

Abstract
Epidermolysis bullosa simplex (EBS) is a rare genetic condition typified by superficial bullous lesions following incident frictional trauma to the skin. Most cases of EBS are due to dominantly-acting mutations in keratin 14 (K14) or K5, the type I and II intermediate filament (IF) proteins that co-polymerize to form a pan-cytoplasmic network of 10nm filaments in basal keratinocytes of epidermis and related epithelia. Defects in K5–K14 filament network architecture cause basal keratinocytes to become fragile, and account for their rupture upon exposure to mechanical trauma. The discovery of the etiology and pathophysiology of EBS was intimately linked to the quest for an understanding of the properties and function of keratin filaments in skin epithelia. Since then, continued cross-fertilization between basic science efforts and clinical endeavors has highlighted several additional functional roles for keratin proteins in the skin, suggested new avenues for effective therapies for keratin-based diseases, and expanded our understanding of the remarkable properties of skin as an organ system.
Discovery of the genetic basis for the inherited bullous disease epidermolysis bullosa simplex (EBS), made twenty years ago, provided a new and paradigm-shifting blueprint for the characterization and understanding of a large number of monogenic disorders with a significant impact on skin. Since then EBS as well as related genetic conditions have continued to provide a breeding ground for the cross-fertilization of ideas and successes between basic science endeavors and clinical findings. After a brief introduction to keratins, we here review the clinical features of EBS, the discovery of its genetic basis, and our current understanding of phenotype-genotype correlations in this genodermatosis. We briefly discuss the numerous roles assigned so far to keratin proteins and/or filaments in skin epithelia, address the challenge of devising effective therapies for this condition and, finally, identify a few important priorities for the future.
A primer on keratin intermediate filaments
Keratins are the most abundant structural proteins in the cytoplasm of epithelial cells, in which they form a network of 10–12 nm wide intermediate filaments (IFs) (Figure 1a–c) (Fuchs and Cleveland, 1998; Kim and Coulombe, 2007). Keratin proteins are encoded by a large family of conserved genes, numbering ~54 in the human genome (Schweizer et al., 2006), that partition to the type I and II subgroupings of IF-encoding genes. There are 28 type I (K9–K28; K31–K40) and 26 type II (K1–K8; K71–K86) genes, each coding for one polypeptide chain (Schweizer et al., 2006). Type I proteins tend to be smaller (40–64 kDa) and more acidic (pI ~4.7–6.1) than the larger (52–70 kDa) and basic-neutral (pI ~5.4–8.4) type II proteins (Moll et al., 1982; Schweizer et al., 2006). Keratin polymerization obligatorily begins with formation of coiled-coil heterodimers involving one type I and one type II protein (Fuchs and Cleveland, 1998; Kim and Coulombe, 2007; Steinert et al., 1976). This requirement underlies the pairwise transcriptional regulation of keratin genes in vivo (Fuchs, 1995; Moll et al., 1982; O’Guin et al., 1990). Further, the regulation of keratin genes, individually or as pairs, depends upon the type of epithelia, stage of cellular differentiation, and context (such as disease) (Freedberg et al., 2001; Fuchs, 1995; Moll et al., 1982; O’Guin et al., 1990). Keratin genes and proteins thus provide unparalleled molecular markers to track the status of epithelial cells in health and disease.
Figure 1. Keratin filaments: Structure, organization, and expression in the interfollicular epidermis.

