

Abstract
Applications of recombinant DNA technology, chemical synthesis on biological templates and fluorescence detection of organophosphorylation provide unexplored avenues for development of antidotes and approaches for remote detection of organophosphate nerve agents and pesticides. We discuss here how acetylcholinesterase (AChE), through appropriate mutations, becomes more susceptible to oxime reactivation. Since the reaction between organophosphate and the mutated enzyme remains rapid, regeneration of active enzyme by oxime becomes the rate-limiting step in the process to complete a catalytic cycle for generation of active enzyme. Accordingly, “Oxime-assisted Catalysis” by AChE provides a potential means for catalyzing the hydrolysis of organophosphates in plasma prior to their reaching the cellular target site. In turn, AChE, when conjugated with organophosphate, can be employed as a template for ‘click-chemistry, freeze-frame’ synthesis of new nucleophilic reactivating agents that could potentially prove useful in AChE reactivation at the target site as well as in catalytic scavenging in plasma. Finally, substituted AChE molecules can be conjugated to fluorophores giving rise to shifts in emission spectra for detection of dispersed organophosphates. Since external reagents do not have to be added to detect the fluorescence change, the modified enzyme would serve as a remote sensor.
Keywords: Organophosphate antidotes, oxime catalysis, catalytic scavenging, organophosphate detection, reactivation chemistry
1. Introduction
Remote detection of chemical toxicant exposure, development of improved antidotes and countermeasures to chemical terrorism threats have received considerable attention of late. In contrast to many of the substances that constitute a chemical threat when illicitly employed in threatened or actual terrorism, the organophosphates have a well-characterized and discrete site of action in producing acute toxicity (Giacobini, 2000). Accordingly, through studies conducted for more than a half-century, investigators have developed antidotes to the organophosphate cholinesterase inhibitors (Wilson, 1951; Wilson et al., 1959; Eyer et al., 2006) and devised means for detection of organophosphate exposure by inhibition of cholinesterase activity (Wilson, 2005). Such approaches to antidotal and prophylactic therapy, while invaluable, have limitations in protection capacity, incurring side effects from the treatment modalities and ease of administration of the antidote. Similarly, detection methods may require on-site addition of reagents or analysis of decreases in variable amounts of cholinesterase activity in the plasma of exposed subjects.
In considering prospective antidotal schemes for organophosphate poisoning, we might draw parallels between progress and challenges encountered in the development of anti-microbial agents for infectious disease. Both the anti-microbial agents and organophosphate antidotes have well defined biological targets. The development of effective agents for infectious disease is seen as a stepwise process where second and third generations of anti-microbials show enhanced efficacy, a wider spectrum of activity and activity towards organisms with increased resistance. Similarly, if a similar paradigm is followed for treating nerve agent exposure, new generations of antidotes to organophosphate toxicity should be staged from our knowledge of existing antidotal therapy and our future antidote design should be patterned to exhibit enhanced efficacy towards the range of existing or newly emerging organophosphate nerve agents.
In this presentation we will consider new approaches to improving organophosphate detection capabilities and antidotal therapy. The approach relies on simple modification of the very target of organophosphate toxicity, acetylcholinesterase (AChE), so that it: (a) is employed in vivo as a catalytic, rather than a stoichiometric, scavenging agent for the organophosphate, (b) serves as a template for the synthesis and selection of more effective reactivating agents, and (c) becomes diagnostic in the detection of organophosphate exposure. The substantive advances in recombinant DNA techniques and the delineation of the structures of AChE and its ligand complexes and conjugates should facilitate this approach.
