

Abstract
Protein function is often modulated by protein-protein interactions (PPIs) and therefore defining the partners of a protein helps to understand its activity. PPIs can be detected through different experimental approaches and are collected in several expert curated databases. These databases are used by researchers interested in examining detailed information on particular proteins. In many analyses the reliability of the characterization of the interactions becomes important and it might be necessary to select sets of PPIs of different confidence levels. To this goal, we generated HIPPIE (Human Integrated Protein-Protein Interaction rEference), a human PPI dataset with a normalized scoring scheme that integrates multiple experimental PPI datasets. HIPPIE's scoring scheme has been optimized by human experts and a computer algorithm to reflect the amount and quality of evidence for a given PPI and we show that these scores correlate to the quality of the experimental characterization. The HIPPIE web tool (available at http://cbdm.mdc-berlin.de/tools/hippie) allows researchers to do network analyses focused on likely true PPI sets by generating subnetworks around proteins of interest at a specified confidence level.
Introduction
Protein function occurs or is regulated by protein interactions and therefore knowledge on the partners of a given protein can give us important information regarding its activity. For instance, specific protein-protein interactions (PPIs) can be involved in diseases (see e.g. [1]). PPIs can be evaluated by many experimental methodologies, which have hugely different degrees of confidence and different experimental set-ups. For instance, while yeast two hybrid (Y2H) identifies direct physical interactions between two proteins, mass spectrometry (MS) based datasets report components of protein complexes, which may or may not be in direct physical contact. In addition to experimental methods, computational methods propose protein interactions based, for example, on orthology, protein domains known to interact, co-expression and functional annotations [2], [3].
PPIs are collected in several databases that make the data and the evidence behind it easily accessible and allow different mechanisms to query and display the data [4], [5], [6], [7], [8], [9], [10]. These resources are very useful for researchers interested in checking a small number of particular proteins of interest. However, PPI data can also be used globally for systematic network analyses, prediction of protein properties, and evaluation of novel datasets of PPIs produced in a high-throughput fashion.
Computational use of PPI datasets often requires selecting PPIs at particular levels of confidence. For example, the quality of a novel PPI dataset may be evaluated by its overlap with known interactions defined with high reliability, whereas a statistical analysis might require a large number of interactions therefore benefiting from a less restricted set of PPIs. The flexible selection of PPI datasets at various confidence levels requires a continuous scoring scheme for PPIs reflecting the reliability of their experimental characterization.
With the objective of creating a resource allowing the selection of PPIs by experimental confidence cut-offs, we generated HIPPIE (Human Integrated Protein-Protein Interaction rEference), a scored human PPI collection integrated from multiple sources. Following [8], we developed an expertly curated scoring scheme that takes into account the reliability of different experimental evidence in the definition of a PPI combining three types of information: experimental techniques used, number of studies finding the PPI, and reproducibility in model organisms.
A web tool to browse the data as well as the scored PPI dataset are provided at http://cbdm.mdc-berlin.de/tools/hippie. The scored dataset includes information on the data we used to build it so that modifications of the scoring mechanism can be easily achieved. We illustrate the usefulness of HIPPIE in increasing the coverage of novel PPI datasets and demonstrate that its scoring scheme reflects the reliability of the reported interactions.
Methods
2.1 Sources
Interactions were retrieved from the following public databases: BioGRID (version 2.0.62; release date: March 16, 2010) [4], DIP (release date: December 30, 2009) [5], HPRD (version 8; release date: July 6, 2009) [6], IntAct (release date: March 29, 2010) [7], MINT (release date: 9 November 2009) [8], BIND (2004 release [11]), and MIPS (published: November 5, 2004) [10]. Genetic interactions were removed from BioGRID. Additionally, we integrated interactions from manually selected studies [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22]; interactions from these studies were integrated that were not contained in the public databases at the time of integrating the sources. All resources integrated in HIPPIE are summarized in Table 1.
