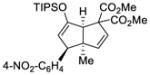 72 |
81 | 89 |
| 5 |
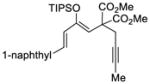 66 |
 73 |
72 | 91 |
| 6 |
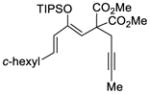 67 |
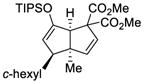 74 |
68 | 81 |
| 7 |
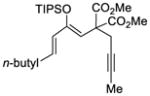 68 |
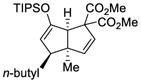 75 |
82 | 73 |
Reactions performed at 0.1 M using 1 equiv of substrate, 5 mol % (R)-DTBM-SEGPHOS(AuCl)2 and 10 mol % of NaBARF for 16 h.
The diastereoselectivity was >20:1 as determined by 1H NMR of the crude reaction mixture.
Isolated yields.
Determined by chiral HPLC.
Acknowledgments
We gratefully acknowledge NIHGMS (RO1 GM073932), Amgen, and Novartis for financial support. J.-F.B. thanks the Fonds Québecois de la Recherche sur la Nature et les Technologies (FQRNT) for a postdoctoral fellowship. S.Y. and I.C.U. acknowledge the Groningen University Fund (GUF) and the Spanish MCI, respectively. We would also like to thank Dr. Antonio DiPasquale for his assistance in collecting and analyzing crystallographic data. Solvias and Takasago are acknowledged for the generous donation of phosphine ligands and Johnson Matthey for a gift of AuCl3.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures and compound characterization data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selected examples catalytic enantioselective synthesis of ketones:. Evans PA, Leahy DK. J Am Chem Soc. 2003;125:8974. doi: 10.1021/ja035983p.Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x.Doyle AG, Jacobsen EN. J Am Chem Soc. 2005;127:62. doi: 10.1021/ja043601p.Trost BM, Xu J. J Am Chem Soc. 2005;127:2846. doi: 10.1021/ja043472c.Yan XX, Liang CH, Zhang Y, Hong W, Cao BX, Dai LX, Hou XL. Angew Chem Int Ed. 2005;44:6544. doi: 10.1002/anie.200502020.Weix DJ, Hartwig JF. J Am Chem Soc. 2007;129:7720. doi: 10.1021/ja071455s.Lundin PM, Fu GC. J Am Chem Soc. 2010;132:11027. doi: 10.1021/ja105148g.Mastracchio A, Warkentin AA, Walji AM, MacMillan DWC. Proc Nat Acad Sci USA. 2010;107:20648. doi: 10.1073/pnas.1002845107.Cheon CH, Kanno O, Toste FD. J Am Chem Soc. 2011;133:13248. doi: 10.1021/ja204331w.For examples of enantioselective α-vinylation of carbonyl compounds see: Taylor AM, Altman RA, Buchwald SL. J Am Chem Soc. 2009;131:9900. doi: 10.1021/ja903880q.Kim K, MacMillan DWC. J Am Chem Soc. 2008;130:398. doi: 10.1021/ja077212h.
- 2.For a review of metal enolate additions to carbon-carbon multiple bonds, see: Denes F, Perez-Luna A, Chemla F. Chem Rev. 2010;110:2366. doi: 10.1021/cr800420x.
- 3.For metal-catalyzed cyclization reactions of alkynyl silyl enol ethers, see: Mercury: Drouin J, Boaventura MA, Conia JM. J Am Chem Soc. 1985;107:1726.Drouin J, Boaventura MA. Tetrahedron Lett. 1987;28:3923.Huang H, Forsyth CJ. J Org Chem. 1995;60:2773.Imamura K, Yoshikawa E, Gevorgyan V, Yamamoto Y. Tetrahedron Lett. 1999;40:4081.Tungsten Maeyama K, Iwasawa N. J Am Chem Soc. 1998;120:1928.Iwasawa N, Maeyama K, Kusama H. Org Lett. 2001;3:3871. doi: 10.1021/ol016718g.Kusama H, Yamabe H, Iwasawa N. Org Lett. 2002;4:2569. doi: 10.1021/ol026202c.Iwasawa N, Miura T, Kiyota K, Kusama H, Lee K, Lee PH. Org Lett. 2002;4:4463. doi: 10.1021/ol026993i.Miura T, Kiyota K, Kusama H, Lee K, Kim H, Kim S, Lee PH, Iwasawa N. Org Lett. 2003;5:1725. doi: 10.1021/ol034365a.Kusama H, Onizawa Y, Iwasawa N. J Am Chem Soc. 2006;128:16500. doi: 10.1021/ja0671924.Grandmarre A, Kusama H, Iwa-sawa N. Chem Lett. 2007;36:66.Rhodium Dankwardt JW. Tetrahedron Lett. 2001;42:5809.Rhenium Kusawa H, Yamabe H, Onizawa Y, Hoshino T, Iwa-sawa N. Angew Chem Int Ed. 2005;44:468. doi: 10.1002/anie.200461559.Saito K, Onizawa Y, Kusama H, Iwasawa N. Chem Eur J. 2010;16:4716. doi: 10.1002/chem.200903586.Platinum Nevado C, Cardenas DJ, Echavarren AM. Chem Eur J. 2003;9:2627. doi: 10.1002/chem.200204646.Silver Godet T, Belmont P. Synlett. 2008:2513.