2. Oxime-assisted Catalysis of Organophosphates by AChE
The reaction steps leading to inactivation of AChE by organophosphates, subsequent spontaneous reactivation, formation of an aged enzyme conjugate resistant to reactivation and an oxime facilitated reactivation reaction are shown in Fig. 1. Organophosphates have long been known to function as hemisubstrates where the initial inactivation is facilitated by an SN2 attack by a nucleophilic serine on the phosphorus atom properly positioned within the active center and rendered more electrophilic by insertion of its phosphoryl or phosphonyl oxygen in the oxyanion hole. These requirements became well established by examining the stereospecificity of the reaction in relation to the structure of the organophosphate conjugates (Hosea et al., 1995; Taylor et al., 1997; Ordentlich et al., 1999). Locking of the tetrahedral phosphorate or phosphonate in this position contributes to the slow spontaneous rate of H2O reactivation. Addition of a nucleophile, particularly when site-directed, should enhance reactivation, and this is the principle of oxime therapy introduced some 50 years ago by Irwin Wilson and his colleagues (Wilson 1951; Wilson and Ginsberg, 1955; Wilson et al., 1961). The historical development of this concept and the emergence of newer oxime compounds are presented in another chapter in this series (Eyer et al., 2006).
Figure 1.

Reaction of Alkyl Methylphosphonates with the Active Center Serine (203) of Acetylcholinesterase to Form a Conjugate with Three Subsequent Fates: Hydrolysis, Oxime Reactivation and Aging.
In studies where the active center gorge structure of AChE was modified through mutagenesis, it became apparent that both rates of inhibition (Hosea et al., 1995; 1996; Ordentlich et al., 1999; Kovarik et al., 2003) and reactivation (Wong et al., 2000; Kovarik et al., 2004) could be affected by altering the active center gorge configuration of AChE through site-directed mutagenesis. Moreover, these parameters could be influenced independently since the inactivation reaction could be treated as the SN2 reaction alluded to above, whereas optimal geometry for the reactivation reaction can not be achieved, since impaction within the narrow confines of the gorge limits the accessibility of the oxime or other nucleophiles to the conjugated phosphorus atom. Accordingly, modifying gorge dimensions should differentially affect the inactivation and reactivation processes.
By examining enantiomeric pairs of methylphosphonates, rates of inhibition by the more reactive Sp-enantiomer, were not appreciably affected by alterations in AChE active center gorge configuration, and only with the less reactive Rp-enantiomer inhibition could be enhanced through mutagenesis (Hosea et al., 1995, Kovarik et al., 2003). By contrast, reactivation of the Sp conjugates by oxime could be enhanced through mutagenesis to enlarge the acyl pocket of the active center and to remove a confining phenylalanine side chain from the choline subsite (Wong et al., 2000; Kovarik et al., 2004). Accordingly, it is possible to maintain rapid reactivity with organophosphate in the mutant enzyme, yet enhance the capacity of the oxime to reactivate the organophosphate conjugated enzyme. Hence, one could favor the rate of organophosphate turnover by oxime through mutation of the AChE structure. A series of mutations were examined in mouse AChE where the two phenylalanines outlining the acyl pocket are changed to leucine and isoleucine and a contributing phenylalanine side chain on the choline side of the active center was converted to alanine. The permutation that gave the greatest enhancement of the reactivation rate for the most efficient oxime, HI-6, was F295L/Y337A (Kovarik et al., 2004); this enhancement increased reactivation some 120 fold (Table 1). A comparison of the reactivation rates for cycloheptyl methylphosphono-AChE, 3,3-dimethylbutyl methylphosphono-AChE and isopropyl methylphosphono-AChE shows the rate enhancement to be the greatest for the cycloheptyl methylphosphono-AChE suggesting that increasing clearance within the space-confining gorge is the predominant factor in enhancing catalytic rates. Interestingly, the pattern of mutations for enhancing 2-PAM reactivation rates differed from HI-6 reactivation rates for the three phosphonates studied here (Kovarik et al., 2004). Accordingly, a rather complex set of structural relationships emerges where antidotal efficacy will be a function of the oxime structure, the conjugate to be reactivated, and the residue replacements in the mutated enzyme.
Table 1.