Table 1. PPI data sources integrated in HIPPIE.
| PPI dataset | Reference | Size |
| HPRD | [6] | 40110 |
| BioGRID | [4] | 30027 |
| IntAct | [7] | 28073 |
| MINT | [8] | 14094 |
| Rual05 | [19] | 6946 |
| Lim06 | [17] | 5579 |
| Bell09 | [13] | 3300 |
| Stelzl05 | [20] | 3232 |
| DIP | [5] | 1618 |
| BIND | [11] | 1415 |
| Colland04 | [21] | 882 |
| Lehner04 | [16] | 385 |
| Albers05 | [12] | 290 |
| MIPS | [10] | 252 |
| Venkatesan09 | [22] | 239 |
| Kaltenbach07 | [15] | 227 |
| Nakayama02 | [18] | 84 |
| HIPPIE | 72916 |
Where available, we retrieved the information on the originating study and the experimental methodology used to measure each interaction from the source databases and also assigned an experimental category to interactions from the additionally included studies. As a result, more than 99% of all interactions in HIPPIE are associated to at least one of the methods listed in Table 2 and are annotated with the studies in which they were detected.
Table 2. Scores for experiment types.
| Technique | PSI | score | Technique | PSI | Score |
| 3 hybrid method | MI:0588 | 5 | footprinting | MI:0417 | 3 |
| acetylation assay | 7.5 | FRET | 6 | ||
| Affinity Capture-Luminescence | 5 | gal4 vp16 complementation | MI:0728 | 5 | |
| Affinity Capture-MS | 5 | genetic interference | MI:0254 | 0 | |
| Affinity Capture-RNA | 2 | gst pull down | MI:0059 | 5 | |
| Affinity Capture-Western | 5 | gtpase assay | MI:0419 | 7.5 | |
| affinity chromatography technology | MI:0004 | 5 | his pull down | MI:0061 | 5 |
| affinity technology | MI:0400 | 5 | homogeneous time resolved fluorescence | MI:0510 | 7 |
| anti bait coimmunoprecipitation | MI:0006 | 5 | imaging technique | MI:0428 | 1 |
| anti tag coimmunoprecipitation | MI:0007 | 5 | in vitro | MI:0492 | 1 |
| antibody array | MI:0678 | 5 | in vivo | MI:0493 | 1 |
| array technology | MI:0008 | 3 | in-gel kinase assay | MI:0423 | 7.5 |
| atomic force microscopy | MI:0872 | 9 | inferred by curator | MI:0364 | 1 |
| beta galactosidase complementation | MI:0010 | 5 | ion exchange chromatography | MI:0226 | 3 |
| beta lactamase complementation | MI:0011 | 5 | isothermal titration calorimetry | MI:0065 | 10 |
| bimolecular fluorescence complementation | MI:0809 | 6 | kinase homogeneous time resolved fluorescence | MI:0420 | 7.5 |
| Biochemical | MI:0401 | 1 | lambda phage display | MI:0066 | 6 |
| Biochemical Activity | 5 | lex-a dimerization assay | MI:0369 | 5 | |
| bioluminescence resonance energy transfer | MI:0012 | 6 | light microscopy | MI:0426 | 1 |
| Biophysical | MI:0013 | 1 | light scattering | MI:0067 | 10 |
| blue native page | MI:0276 | 3 | mammalian protein protein interaction trap | MI:0231 | 6 |
| chromatin immunoprecipitation assay | MI:0402 | 2 | mass spectrometry studies of complexes | MI:0069 | 5 |
| chromatography technology | MI:0091 | 1 | methyltransferase assay | MI:0515 | 7.5 |
| circular dichroism | MI:0016 | 9 | methyltransferase radiometric assay | MI:0516 | 7.5 |
| classical fluorescence spectroscopy | MI:0017 | 7.5 | molecular sieving | MI:0071 | 2 |
| Co-crystal Structure | 10 | no experiment assigned | 0 | ||
| Co-fractionation | 1 | nuclear magnetic resonance | MI:0077 | 10 | |
| Co-localization | 1 | peptide array | MI:0081 | 5 | |
| Coimmunoprecipitation | MI:0019 | 5 | phage display | MI:0084 | 6 |
| colocalization by fluorescent probes cloning | MI:0021 | 1 | phosphatase assay | MI:0434 | 7.5 |
| colocalization by immunostaining | MI:0022 | 1 | phosphotransfer assay | 7.5 | |
| colocalization/visualisation technologies | MI:0023 | 1 | polymerization | MI:0953 | 5 |