A) Schematic representation of the common domain structure shared by IF proteins including keratins. A central domain, comprised of α-helical coils 1A, 1B, 2A, and 2B separated by non-helical linkers L1, L12 and L2, is flanked by head and tail domains of unknown structure at its N- and C-termini. The boundaries of the rod domain (see red boxes) are highly conserved in primary structure. B) Visualization of filaments, reconstituted in vitro from purified human K5 and K14, by negative staining and electron microscopy. Bar: 150 nm. C) Double-labeling for keratin (red chromophore) and desmoplakin, a desmosome component (green chromophore), epitopes in human epidermal keratinocytes in culture. Keratin IFs are organized in a network that spans the entire cytoplasm and are attached at desmosomal cell-cell contacts between cells (arrowheads). n, nucleus. Bar: 50 μm. Micrograph courtesy of Dr. K. Green (Northwestern Univ., Chicago, IL). D) Histological cross-section of resin-embedded human trunk epidermis, revealing the basal (B), spinous (S), granular (G), and cornified (C) cell layers. Bar: 50 μm. n, nucleus. E, F) Differential distribution of keratin epitopes in the human epidermis. K14 occurs in the basal layer (E), where the epidermal progenitor cells reside. K10 occurs in the suprabasal, differentiating keratinocytes of epidermis (F). Basal lamina is depicted by a dashed line. Bar: 50μm. G) Ultrastructure of the boundary between the basal and suprabasal layer in mouse trunk epidermis, as seen in cross-section by routine transmission electron microscopy. Organization of keratin filaments as loose bundles correlates with the expression of K5–K14 in basal cells (see brackets), whereas the formation of denser, electron-dense filament bundles reflects the onset of K1–K10 expression in early differentiating cells (see arrowheads). Arrows point to desmosomes. Bar: 2 μm. n, nucleus. Adapted from (Miller et al., 2008).
In homo sapiens, the functional type I and type II keratin genes are clustered on the long arms of chromosomes 17 and 12, respectively, the sole exception being the K18 locus (located next to its partner gene K8 on the type II cluster). Whole genome sequencing has confirmed that the genomic organization, sequence, and regulation of keratin genes are conserved among mammals (Coulombe et al., 2004; Hesse et al., 2004; Schweizer et al., 2006). In addition to the large number of keratin genes (Schweizer et al., 2006), the promiscuity with which type I and II keratin proteins pair up and polymerize with one another (Hatzfeld and Franke, 1985) generates a tremendous potential for diversity in the surface chemistry of keratin filaments in living cells. The latter remains underappreciated as there is yet no high-resolution structural insight into the core architecture of keratins filaments.
An astounding two-thirds of known keratin genes are expressed in skin epithelia alone. In normal interfollicular epidermis, progenitor basal cells express the K5–K14 pair, and small amounts of K15 protein (Lloyd et al., 1995; Nelson and Sun, 1983; Porter et al., 2000). Upon commitment to differentiation, keratinocytes rapidly turn on the expression of K1–K10 (Fuchs and Green, 1980; Woodcock-Mitchell et al., 1982) as they switch off K5–K14 gene transcription (though the proteins persist) (Figure 1D–F) (Fuchs, 1995). At a later stage, i.e., in the granular layer, K2e gene expression occurs (Collin et al., 1992). These specific switches in keratin gene expression have been highly conserved throughout evolution, and are paralleled by striking changes in the density and organization of keratin IFs in keratinocytes (Figure 1G). This basic blueprint can be altered to reflect regional specialization (e.g., palmo-plantar epidermis) or a departure from normal differentiation. For instance, K9 is prominently expressed post-mitotically in the thick “primary epidermal ridges” of palmar and plantar epidermis, whereas K6, K16 and K17 occur in the thinner, “secondary ridges” connecting them (Swensson et al., 1998). Otherwise, K6, K16 and K17 are induced, generally at the expense of the K1–K10 pair, in the post-mitotic layers of interfollicular epidermis under conditions of environmental challenges (e.g., tissue injury, UV exposure, viral infection) and in specific diseases (e.g., psoriasis, carcinoma) (Freedberg et al., 2001; McGowan et al., 1998; O’Guin et al., 1990). Evidence in hand suggests that while keratin proteins do not impact the execution of differentiation, the maintenance of a specific complement of keratin proteins plays an important role towards cytoarchitecture and several cellular functions in differentiated or specialized epithelial states.