Enhancement of Oxime Elicited Rates of Reactivation of the SP- Methylphosphonyl Conjugates of Mouse Acetylcholinesterase with Various Mutations.1
| kr (mutant) / kr (wild type) | ||||
|---|---|---|---|---|
| Oxime | Mutant |  |
 |
 |
| HI-62 | F295L | 20 | 12 | 2.5 |
| F297I | 2.9 | 3.1 | 1.8 | |
| Y337A | 18 | 12 | 0.18 | |
| F295L/Y337A | 118 | 13 | 0.55 | |
| F297I/Y337A | 21 | 7.1 | 1.8 | |
| Y337A/F338A | 0.91 | 0.46 | 0.13 | |
|
| ||||
| 2-PAM3 | F295L | 2.1 | 6.1 | 0.11 |
| F297I | 14 | 167 | 0.72 | |
| Y337A | 6.2 | 0.22 | 0.076 | |
| F295L/Y337A | 2.6 | 3.7 | 0.0032 | |
| F297I/Y337A | 11 | 1.5 | 0.49 | |
| Y337A/F338A | 0.71 | 2.3 | 0.043 | |
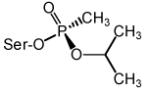
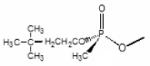
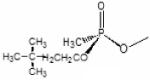
Data computed from Wong et al., 2000 and Kovarik et al., 2004. Structures of SP-cycloheptyl, 3,3-dimethylbutyl and isopropyl methylphosphonate conjugates with AChE are shown.
kr (wild type) was 122, 102 and 1330 min−1M−1 for SP-cycloheptyl, 3,3-dimethylbutyl and isopropyl methylphosphonate conjugates, respectively (Kovarik et al., 2004).
kr (wild type) was 0.66, 0.18 and 1080 min−1M−1 for SP-cycloheptyl, 3,3-dimethylbutyl and isopropyl methylphosphonate conjugates, respectively (Kovarik et al., 2004).
Because of the multiple structural permutations, we are developing a higher through-put, multiple reaction well system that can measure organophosphate turnover colorimetrically (Kovarik et al., 2006). With this system we can detect directly the enhancement of rate (kr) using organophosphates with a thiocholine or a thiomethyl leaving group (Fig. 2). These studies show the dependence of the overall rate of reaction on oxime. The pH dependence also documents the expected dependence of the rate of formation of the nucleophilic oxime anion (Kovarik et al., 2006).
Figure 2.

Reaction Cycle of Alkyl Methylphosphonfluoridate Hydrolysis Mediated by Acetylcholinesterase (E-OH) and an Oxime (OX). The Resulting Phospho-oxime Hydrolyzes to the Phosphonate Acid and Oxime.
Reports at this and other conferences have shown the utility of BuChE as a stoichiometric scavenger and its potential use as an antidote in the treatment of organophosphate toxicity in experimental animals (Lenz et al., 2006; Doctor et al. 2003). While this is a critical lead into scavenging inhibitors in vivo, developing catalytic capacity toward organophosphate hydrolysis will substantially reduce dose requirements.
An ideal catalytic system should not only have a high turnover rate, but also a rapid rate of reaction with the offending organophosphate. Scavenging in the plasma space requires that catalyzed hydrolysis occurs rapidly in the initial distribution phase of pharmacokinetics. Hence, to preserve the tissue localized AChE, the offending agent should be hydrolyzed within a minute or two and prior to its movement from the plasma to extra-vascular space. The kinetics reveal that the transphosphorylation step in going from the alkylmethylphosphonothioester to the alkylmethylphosphono-AChE for the mutant enzymes is rapid and proceeds at the efficient rate seen with nerve agent phosphonates or insecticide phosphorates when they react with AChE in situ (Hosea et al., 1995; Kovarik et al., 2003). Hence, regeneration of active enzyme depends on the second step or the “oxime nucleophile-assist” in the reaction. Details may be found in a companion study presented in this volume (Kovarik et al., 2006).
A second advantage of this system is that the oxime distributed in the body will have a dual mode of action. In the plasma space and in the presence of a mutant AChE retained in that location, oximes will enhance the second step in the catalytic turnover reaction (Fig. 1). In tissues, it will reactivate the already phosphorylated enzyme. Since oximes are well distributed in extracellular space, they have the pharmacokinetic and bioavailability parameters to serve this function. Repeated and prolonged oxime administration would confer maximum benefit to this mode of administration, since hydrophobic and volatile organophosphates are expected to be retained in and then slowly leeched from body fat.