| comigration in gel electrophoresis | MI:0807 | 3 | protease assay | MI:0435 | 7.5 |
| comigration in non denaturing gel electrophoresis | MI:0404 | 3 | protein array | MI:0089 | 5 |
| comigration in sds page | MI:0808 | 3 | protein complementation assay | MI:0090 | 5 |
| competition binding | MI:0405 | 5 | protein cross-linking with a bifunctional reagent | MI:0031 | 5 |
| confocal microscopy | MI:0663 | 1 | protein kinase assay | MI:0424 | 7.5 |
| Copurification | MI:0025 | 2 | protein tri hybrid | MI:0437 | 5 |
| Cosedimentation | MI:0027 | 2 | Protein-peptide | 5 | |
| cosedimentation in solution | MI:0028 | 2 | Protein-RNA | 0 | |
| cosedimentation through density gradient | MI:0029 | 2 | pull down | MI:0096 | 2.5 |
| cross-linking study | MI:0030 | 5 | pull-down/mass spectrometry | 5 | |
| deacetylase assay | MI:0406 | 7.5 | Reconstituted Complex | 10 | |
| demethylase assay | MI:0870 | 7.5 | reverse phase chromatography | MI:0227 | 1 |
| dihydrofolate reductase reconstruction | MI:0111 | 6 | reverse two hybrid | MI:0726 | 5 |
| dynamic light scattering | MI:0038 | 9 | ribonuclease assay | MI:0920 | 7.5 |
| electron microscopy | MI:0040 | 5 | saturation binding | MI:0440 | 7.5 |
| electron paramagnetic resonance | MI:0042 | 9 | scintillation proximity assay | MI:0099 | 7.5 |
| electron tomography | MI:0410 | 9 | solid phase assay | MI:0892 | 1 |
| electrophoretic mobility shift assay | MI:0413 | 2 | surface plasmon resonance | MI:0107 | 10 |
| electrophoretic mobility supershift assay | MI:0412 | 2 | t7 phage display | MI:0108 | 6 |
| enzymatic study | MI:0415 | 1 | tandem affinity purification | MI:0676 | 5 |
| enzyme linked immunosorbent assay | MI:0411 | 5 | tox-r dimerization assay | MI:0370 | 5 |
| experimental interaction detection | MI:0045 | 1 | transcriptional complementation assay | MI:0232 | 5 |
| far western blotting | MI:0047 | 5 | transmission electron microscopy | MI:0020 | 5 |
| filamentous phage display | MI:0048 | 6 | two hybrid fragment pooling approach | MI:0399 | 5 |
| filter binding | MI:0049 | 5 | Two-hybrid | MI:0018 | 5 |
| fluorescence correlation spectroscopy | MI:0052 | 10 | ubiquitin reconstruction | MI:0112 | 5 |
| fluorescence microscopy | MI:0416 | 1 | x ray scattering | MI:0826 | 9 |
| fluorescence polarization spectroscopy | MI:0053 | 10 | x-ray crystallography | MI:0114 | 10 |
| fluorescence technology | MI:0051 | 1 | x-ray fiber diffraction | MI:0825 | 9 |
| fluorescence-activated cell sorting | MI:0054 | 1 | yeast display | MI:0115 | 5 |
| fluorescent resonance energy transfer | MI:0055 | 6 |
To add to the confidence scoring of experimentally verified human PPIs a component based on experimental evidence in non-human organisms we included data from three databases that map interactions between non-human protein pairs to their human orthologs: HomoMINT (release date: March 5, 2009) [23], I2D (release date: January 7, 2010) [2] and the PPI dataset from [24].
2.2 Identifier mapping
Different public PPI databases and datasets use different types of gene or protein identifiers. We aimed at mapping all protein pairs listed in HIPPIE to Entrez Gene and UniProt identifiers. For this purpose we applied the database identifier mapping tables curated by UniProt [25] and the HUGO Gene Nomenclature Committee (HGNC) [26]. We mapped all database entries to their canonical representatives and did not consider splicing forms. In the web interface the data can be queried either by protein (UniProt id or accession) or by gene identifier (Entrez Gene id or gene symbol). Interactions containing identifiers that could not be mapped to human Entrez Gene ids or UniProt ids were not included in HIPPIE.