Epidermolysis Bullosa Simplex: A rare, devastating, but instructive genodermatosis
Epidermolysis Bullosa (EB) corresponds to a group of rare genetic conditions in which bullous lesions affecting primarily the skin arise after exposure to mechanical trauma. Three major forms of EB have been defined. The dystrophic, junctional, and simplex forms of EB are characterized by loss of tissue integrity in the upper dermis, at the dermo-epidermal interface, and within the epidermis, respectively (Figure 2A) (Fine et al., 2008). EB Simplex (EBS) (Figure 2B) is the most frequently occurring, and least severe, form of EB disease (~1 case per 25,000 live births), and is inherited in an autosomal dominant fashion with rare exceptions (Coulombe et al., 2009; Fine et al., 2008; Szeverenyi et al., 2008). In EBS, trauma-induced loss of tissue integrity occurs within the basal layer of epidermal keratinocytes (Figure 2A; Figure 3). The genetic defect renders basal keratinocytes fragile, causing them to “rupture” when the epidermis (and, in some cases, other stratified epithelia) is subjected to mechanical trauma. The rupture of basal keratinocytes often occurs in a specific area of the cell, between hemidesmosomes and the nucleus, in the epidermis of EBS individuals (Haneke and Anton-Lamprecht, 1982) and in relevant mouse models (Coulombe et al., 1991a) (Figure 3), a phenomenon that remains unexplained. Skin pigmentation anomalies can occur in EBS (Fine et al., 2008; Gu and Coulombe, 2007), but terminal keratinocyte differentiation and epidermal barrier function are reportedly normal in the untraumatized skin of EBS patients.
Figure 2. Introduction to Epidermolysis Bullosa Simplex.
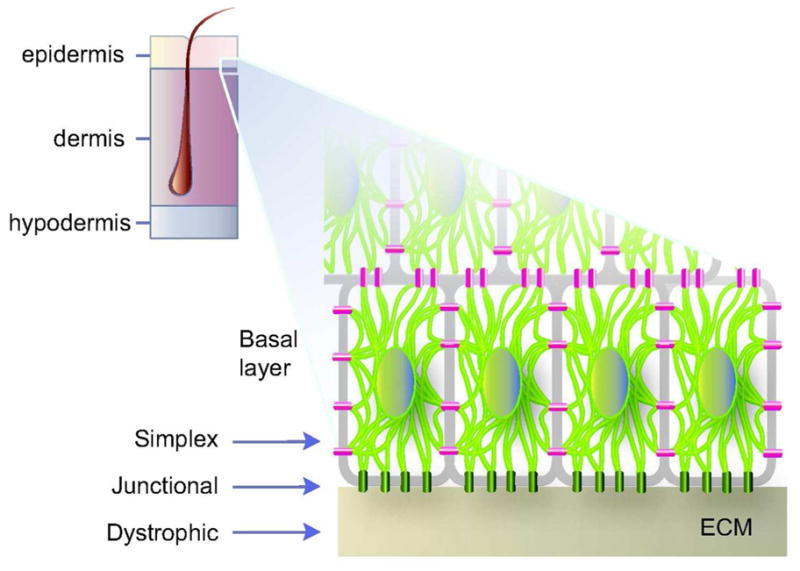
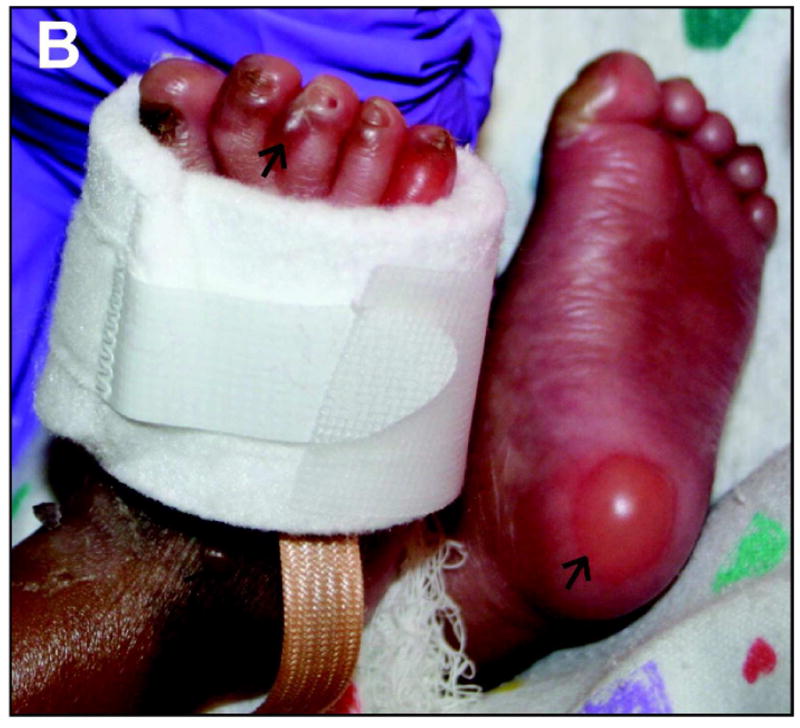
A) Schematic representation of skin tissue (left) and detailed view of the bottom portion of the epidermis (right), highlighting the cytoplasmic network of keratin IFs (light green) attached to hemidesmosome cell-ECM (green) and desmosome cell-cell (magenta) contacts in basal keratinocytes. Arrows depict the plane of tissue rupture seen in the “simplex”, “junctional”, and “dystrophic” forms of EB. B) Example of trauma-induced bullous skin lesions (arrows) in the feet of a 2-month old child diagnosed with EBS. Picture kindly provided by Dr. Bernard Cohen (Johns Hopkins School of Medicine; see http://dermatlas.med.jhmi.edu). Adapted from (Coulombe et al., 2009).