Much more should be learned of the catalytic potential and selectivity of the mutant AChE’s. AChE, itself, may have other advantages over butyrylcholinesterase (BuChE) as a scavenging entity in that it will have virtually identical stereoselectivity as the target itself. Accordingly, the mouse AChE mutation sequence described above establishes proof of principle, in vitro, and we are developing the same mutation sequence for human AChE to achieve enhancement of organophosphate catalysis. It is likely that we have not yet maximized catalytic potential in the limited mutation sequence we have developed to date.
Accordingly, there are two ways to advance mutagenesis for enhancing organophosphate hydrolysis rates further. One approach relies on structure-guided design where we track efficiencies of oxime reactivation in relation to active center gorge configuration and dimensions, attempting to enhance access of the oxime to the electrophilic phosphorus atom and optimize the angle of oxime access. Our studies showing rather limited reactivation rates of BuChE (Kovarik et al., 2004), compared to some of the AChE mutants with BuChE residue substitutions, reveal that simply an open gorge is not the answer (Table 1). Conjugated phosphonates may associate with the gorge wall making them inaccessible to oxime attack. Fine detailing of the mutations should reveal means for improving the catalytic constants.
A second means to enhance catalysis entails the development of a random mutagenesis sequence in which we select mutant enzymes for enhanced catalytic rates. To achieve this, a multi-well, mid-range through-put system will be necessary for a full analysis (Kovarik et al., 2006). We propose somatic cell hypermutation (Wang et al., 2004; Wang, C.L., 2004) as an attractive approach to optimizing the mutants for achieving AChE-catalyzed hydrolysis of particular organophosphate conjugates.
It should be recognized that such structure-activity considerations will be extensive. First, one will have to direct the particular oxime and mutant enzyme to the offending organophosphate that forms the near irreversible complex. In the case of nerve agents, these will be methylphosphonates of the G series, but also the phosphoramidates, such as tabun, will be examined. Second, the efficiency of the oximes will have to be ranked with respect to the offending organophosphate and mutant enzyme. In the case of pairing oximes and nerve agents, the data of Szinicz and co-workers (Worek et al., 2004a,b) provide a basis for oxime selection, and we might assume initially a general relationship between their efficiency as reactivators and as assisting agents in catalytic scavenging (Kovarik et al., 2006). Finally, AChE mutants will have to be assessed for catalytic efficiency with the particular oxime couple or partner optimized to the organophosphate conjugate. Again we can draw upon the development of antimicrobial agents as predictors of what we may find. It is unlikely that any one combination will prove to be the “panaceamycin” that will cover all organophosphates. Some combinations will exhibit greater spectrum of antidotal potential than others, but we may find only certain oxime mutant-AChE combinations work well for particular organophosphates. For example, ortho-substituted aldoximes are rather ineffective as reactivators of tabun, but more efficient reactivation can be achieved with para- substituted aldoximes (Čalić et al., 2006).
Finally, we should consider the prospects for “Oxime-assisted Catalysis” therapy for rapidly aging compounds such as soman. Oxime-assisted catalysis cannot reverse or reactivate aged AChE. On the other hand, the organophosphate must get to the target tissue for aging to be deleterious. Hence, prior exposure prophylactic scavenging in the plasma should be beneficial even for compounds with a high propensity for aging. It is also possible through mutations to minimize aging rates in recombinant AChEs (Saxena et al., 1993; Shafferman et al., 1996).
3. Design of a Second Generation of Nucleophilic Reactivators
The original studies showing reactivation potential of the oximes were based on nucleophilic reactivation with hydroxylamine and site-directing the nucleophicity using the methylpyridinium cation (Wilson et al., 1961). The early studies also showed hydroxamates to be reactivating agents. Work to date has largely been confined to the aldoximes, and as detailed in other chapters (Eyer et al., 2006), bis-quaternary oximes have shown enhanced therapeutic efficacy in many cases.
However, despite extensive work, cloning the cholinesterase genes, determining AChE primary and tertiary structures and assigning function and selectivity of action to particular domains and side chains in the molecule, little has been done to develop other nucleophilic reactivators. Accordingly, should oxime-assisted catalysis continue to prove a productive avenue for investigation, optimizing the nucleophilic reaction to the AChE gorge would serve a dual purpose.