Mapping PPIs to the genes encoding the interacting proteins is affected by certain ambiguity since the same protein sequence may be encoded by duplicated genomic loci. In the flat file version of HIPPIE these ambiguous PPIs are expanded such that a given PPI is represented by all possible combinations of gene identifiers.
2.3 Score calculation
For each interaction a score S between 0 and 1 was calculated reflecting the reliability of its combined experimental evidence. This score was calculated as a weighted sum of three different subscores which are ss (a function of the number of studies in which an interaction was detected), st (a function of the number and quality of experimental techniques used to measure an interaction; see below for details) and so (a function of the number of non-human organisms in which an interaction was reproduced). Each of these three subscores si was calculated with a non-linear saturating function of the form:
such that si(0) = 0 and si(∞) = 1, where the a i are constants that control the steepness of the function.
For subscore ss, n is the number of different studies where the interaction was reported (number of PubMed identifiers associated), regardless of whether multiple experimental evidence was provided in each study.
For subscore so, n is the number of species where orthologs of the interacting proteins could be defined and were found experimentally to interact (currently Bos taurus, Caenorhabditis elegans, Canis familiaris, Drosophila melanogaster, Gallus gallus, Mus musculus, Rattus norvegicus, Saccharomyces cerevisiae, and Sus scrofa).
For subscore st, n is a sum of scores from different experimental techniques by which an interaction was verified (even if used in the same study). Most PPI databases use controlled vocabulary descriptors for these experimental techniques as defined by the PSI-MI ontology [27], however for some terms we could not find an equivalent ontology term. Manual curation was used to assign a score to each PPI detection method ranging from 0 (no experiment assigned, less than 1% of PPIs) to 10. Scores and corresponding PSI-MI codes are displayed in Table 2. Methods that can ascertain interactions with the highest reliability, such as in vitro techniques like X-ray crystallography, were assigned the highest scores. Complementation-based assays and affinity based technologies were roughly equally scored with an average value of 5, slightly increased for those methods that are used generally in homologous, more physiological setups, such as FRET. Methodologies that do not directly provide evidence for interaction, such as colocalization or cosedimentation, are scored with the lowest values. The total score S was calculated as a weighted sum of the three subscores:
with  .
.
It is important to note that our dataset does not include interactions not experimentally verified with human proteins: no interaction received a score alone from its verification in non-human organisms. We also remark that this scoring scheme does not consider computational evidence other than the definition of orthology relations from human proteins to proteins in other organisms.
2.4 Parameter selection
The six free parameters of the scoring formula (as, ao, at, ws, wo and wt) were optimized by performing a grid search in the parameter space. We performed the search in the range [0, 3] for the ai and in the range [0, 1] for the wi. We chose a step width of 0.1 for both ai and the wi. The step width was chosen sufficiently small such that selecting neighboring parameter combinations resulted only in small changes in the interaction scores which decreased the probability of missing an optimal solution. Constraints were set on the weights wi by requiring that they sum up to 1. Parameter combinations leading to only few discrete scores were excluded (this happened, for example, when wt was set to 0, since the different experimental weights account for a large fraction of the score's granularity).
PPIs are sometimes reported in multiple studies. We reasoned that we could use this property to assess the performance of a parameter combination. To do this evaluation we used the IntAct dataset, which currently consists of 28 073 interactions (38.5% of HIPPIE). This dataset has explicit associations between studies and experiments, and the experimental information is annotated following the PSI-MI format.
The assessment of performance of a parameter set was done by successively removing each one of the 109 studies in IntAct that contain at least 10 interactions and more than two PPIs found in multiple studies. For each study j, we recalculated the scores of the remaining dataset, IntActred, found the set of PPIs described both in the study j and in IntActred, 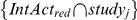 , and computed the deviation from random expectation of the number of highly scored interactions among the overlap:
, and computed the deviation from random expectation of the number of highly scored interactions among the overlap:
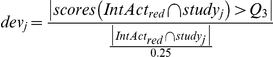 |
where Q3 is the upper quartile of the score distribution of IntActred.
To measure the overall performance of a parameter combination we chose a function f of the weighted mean of the logarithm of devi over all studies:
where the weights vi were chosen proportional to the overlap size between IntActred and studyj and n is the number of studies. The best parameter combination maximizes f.