Figure 3. Loss of keratin 14 elicits EBS-like features in newborn mouse skin.

A, B) Picture of newborn mouse littermates, comparing K14 null and wildtype. A) The K14 null neonate exhibits massive skin blistering. Front paws and facial area are severely affected (see arrows). B) A wildtype littermate exhibits intact skin. C, D) Micrographs from hematoxylin/eosin-stained histological sections prepared from front paws of 2-day old K14 null (C) and wildtype (D) mice. Epidermal cleavage is obvious in the K14 null sample. Loss of epidermal integrity occurs “near” the basal layer of keratinocytes (see “blister”), the defining characteristic of EBS. Three basal keratinocytes are boxed in frame D. Abbreviations: epi, epidermis; hf, hair follicles. Bar: 100 μm. Adapted from (Coulombe et al., 2009).
Several clinical variants of EBS have been described (note: the new nomenclature for EBS is being used here; Fine et al., 2008). The most frequent, and better understood, variants are EBS-generalized (in which blistering is “generalized” over the body), EBS-localized (in which blistering is largely restricted to hands and feet), and EBS-Dowling-Meara (EBS-DM, in which blisters are generalized as well but show a distinct “herpetiform” or clustered pattern). The main clinical findings distinguishing these three variants are the distribution, frequency, and severity of skin blistering over the body (Table 1), the ultrastructural features of basal keratinocytes, involvement of other epithelia, and prognosis (Table 1). Additional forms of EBS are not only less frequent but also tend to exhibit additional clinical features (Table 1; see also Fine et al., 2008). For instance, EBS-AR resembles the generalized form but is recessively inherited, EBS with mottled pigmentation (EBS-MP) feature anomalies in skin pigmentation, and EBS with muscular dystrophy (EBS-MD) is accompanied by a progressive, limb-girdle type of muscular dystrophy (Table 1) (Fine et al., 2008; Omary et al., 2004).
Table 1.