Major considerations emerging from structural studies have been the sterically confined environment of the active center gorge of AChE (Sussman et al., 1991; Bourne et al., 1995), and that impaction can be more severe when bulky organophosphates are conjugated to the active center serine. One avenue of pursuit should be the development of soft cations, analogous to the alkylpyridiniums, where the cationic charge is distributed over several atoms by virtue of the resonance states of the molecule. Also, one could reduce steric limitations by going to five (imidazoliums) or four membered (tetrazolium) ring systems. Several other heterocyclic ring systems are known and should also be considered. Linear cations such as the amidines, formamidine and acetamidine, and the hydroxamates should be considered since they would provide a soft cation with fewer steric constraints. Certain amidines have been designed to be nicotinic agonists (Lewis, 2005), and when the position of the cationic amidine and nucleophilic moieties are optimized, a prospective reactivating agent is generated.
Finally, freeze-frame, click chemistry (Fig. 3), where the target itself is used to synthesize triazoles from acetylenic and azide building blocks, offers additional potential as a site directed, combinatorial approach to developing new reactivating agents (Lewis et al, 2002; Bourne et al., 2004; Manetsch et al., 2004). In the AChE case the organophosphate conjugate and the apo-enzyme could be used comparatively as the building blocks. Should the conjugate be unstable, other conjugates formed from sulfonate esters or sulfonyl fluorides should be considered. Hence, several avenues exist for the design not only of new oximes, but also other novel nucleophilic structures that minimize impaction constraints within the space-limited gorge.
Figure 3.

Proposed Reaction of a Peripheral Site Propidium Analogue with an Exocyclic Chain of Variable Chain Length and an Azido Alkyl Pyridine Aldoxime, also with variable Methylene Chain Length, (CH2)n and (CH2)m. Shown above is the proposed reaction in the mouse AChE active center gorge (Bourne et al., 1995) for the six membered methylene chain connected to propidium and three membered chain connected to the oxime.
In the case of design of new nucleophiles to assist the second step in organophosphate catalysis, formation of high affinity ligands is likely not to be the ultimate goal. If the affinity of the reactivating agent is too high, the reaction may result in formation of high affinity phospho-oxime complexes that reversibly inhibit the enzyme. Rather reactivating agents with intermediate affinities would be desired. Should phospho-oximes form and persist, trace amounts of phosphotriesterases or paraoxonases should ensure their removal (Kiderlen et al., 2005).
4. Remote Detection of Organophosphorus Compounds
A problem perhaps only secondary to antidotal therapy in chemical terrorism with organophosphates is detection of these agents in various settings. Here again, a modified AChE may prove useful in the remote sensing of organophosphorus compounds. The target, itself, has been modified by mutagenesis producing a single free cysteine in the molecule and conjugation to the cysteine to form a fluorescent side chain. The fluorescent side chain is inserted at a position where conformation is sensitive to organophosphate association, and thus changes in fluorescence only require reaction with the offending organophosphate ligand and obviate the requirement of adding reagents for detection of signal changes.
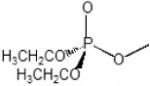
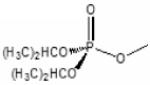
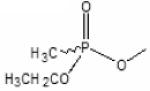
Previous studies have shown that, when ligands bind to the active center of AChE, fluorophores linked to positions 81 and 84 through substituted cysteines on the omega loop exhibit specific changes in fluorescence (Boyd et al., 2000; Shi et al., 2002). Since these residues are distant from the active center, the effect is allosteric in nature, reflecting a change in conformation in the omega loop, presumably loop closure or capping of the gorge. When Cys is substituted for Glu 81 in mammalian AChE and an acrylodan molecule is coupled to the cysteine, a substantial change in fluorescence quantum yield and emission maximum is observed upon binding of substrates, reversible inhibitors and conjugating inhibitors such as the organophosphates and carbamoylating agents. For the organophosphate conjugates, the emission spectrum is sensitive to the fit of the conjugating ligand in the active center. The SP-conjugates of the methylphosphonates cause a red or bathochromic shift in emission maximum whereas the RP organophosphates cause a blue or hypsochromic shift (Table 2, Fig. 4). Shifts in the blue direction reflect the fluorophore residing in a hydrophilic environment, whereas the red shift denotes exposure to the aqueous solvent. A comparison of a wide range of reversible inhibitors, carbamoylating agents and phosphorylating agents reveals that those ligands that fit the gorge and their carbonyl or phosphoryl oxygen can occupy the oxyanion hole show a red shift, while those ligands whose acyl or phosphoryl chain are too large to be accommodated in the acyl pocket effect a blue shift (Shi et al.,2001; 2002; 2003). Accordingly, the fluorescent AChE not only elicits a signal change with the binding of ligands, but can distinguish between certain types of ligands, ie: SP and RP-methylphosphonates with a larger alkoxy group; dimethoxyphosphorates from diethoxy- and larger alkylphosphorates; and carbamoylating agents with short, methyl and ethyl versus propyl and longer chains as the carbamoyl moiety. These differences reflect the limitations on acyl pocket size and are consistent with the well known cut off of AChE activity between propionylcholine and butyrylcholine (Augustinsson,1948). Thus, such fluorescence modifications not only show the potential for remote detection of organophosphates reacting with AChE, but also shed light on the determinants of specificity of ligands directed to the active center serine.