We found several parameter combinations (several thousand optimal combinations out of more than 700 000 different parameter combinations tested) maximizing the function f (max(f) = 1.023). From the equally well performing parameter combinations we chose the set of parameters that resulted in the largest spread of the distribution of scored interactions. For that purpose the scores of the entire HIPPIE were repeatedly calculated for each of the optimal parameter combination and for each score distribution the interquartile range (iqr) was determined. We found that the parameter set [as = 2.3, ao = 1.6, at = 0.2, ws = 0.6, wo = 0.1, wt = 0.3] maximized both f and iqr.
Results
HIPPIE is a dataset of experimentally measured human PPI derived from several publicly available PPI datasets (Table 1). For reference, we distribute a stable release of HIPPIE consisting of 72 916 interactions, which was used in this manuscript for several descriptive analyses (Table S1; HIPPIE version 1.2). The live version of HIPPIE is monthly updated making use of the web query interface PSICQUIC [28], which allows us to automatically retrieve the newest interaction data from most of the manually curated source databases (BioGrid, IntAct, MINT, DIP and BIND) and integrate the new interactions and updated evidence records into HIPPIE.
The network is accessible via a web tool (http://cbdm.mdc-berlin.de/tools/hippie) that allows for querying the interactions by a gene symbol, Entrez gene id or UniProt identifier (id and accession). On the result page a confidence score is listed with each interaction partner of the query protein and detailed information on the evidence contributing to the confidence score can be accessed. Links to the original studies are provided.
A typical problem after generation of experimental results producing a list of genes, proteins and/or interactions between them, is the evaluation of the results in relation to the known PPI data. For example, a researcher may have obtained proteomics data for a few proteins of interest and wants to evaluate the novelty of the interactions, or the possible relation of the interactors with a disease protein of interest.
To facilitate this analysis, HIPPIE can be queried with a set of proteins and/or interactions between them from which a network of known data around the proteins of interest is constructed. The online tool will identify interactions between the proteins submitted (layer 0 network), or their interactors not contained in the query set (layer 1 network). The computation of networks with more layers might be lengthy if hundreds of protein partners have to be analysed. For this we provide a Java command line tool (available from http://cbdm.mdc-berlin.de/tools/hippie and also deposited at the SourceForge open software archive: https://sourceforge.net/projects/hippiecbdm) that will do the computation on the local machine of the user for large input sets or neighbours of neighbours. A confidence threshold to control the reliability and size of the constructed network can be also applied. Additionally, we provide a filter option for the PSI-MI interaction type annotation provided by most of HIPPIE's source databases. This feature allows for selecting direct physical interactions from HIPPIE. The thereby generated HIPPIE subnetworks can be exported from HIPPIE for further analyses or can be visualized using the tool Cytoscape Web [29], which has been integrated into HIPPIE.
The web site also offers the entire HIPPIE dataset for download in two different formats: in PSI-MI TAB 2.5 format as defined by the Protein Standard Initiative [27] and in our own tab delimited flat file format. Currently we distribute a freeze version (version 1.2) used in this manuscript for analyses, and the monthly updated version.
While merging the different data sources we kept track of the information about which experimental system type was used to detect each single interaction and whether there were several studies where the interaction was found. Additionally we retrieved the interaction data from PPI databases that link interactions in non-human model organisms to their human orthologs. From these different types of information (experimental systems, number of studies and reproducibility in other organisms) we calculated an overall score reflecting the reliability of each interaction (See Methods for details and Table 2).
We note that the different experimental methodologies behind the PPIs in HIPPIE are able to detect direct physical interactions between proteins to a varying degree. Even though some of them are in fact measuring co-membership in larger protein-complexes we will refer to all types of associations detected by these methods as interactions or PPIs. The HIPPIE score tries to reflect both the reliability of the various methods as well as the ability to detect direct rather than indirect interactions.