- 4.For gold-catalyzed cyclization reactions alkynyl silyl enol ethers, see: Staben ST, Kennedy-Smith JJ, Huang D, Corkey BK, LaLonde RL, Toste FD. Angew Chem Int Ed. 2006;45:5991. doi: 10.1002/anie.200602035.Lee K, Lee PH. Adv Synth Catal. 2007;349:2092.Minnihan EC, Colletti SL, Toste FD, Shen HC. J Org Chem. 2007;72:6287. doi: 10.1021/jo071014r.Barabe F, Betournay G, Bellavance G, Barriault L. Org Lett. 2009;11:4236. doi: 10.1021/ol901722q.Kusama H, Karibe Y, Onizawa Y, Iwasawa N. Angew Chem Int Ed. 2010;49:4269. doi: 10.1002/anie.201001061.Ito H, Ohmiya H, Sawamura M. Org Lett. 2010;12:4380. doi: 10.1021/ol101860j.Hashmi ASK, Yang W, Rominger F. Angew Chem, Int Ed. 2011;50:5762. doi: 10.1002/anie.201100989.Barabe F, Levesque P, Korobkov I, Barriault L. Org Lett. 2011;13:5580. doi: 10.1021/ol202314q.See also Shapiro ND, Toste FD. Proc Natl Acad Sci. 2008;105:2779.
- 5.(a) Maeyama K, Iwasawa N. J Am Chem Soc. 1998;120:1928. [Google Scholar]; (b) Iwasawa N, Maeyama K, Kusama H. Org Lett. 2001;3:3871. doi: 10.1021/ol016718g. [DOI] [PubMed] [Google Scholar]; (c) Kusama H, Yamabe H, Iwasawa N. Org Lett. 2002;4:2569. doi: 10.1021/ol026202c. [DOI] [PubMed] [Google Scholar]; (d) Iwasawa N, Miura T, Kiyota K, Kusama H, Lee K, Lee PH. Org Lett. 2002;4:4463. doi: 10.1021/ol026993i. [DOI] [PubMed] [Google Scholar]
- 6.For recent reviews on cycloisomerization reactions: Buisine O, Aubert C, Malacria M. Chem Rev. 2002;102:813. doi: 10.1021/cr980054f.Lloyd-Jones G. Org Biomol Chem. 2003;1:215. doi: 10.1039/b209175p.Fairlamb IJS. Angew Chem Int Ed. 2004;43:1048. doi: 10.1002/anie.200301699.Zhang L, Sun J, Kozmin S. Adv Synth Catal. 2006;348:2271.Ma S, Yu S, Gu Z. Angew Chem, Int Ed. 2006;45:200. doi: 10.1002/anie.200502999.Michelet V, Toullec PY, Genêt JP. Angew Chem Int Ed. 2008;47:4727. doi: 10.1002/anie.200701589.Jiménez-Nùñez E, Echavarren AM. Chem Rev. 2008;108:3326. doi: 10.1021/cr0684319.Furstner A. Chem Soc Rev. 2009;38:3208. doi: 10.1039/b816696j.Belmont P, Parker E. Eur J Org Chem. 2009;35:6075.Lee SI, Chatani N. Chem Commun. 2009:371. doi: 10.1039/b812466c.Trost BM, Gutierrez AC, Ferreira EF. J Am Chem Soc. 2010;132:9206. doi: 10.1021/ja103663h.Toullec PY, Michelet V. Top Curr Chem. 2011;302:31. doi: 10.1007/128_2010_116.