Table 2.
Distinctions of the Spectral Fluorescence Emission Shifts for Acrylodan Conjugated to E81C Acetylcholinesterase.
| Blue Spectral Shift | Red Spectral Shift | ||
|---|---|---|---|
|
Diethylphosphoryl | Dimethylphosphoryl |

|
|
Diisopropylphosphoryl | Ethyl methylphosphonyl |
|
|
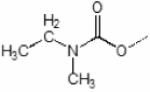
Rp-Dimethylbutyl methylphosphonyl | Sp-Dimethylbutyl methylphosphonyl |
|

|
Dimethylcarbamoyl | Sp-Isopropyl methylphosphonyl |
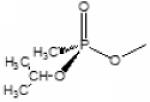
|
|
Ethyl methylcarbamoyl | SP- Cycloheptyl methylphosphonyl |
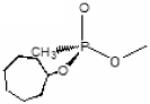
|
Organophosphates and carbamates whose alkyl group cannot be accommodated in the acyl pocket produce a blue shift with E81C whereas those with a small alkyl group induce a red shift.
Figure 4.

A) Emission Spectra of Conjugated Acrylodan for a 81 Glu (E) to Cys (C) mutation prior to and after reaction with SP-and RP -3,3-dimethylbutyl methylphosphono thiocholine (DMBMP-TCh). B) The actual color of cuvettes containing the conjugated AChE with fluorophore and organophosphate DDVP (O,O-dimethyl O-(2,2-dichlorovinyl)phosphate) or SP-and RP - enantiomers of DMBMP-TCh.
Summary
We describe here how AChE can serve as a template for the synthesis of inhibitors and in the future potential reactivating agents. The AChE molecule, which is the target of toxicity, can be modified in subtle ways through site-directed mutagenesis and chemical modification to become useful in the prophylaxis of and as an antidote for toxic exposure. Finally through conjugation of fluorophores to a cysteine substituted enzyme, AChE may serve as a detector of organophosphorus compounds.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augustinsson KB. Cholinesterases: a study in comparative enzymology. Acta Physiol. Scand. Suppl. 1948;52:1–182. [Google Scholar]
- Bourne Y, Taylor P, Marchot P. Acetylcholinesterase inhibition by fasciculin: crystal structure of the complex. Cell. 1995;83:493–506. doi: 10.1016/0092-8674(95)90128-0. [DOI] [PubMed] [Google Scholar]
- Bourne Y, Kolb HC, Radić Z, Sharpless KB, Taylor P, Marchot P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA. 2004;101:1449–1454. doi: 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd AE, Marnett AB, Wong L, Taylor P. Probing the active center gorge on acetylcholinesterase by fluorophores linked to substituted cysteines. J. Biol. Chem. 2000;275:22401–22408. doi: 10.1074/jbc.M000606200. [DOI] [PubMed] [Google Scholar]
- Čalić M, Vrdoljak A. Lucić, Radić B, Jelić D, Jun D, Kuča K, Kovarik Z. In vitro and in vivo evaluation of pyridinium oximes: mode of interaction with acetylcholinesterase, effect on tabun- and soman-poised mice and their cytotoxicity. Toxicology. 2006;219:85–96. doi: 10.1016/j.tox.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Doctor BP. Butyrylcholinesterase: its use for prophylaxis of organophosphate exposure. In: Giocobini E, editor. Butyrylcholinesterase: Its Function and Inhibitors. Martin Dunitz; London: 2003. pp. 163–177. [Google Scholar]
- Eyer P, Szinicz L, Thiermann H, Worek F, Zilker T. Testing of antidotes for organophosphorus compounds: Experimental procedures and clinical Reality. Medical Chemical Defense Special Issue, Toxicology. 2006 doi: 10.1016/j.tox.2006.08.033. submitted. [DOI] [PubMed] [Google Scholar]