The number of PPIs derived from different experimental system types was very variable. HIPPIE integrates various datasets dealing with different experimental systems and thus contains a larger amount of interactions than each of those sets separately (Table 1). Values for three well populated and meaningful sources of PPIs, Y2H, anti-bait coimmunoprecipitation (Coprep), and tandem affinity purification (TAP) are shown in Figure 1 that cover 78% of the total amount of proteins in the current version of HIPPIE, but only around 50% of its interactions. Coprep and TAP share relatively many PPIs between each other (139 PPIs) compared to the other pairwise overlaps between methods. For example, TAP shares 95 interactions with Y2H despite the much higher amount of Y2H interactions as compared to Coprep. This higher overlap between Coprep and TAP in comparison with the Y2H data might reflect the similarity between the first two approaches in comparison with the latter, as Coprep and TAP are both based on antibody capture of a protein complex while Y2H is based on the reconstitution of a binary interaction inside of a heterologous system (yeast).
Figure 1. Coverage of HIPPIE and overlap by three technique specific datasets.

Left: proteins. Right: PPIs. Y2H is yeast-two-hybrid, Coprep is anti-bait coimmunoprecipitation and MS is affinity capture mass spectrometry. The protein numbers show that Y2H can focus on many proteins that have not been targeted by the other two techniques. Together the three techniques already cover 80% of all proteins currently considered in HIPPIE (i.e. 80% of all proteins in HIPPIE participate in at least one Y2H, Coprep or MS experiment). However, the overlap in PPIs between these datasets and to the remainder of HIPPIE is much smaller indicating that PPI detection is technique specific. Nevertheless, one can appreciate that similar techniques have a bias towards detecting similar PPIs, here illustrated by the significant overlap between Coprep and MS and by the little overlap of Y2H to the other two techniques.
To illustrate the benefit of using a large dataset such as HIPPIE, we compared it with novel high-throughput PPI datasets not used for its production. We chose two high-throughput PPI datasets from the recent literature: a Y2H dataset, Y2He [30], containing 551 PPIs between 434 proteins and a MS dataset, MSe [31], containing 711 PPIs between 424 proteins. The coverage of the Y2He and MSe datasets by HIPPIE was of 120 (21.8%) and 73 (10.3%) PPIs, respectively.
We evaluated the usefulness of the HIPPIE score using the two novel datasets. The HIPPIE database was divided in a high quality subset containing the top 25% highest scoring interactions (score > = 0.73) and a lower quality subset (score <0.73; see Figure 2). Then, we compared the fraction of PPIs in each HIPPIE subset that was recalled by the novel dataset. If the scores are meaningful one would expect better recall of the set with high-confidence scores.
Figure 2. Distribution of HIPPIE confidence scores.

Interactions with scores above 0.73 (black bars) constitute only 25% of all and could be considered high-confidence interactions. According to the design of the scoring function, such score implies that the interaction is supported by multiple evidence.
To measure the recall of HIPPIE by an external dataset of PPIs one has to consider that some HIPPIE PPIs may not be detectable by the experimental setup used to produce the external dataset. In the case of Y2H and MS datasets a number of proteins are used as baits. Therefore, we considered for each of these studies that the “detectable PPIs” from HIPPIE were those where at least one of the interacting proteins was a bait in the study considered (Table 3). The values of detectable PPIs and recall were used to calculate one-sided Fisher's exact tests to assess the significance of the differences in recall between high and low confidence HIPPIE subsets. The high quality subset had the largest overlaps in percentage with the PPIs of the novel datasets and these overlaps were significant (Table 3; p-values of 6.40e-15 and 1.75e-6 for Y2He and MSe, respectively) suggesting that the PPI score correlates with experimental reproducibility.
Table 3. Coverage of HIPPIE by novel datasets.
| HIPPIE subset | HIPPIE subset size | Y2He | MSe | ||
| detectablePPIs | Overlap PPIs(recall) | detectablePPIs | Overlap PPIs (recall) | ||
| score > = 0.73 | 18592 | 2239 | 75 (3.3%) | 322 | 41 (12.7%) |
| score <0.73 | 54324 | 5760 | 45 (0.8%) | 806 | 32 (4.0%) |
Discussion
In this work we presented HIPPIE, an integrated dataset of human protein interaction data scored according to experimental evidence. This resource has been created for those researchers that need to use globally the complete knowledge on human protein interactions. This is required in systems biology studies and in the evaluation of high-throughput results (e.g. novel PPI datasets) that require contrasting results with interactions selected for a particular level of reliability.