- 7.Munoz MP, Adrio J, Carrtero JC, Echavarren AM. Organometallics. 2005;24:1293. [Google Scholar]
- 8.Hatano M, Terada M, Mikami K. Angew Chem Int Ed. 2001;40:249. [PubMed] [Google Scholar]
- 9.(a) Charruault L, Michelet V, Taras R, Gladiali S, Genêt JP. Chem Commun. 2004:850. doi: 10.1039/b400908h. [DOI] [PubMed] [Google Scholar]; (b) Toullec PY, Chao CM, Chen Q, Gladiali S, Genet JP, Michelet V. Adv Synth Catal. 2008;350:2401. [Google Scholar]
- 10.Enantioselective gold-catalyzed 6-exo-dig cyclizations: Sethofer SG, Staben ST, Hung OY, Toste FD. Org Lett. 2008;10:4315. doi: 10.1021/ol801760w.Sethofer SG, Mayer T, Toste FD. J Am Chem Soc. 2010;132:8276. doi: 10.1021/ja103544p.
- 11.(a) Chao CM, Viatle MR, Toullec PY, Genêt JP, Michelet V. Chem Eur J. 2009;15:1319. doi: 10.1002/chem.200802341. [DOI] [PubMed] [Google Scholar]; (b) Chao CM, Genin E, Toullec PY, Genêt JP, Michelet V. J Organomet Chem. 2009;694:538. [Google Scholar]; (c) Chao CM, Beltrami D, Toullec PY, Michelet V. Chem Comm. 2009:6988. doi: 10.1039/b913554e. [DOI] [PubMed] [Google Scholar]; (d) Pradal A, Chao C-M, Vitale MR, Toullec PY, Michelet V. Tetrahedron. 2011;67:4371. [Google Scholar]
- 12.Martínez A, García-García P, Fernández-Rodríguez MA, Rodríguez F, Sanz R. Angew Chem Int Ed. 2010;49:4633. doi: 10.1002/anie.201001089. [DOI] [PubMed] [Google Scholar]
- 13.For recent reviews on gold catalysis: Gorin DJ, Sherry BD, Toste FD. Chem Rev. 2008;108:3351. doi: 10.1021/cr068430g.Jimenez-Nunez E, Echavarren AM. Chem Rev. 2008;108:3326. doi: 10.1021/cr0684319.Arcadi A. Chem Rev. 2008;108:3266. doi: 10.1021/cr068435d.Das A, Abu SMA, Liu RS. Org Biomol Chem. 2010;8:960. doi: 10.1039/b923510h.
- 14.For recent reviews on enantioselective gold-catalyzed transformations: Bongers N, Krause N. Angew Chem Int Ed. 2008;47:2178. doi: 10.1002/anie.200704729.Widenhoefer RA. Chem Eur J. 2008;14:5382. doi: 10.1002/chem.200800219.Shapiro N, Toste FD. Synlett. 2010;5:675. doi: 10.1055/s-0029-1219369.Sengupta S, Shi X. ChemCatChem. 2010;2:609.Lee AL. Annu Rep Prog Chem Sect B Org Chem. 2010;106:428.Pradal A, Toullec PY, Michelet V. Synthesis. 2011;10:1501.
- 15.For a preliminary account describing Pd-catalyzed enantioselective cyclization with alkynyl silyl enol ether, see: Corkey BK, Toste FD. J Am Chem Soc. 2007;129:2764. doi: 10.1021/ja068723r.
- 16.Corkey BK, Toste FD. J Am Chem Soc. 2005;127:17168. doi: 10.1021/ja055059q. [DOI] [PubMed] [Google Scholar]
-
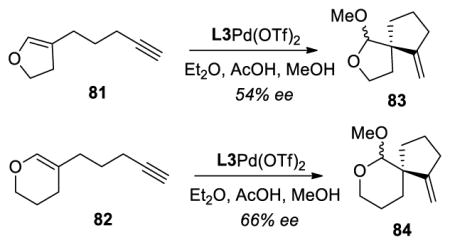
17.We also studied the reactivity of cyclic enol ethers 81 and 82 to generate spiro-cyclic ethers. However, moderate yields and enantioselectivities were noticed under our optimized conditions.