- Giacobini E, editor. Cholinesterases and Cholinesterase Inhibitors. Martin Dunitz; London: 2000. [Google Scholar]
- Hosea NA, Berman HA, Taylor P. Specificity and orientation of trigonal carboxyl esters and tetrahedral alkylphosphonyl esters in cholinesterases. Biochemistry. 1995;34:11528–11536. doi: 10.1021/bi00036a028. [DOI] [PubMed] [Google Scholar]
- Hosea NA, Radić Z, Tsigelny I, Berman HA, Quinn DM, Taylor P. Aspartate 74 as a primary determinant in acetylcholinesterase governing specificity to cationic organophosphates. Biochemistry. 1996;35:10995–11004. doi: 10.1021/bi9611220. [DOI] [PubMed] [Google Scholar]
- Kiderlen D, Eyer P, Worek F. Formation and disposition of diethylphosphoryl-obidoxime, a potent anticholinesterase that is hydrolyzed by human paraoxonase (PON1) Biochem. Pharmacol. 2005;69:1853–1867. doi: 10.1016/j.bcp.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Radić Z, Berman HA, Simeon-Rudolf V, Reiner E, Taylor P. Acetylcholinesterase active center and gorge conformations analysed by combinatorial mutations and enantiomeric phosphonates. Biochem. J. 2003;373:33–40. doi: 10.1042/BJ20021862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik Z, Radić Z, Berman HA, Simeon-Rudolf V, Reiner E, Taylor P. Mutant cholinesterases possessing enhanced capacity for reactivation of their phosphonylated conjugates. Biochemistry. 2004;43:3222–3229. doi: 10.1021/bi036191a. [DOI] [PubMed] [Google Scholar]
- Kovarik Z, Radić Z, Berman HA, Taylor P. Mutation of acetylcholinesterase to enhance oxime assisted catalytic turnover of methylphosphonates. Medical Chemical Defense Special Issue, Toxicology. 2006 doi: 10.1016/j.tox.2006.08.032. submitted. [DOI] [PubMed] [Google Scholar]
- Lenz DE, Yeung D, Smith JR, Sweeney RE, Lumley LA, Cerasoli DM. Stoichiometric and catalytic scavengers as protection against nerve agent toxicity. A mini review. Medical Chemical Defense Special Issue, Toxicology. 2006 doi: 10.1016/j.tox.2006.11.066. submitted. [DOI] [PubMed] [Google Scholar]
- Lewis WG, Green LG, Grynszpan F, Radić Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Chick chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of femtomolar inhibitor from an array of building blocks. Angew Chem. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Lewis W. Ph.D. Thesis. The Scripps Research Institute; La Jolla, USA: 2005. [Google Scholar]
- Manetsch R, Kransinski A, Radić Z, Raushel J, Taylor P, Sharpless KB, Kolb HC. In situ click chemistry: enzyme inhibitors made to their own specifications. J. Amer. Chem. Soc. 2004;126:12809–12818. doi: 10.1021/ja046382g. [DOI] [PubMed] [Google Scholar]
- Ordentlich A, Barak D, Kronman C, Benshop HP, De Jong L, Ariel N, Barak R, Segall Y, Velan B, Shafferman A. Exploring the active center of human acetylcholinesterase with diastereoisomers of an organophosphorus inhibitor with two chiral centers. Biochemistry. 1999;38:3055–3066. doi: 10.1021/bi982261f. [DOI] [PubMed] [Google Scholar]
- Saxena A, Doctor BP, Maxwell DM, Lenz DE, Radić Z, Taylor P. The role of glutamate-199 in the aging of cholinesterase. Biochem Biophys Res Commun. 1993;197:343–349. doi: 10.1006/bbrc.1993.2481. [DOI] [PubMed] [Google Scholar]