HIPPIE currently integrates 72 916 interactions from several public PPI resources scored according to confidence. For comparison, the complete human interactome map has been estimated to contain between 200 000 and 400 000 interactions (according to [32] and [33], respectively) suggesting that our knowledge of the human interactome is still incomplete. Nevertheless, producing a large collection of integrated PPI data is critical for its usability because novel high-throughput PPI datasets often contain just hundreds of PPIs and might have little overlap with smaller existing PPI resources integrated in HIPPIE.
Several resources have been created that, like HIPPIE, integrate PPI data from multiple sources but do not have a focus on distributing a simple scored dataset, while offering excellent tools to examine evidence behind each PPI (e.g. iRefWeb [34]) or do not focus on experimentally verified interactions (e.g. STRING [35]). Some other databases offer a continuous confidence scoring scheme, e.g. MINT [8] and HAPPI [36], but they do not allow batch scoring of PPI sets or the exclusive retrieval of high confidence interactions and lack the integration of several important high-throughput experimental datasets. The scoring system of MINT is closer to the one we use as it considers levels of technical evidence, number of studies and orthology [8]; however, as the PPI data from MINT is manually curated, the amount of human PPIs in MINT is currently less than a third of those in HIPPIE, limiting its use in the evaluation of novel datasets. Finally, in contrast with MINT and HIPPIE, HAPPI contains only a small fraction of PPIs experimentally derived in human while the majority are either computationally predicted or inferred from other species.
We are aware that any assignment of reliability scores to experimental techniques necessarily reflects the individual belief of researchers. We tried however to base our selection of parameters and weights in the scoring formula on objective criteria by optimizing the performance of our scoring scheme in assigning high values to reproducible interactions. For researchers who nevertheless wish to modify either the selected parameters or the scores assigned to the different techniques we offer a tool at our homepage that allows the scoring of HIPPIE using an altered set of these values.
HIPPIE has been used for the evaluation of existing novel PPI datasets showing that it increases their coverage over individual resources and that its scoring scheme correlates with the ability to find a PPI in experimental data not included in the database (Table 3). A web tool to query the data, the scored PPI dataset as well as the raw data are available at http://cbdm.mdc-berlin.de/tools/hippie. The tool allows batch annotation of datasets of PPIs. Future work on HIPPIE will be directed towards the inclusion of novel datasets and versions for major model organisms.
Supporting Information
Scored dataset of PPIs. The columns indicate (1) UniProt identifier and (2) Entrez Gene identifier of the first protein partner, (3) UniProt identifier and (4) Entrez Gene identifier of the second protein partner, (5) score and (6) a comment field summarizing the origin of the evidence. Evidence is arranged in three types: experiments, pmids, and sources. Experiment types are indicated in Table 2. Pmids are the PMID of manuscripts reporting the interaction. Sources are the datasets where the interaction was found and are indicated in Table 1. Multiple evidences for each type are separated by semicolon and multiple evidence codes for each type are separated by comma. If one protein maps to several genes, each combination of genes is listed in a separate line. This table is available from: http://cbdm.mdc-berlin.de/tools/hippie/hippie_v1_2.txt.