-
18.Cyclization reaction of silyl enol ether 16 with achiral (phosphine)palladium complexes in the presence of chiral counterions gave 20 with up to 40% ee. For additional example of this strategy see: Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229.Mukherjee A, List B. J Am Chem Soc. 2007;129:11336. doi: 10.1021/ja074678r.LaLonde RL, Wang ZJ, Mba M, Lackner AD, Toste FD. Angew Chem Int Ed. 2010;49:598. doi: 10.1002/anie.200905000.Jaing G, Halder R, Fang Y, List B. Angew Chem Int Ed. 2011;50:9752. doi: 10.1002/anie.201103843.Rauniyar V, Wang ZJ, Burks HE, Toste FD. J Am Chem Soc. 2011;133:8486. doi: 10.1021/ja202959n.Barbazanges M, Augé M, Moussa J, Amouri H, Aubert C, Desmarets C, Fensterbank L, Gandon V, Malacria M, Olliver C. Chem Eur J. 2011;48:13789. doi: 10.1002/chem.201102723.
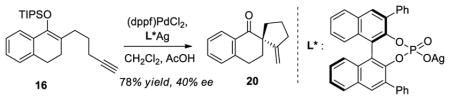
-
19.Various silyl enol ethers having a terminal alkyne were tested under gold catalysis with little success in terms of enantioselectivity. The experiment below with substrate 12 highlights the complementary utility of palladium (Table 2, entry 6) and gold catalysis.
- 20.The cyclization of silyloxyenyne 22 was also tested with chiral phosphoramidite and (acyclic diamino)carbene gold(I) complexes in presence of NaBARF and low enantioselectivities were obtained (21% and 6% ee, respectively). For details of these catalysts: phosphoramidites: Alonso I, Trllo B, López F, Montserrat S, Ujaque G, Caseto L, Lledos A, Masareñas JL. J Am Chem Soc. 2009;131:13020. doi: 10.1021/ja905415r.Gonz!alez AZ, Toste FD. Org Lett. 2010;12:200. doi: 10.1021/ol902622b.Teller H, Flugge S, Goddard R, Furstner A. Angew Chem, Int Ed. 2010;49:1949. doi: 10.1002/anie.200906550.Gonzalez AZ, Benitez D, Tkatchouk E, Goddard WA, III, Toste FD. J Am Chem Soc. 2011;133:5500. doi: 10.1021/ja200084a.(acyclic diamino)carbene: Wang YM, Kuzniewski CN, Rauniyar V, Hoong C, Toste FD. J Am Chem Soc. 2011;133:12972. doi: 10.1021/ja205068j.
- 21.Treatment of 22 with AgOTf or TfOH led to the corresponding hydrolyzed ketone in small amounts.
- 22.See Supporting Information.
- 23.For references on all-carbon quaternary centers, see: Christophers J, Baro A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim: 2006. Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0307113101.Cozzi PG, Hilgraf R, Zimmermann N. Eur J Org Chem. 2007:5969.
- 24.Seidel G, Mynott R, Furstner AF. Angew Chem Int Ed. 2009;48:2510. doi: 10.1002/anie.200806059. [DOI] [PubMed] [Google Scholar]; (b) Benitez D, Shapiro ND, Tkatchouk E, Wang Y, Goddard WA, Toste FD. Nat Chem. 2009;1:482. doi: 10.1038/nchem.331. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hashmi ASK. Angew Chem Int Ed. 2010;49:5232. doi: 10.1002/anie.200907078. [DOI] [PubMed] [Google Scholar]; (d) Xiao J, Li X. Angew Chem Int Ed. 2011;50:7226. doi: 10.1002/anie.201100148. [DOI] [PubMed] [Google Scholar]
- 25.(a) Kusuma H, Yamabe H, Onizawa Y, Hoshino T, Iwasawa N. Angew Chem Int Ed. 2005;44:468. doi: 10.1002/anie.200461559. [DOI] [PubMed] [Google Scholar]; (b) Onizawa Y, Kusama H, Iwasawa N. J Am Chem Soc. 2008;130:802. doi: 10.1021/ja0782605. [DOI] [PubMed] [Google Scholar]; (c) Hiroyuki K, Onizawa Y, Iwasawa N. J Am Chem Soc. 2006;128:16500. doi: 10.1021/ja0671924. [DOI] [PubMed] [Google Scholar]; (d) Kusama H, Karibe Y, Onizawa Y, Iwasawa N. Angew Chem Int Ed. 2010;49:4269. doi: 10.1002/anie.201001061. [DOI] [PubMed] [Google Scholar]; (e) Kusama H, Karibe Y, Imai R, Onizawa Y, Yamabe H, Iwasawa N. Chem–Eur J. 2011;17:4839. doi: 10.1002/chem.201003019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.