- Shafferman A, Ordentlich A, Barak D, Stein D, Ariel N, Velan B. Aging of phosphonylated human acetylcholinesterase: catalytic processes mediated by aromatic and polar residues of the active center. Biochem J. 1996;318:833–840. doi: 10.1042/bj3180833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Boyd A,E, Radić Z, Taylor P. Reversibly bound and covalently attached ligands induce conformational changes in the omega loop, Cys69-Cys96, of mouse acetylcholinesterase. J. Biol. Chem. 2001;276:42196–42204. doi: 10.1074/jbc.M106896200. [DOI] [PubMed] [Google Scholar]
- Shi J, Radić Z, Taylor P. Inhibitors of different structure induce distinguishing conformations in the omega loop, Cys 69-Cys96, of mouse acetylcholinesterase. J. Biol. Chem. 2002;277:43301–43308. doi: 10.1074/jbc.M204391200. [DOI] [PubMed] [Google Scholar]
- Shi J, Tai K, McCammon JA, Taylor P, Johnson DA. Nanosecond dynamics of the mouse acetylcholinesterase Cys69-Cys96 omega loop. J Biol Chem. 2003;278:30905–30911. doi: 10.1074/jbc.M303730200. [DOI] [PubMed] [Google Scholar]
- Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine binding protein. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- Taylor P, Hosea NA, Tsigelny I, Radić Z, Berman HA. Determining ligand orientation and transphosphonylation mechanisms on acetylcholinesterase by RP, SP enantiomer selectivity and site-specific mutagenesis. Enantiomer. 1997;2:249–260. [PubMed] [Google Scholar]
- Wang L, Jackson WC, Steinbach PA, Tsien RY. Evolution of new non-antibody proteins via iterative somatic hypermutation. Proc. Natl. Acad. Sci. USA. 2004;101:16745–16749. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CL, Harper RA, Wabl M. Genome wide somatic hypermutation. Proc. Natl. Acd. Sci. USA. 2004;101:7352–7356. doi: 10.1073/pnas.0402009101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IB. Acetylcholinesterase IX. Reversibility of tetraethylpyrophosphate inhibition. J. Biol. Chem. 1951;190:111–117. [PubMed] [Google Scholar]
- Wilson IB, Ginsberg S. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim. Biophys. Acta. 1955;18:168–170. doi: 10.1016/0006-3002(55)90040-8. [DOI] [PubMed] [Google Scholar]
- Wilson IB. Molecular complementarity and antidotes for alkylphosphate poisoning. Fed. Proc. 1959;18:752–758. [PubMed] [Google Scholar]
- Wilson IB, Harrison MA, Ginsburg S. Carbamyl derivatives of acetylcholinesterase. J. Biol. Chem. 1961;236:1498–1500. [PubMed] [Google Scholar]
- Wilson BW, Arrieta DE, Henderson JD. Monitoring cholinesterases to detect pesticide exposure. Chem.-Biol. Interact. 2005;157:253–256. doi: 10.1016/j.cbi.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Wong L, Radić Z, Bruggeman RJM, Hosea N, Berman HA, Taylor P. Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis. Biochemistry. 2000;39:5750–5757. doi: 10.1021/bi992906r. [DOI] [PubMed] [Google Scholar]
- Worek F, Thiermann H, Szinicz L. Reactivation and aging kinetics of acetylcholinesterase inhibited by organophosphonylcholines. Arch. Toxicol. 2004a;78:212–217. doi: 10.1007/s00204-003-0533-0. [DOI] [PubMed] [Google Scholar]
- Worek F, Thiermann H, Szinicz L, Eyer P. Kinetic analysis of interactions between human acetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem. Pharmacol. 2004b;68:2237–2248. doi: 10.1016/j.bcp.2004.07.038. [DOI] [PubMed] [Google Scholar]