(TXT)
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by the German Ministry of Education and Research (BMBF) [grant number 01GS08170], and by the Helmholtz Alliance in Systems Biology (Germany). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hwang S, Son SW, Kim SC, Kim YJ, Jeong H, et al. A protein interaction network associated with asthma. J Theor Biol. 2008;252:722–731. doi: 10.1016/j.jtbi.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 2.Brown KR, Jurisica I. Online predicted human interaction database. Bioinformatics. 2005;21:2076–2082. doi: 10.1093/bioinformatics/bti273. [DOI] [PubMed] [Google Scholar]
- 3.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, et al. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, et al. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–539. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Isserlin R, El-Badrawi RA, Bader GD. The Biomolecular Interaction Network Database in PSI-MI 2.5. Database (Oxford) 2011;2011:baq037. doi: 10.1093/database/baq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagel P, Kovac S, Oesterheld M, Brauner B, Dunger-Kaltenbach I, et al. The MIPS mammalian protein-protein interaction database. Bioinformatics. 2005;21:832–834. doi: 10.1093/bioinformatics/bti115. [DOI] [PubMed] [Google Scholar]
- 11.Bader GD, Betel D, Hogue CW. BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albers M, Kranz H, Kober I, Kaiser C, Klink M, et al. Automated yeast two-hybrid screening for nuclear receptor-interacting proteins. Mol Cell Proteomics. 2005;4:205–213. doi: 10.1074/mcp.M400169-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Bell R, Hubbard A, Chettier R, Chen D, Miller JP, et al. A human protein interaction network shows conservation of aging processes between human and invertebrate species. PLoS Genet. 2009;5:e1000414. doi: 10.1371/journal.pgen.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell. 2004;15:853–865. doi: 10.1016/j.molcel.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehner B, Sanderson CM. A protein interaction framework for human mRNA degradation. Genome Res. 2004;14:1315–1323. doi: 10.1101/gr.2122004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim J, Hao T, Shaw C, Patel AJ, Szabo G, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 18.Nakayama M, Kikuno R, Ohara O. Protein-protein interactions between large proteins: two-hybrid screening using a functionally classified library composed of long cDNAs. Genome Res. 2002;12:1773–1784. doi: 10.1101/gr.406902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 20.Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 21.Colland F, Jacq X, Trouplin V, Mougin C, Groizeleau C, et al. Functional proteomics mapping of a human signaling pathway. Genome Res. 2004;14:1324–1332. doi: 10.1101/gr.2334104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, et al. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Persico M, Ceol A, Gavrila C, Hoffmann R, Florio A, et al. HomoMINT: an inferred human network based on orthology mapping of protein interactions discovered in model organisms. BMC Bioinformatics. 2005;6(Suppl 4):S21. doi: 10.1186/1471-2105-6-S4-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehner B, Fraser AG. A first-draft human protein-interaction map. Genome Biol. 2004;5:R63. doi: 10.1186/gb-2004-5-9-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seal RL, Gordon SM, Lush MJ, Wright MW, Bruford EA. genenames.org: the HGNC resources in 2011. Nucleic Acids Res. 2011;39:D514–519. doi: 10.1093/nar/gkq892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, et al. The HUPO PSI's molecular interaction format–a community standard for the representation of protein interaction data. Nat Biotechnol. 2004;22:177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]
- 28.Aranda B, Blankenburg H, Kerrien S, Brinkman FS, Ceol A, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat Methods. 2011;8:528–529. doi: 10.1038/nmeth.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, et al. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bandyopadhyay S, Chiang CY, Srivastava J, Gersten M, White S, et al. A human MAP kinase interactome. Nat Methods. 2010;7:801–805. doi: 10.1038/nmeth.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bork P, Jensen LJ, von Mering C, Ramani AK, Lee I, et al. Protein interaction networks from yeast to human. Curr Opin Struct Biol. 2004;14:292–299. doi: 10.1016/j.sbi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Ramani AK, Bunescu RC, Mooney RJ, Marcotte EM. Consolidating the set of known human protein-protein interactions in preparation for large-scale mapping of the human interactome. Genome Biol. 2005;6:R40. doi: 10.1186/gb-2005-6-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner B, Razick S, Turinsky AL, Vlasblom J, Crowdy EK, et al. iRefWeb: interactive analysis of consolidated protein interaction data and their supporting evidence. Database (Oxford) 2010;2010:baq023. doi: 10.1093/database/baq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JY, Mamidipalli S, Huan T. HAPPI: an online database of comprehensive human annotated and predicted protein interactions. BMC Genomics. 2009;10(Suppl 1):S16. doi: 10.1186/1471-2164-10-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scored dataset of PPIs. The columns indicate (1) UniProt identifier and (2) Entrez Gene identifier of the first protein partner, (3) UniProt identifier and (4) Entrez Gene identifier of the second protein partner, (5) score and (6) a comment field summarizing the origin of the evidence. Evidence is arranged in three types: experiments, pmids, and sources. Experiment types are indicated in Table 2. Pmids are the PMID of manuscripts reporting the interaction. Sources are the datasets where the interaction was found and are indicated in Table 1. Multiple evidences for each type are separated by semicolon and multiple evidence codes for each type are separated by comma. If one protein maps to several genes, each combination of genes is listed in a separate line. This table is available from: http://cbdm.mdc-berlin.de/tools/hippie/hippie_v1_2.txt.
(TXT)