Epidermolysis Bullosa Simplex and other diseases caused by mutations in Keratin 5 or Keratin 14
| EBS Variant # | Distinguishing Features # | Onset | Target Genes | Orig. Clinical Description |
|---|---|---|---|---|
| EBS, localized | Formerly known as “Weber-Cockayne”-type EBS. Blistering usually limited to hands and feet, but can occur at sites of repeated trauma; often associated with palmo-plantar keratoderma; worsens in warmer months | Infancy, early childhood, adulthood | Most frequently K5; Less frequently, K14 | F.P. Weber (1926) 1 E.A. Cockayne (1938) 2 |
| EBS, generalized | Formerly known as “Koebner”-type EBS. Blisters predominantly on hands and feet but blistering is often generalized; absence of large tonofilaments in basal keratinocytes on electron microscopy; worsens in warmer months | Birth or infancy | K5 (C-terminal region and L2 linker domain); K14 | H. Koebner (1886) 3 |
| EBS-DM (Dowling-Meara) | This form of EBS retained its original designation. Widespread and severe blistering; herpetiform and hemorrhagic blisters; frequent mucosal involvement; progressive palmoplantar keratoderma; nail dystrophy and milia; hyper- and hypo- pigmentation may occur; may improve with heat or fever | Birth | K5, K14 (at N- or C- terminal ends of the rod domain) | G.B. Dowling & R.H. Meara (1954) 4 |
| EBS-AR (autosomal recessive) | Very rare variant. Largely similar to the generalized form, though the frequency of blistering may be less. | Infancy | K14 | Hovnanian et al., (1993) provided the first mutation report. 5 |
| EBS-MP (with Mottled Pigmentation) | Skin blistering with mottled pigmentation of trunk and limbs; punctate palmo- plantar keratoderma; nail dystrophy. | Birth or infancy | K5 (Pro24->Leu mutation in head domain); K14 | T. Fischer & T. Gedde-Dahl (1979) 6 |
| EBS-MD (with Muscular Dystrophy) | EBS-like skin blistering with progressive limb-girdle muscular dystrophy. Very rare variant. | Infancy | Plectin | K.M Niemi et al. (1988) 7 |
| EBS-Migr (Migratory) | Formerly known as EBS with migratory circinate erythema. Annular migratory multiple erythema circinatum; multiple vesicles on the hands, feet, and legs; lesions heal with brown pigmentation but no scarring. Very rare variant. | Birth | K5 (tail domain) | L.H. Gu et al. (2003) 8 |
| DDD (Dowling-Degos Disease) | Reticular hyperpigmentation on flexure surfaces. | After puberty | K5 haploinsufficiency | G.B. Dowling and W. Freudenthal, (1938) 9 |
| NFJS (Naegeli-Franceschetti- Jadassohn) | Reticular hyper-pigmentation that disappears over time; hypohydrosis; palmoplantar keratoderma; absent fingerprint lines | Birth | K14 (head domain) | O. Naegeli (1927); A. Fransceschetti & W. Jadassohn (1954) 10 |
This table is adapted from Coulombe et al. (J. Clin. Invest. 119: 1784–93, 2009). Additional EBS subtypes, caused by mutations in genes other than K5 and K14, are more rare. See Fine et al. (J. Amer. Acad. Dermatol. 58:931–50, 2008) for a classification of all EBS variants.
References: 1) Weber, F.P. Proc. R Soc. Med. 19:72–5 (1926). 2) Cockayne, E.A. Br. J. Dermatol. 50:358–66 (1938). 3) Koebner, H. Dtsch. Med. Wochenschr. 12:21–22 (1886). 4) Dowling, G.B., and Meara, R.H. Br J Dermatol 66:139–143 (1954). 5) Hovnanian, A., Pollack, E., Hilal, L., Rochat, A., Prost, C., Barrandon, Y., Goossens, M. Nat Genet 3:327–332 (1993). 6) Fischer, T., Gedde-Dahl, T., Jr. Clin Genet 15:228–238 (1979). 7) Niemi, K. M., Sommer, H., Kero, M., Kanerva, L., Haltia, M. Arch Dermatol. 124:551–4 (1988). 8) Gu, L.H., Kim, S. C., Ichiki, Y., Park, J., Nagai, M., Kitajima, Y. J Invest Dermatol 121:482–485 (1983). 9) Dowling, G.B., Freudenthal, W. Br. J. Dermatol. 50:467–71 (1938). 10) Fransceschetti, A., Jadassohn, W. Dermatologica 108:1–28 (1954).
Despite the degree to which clinical presentation varies (Fine et al., 2008; Fine et al., 2000; Horn et al., 2000; Paller, 2004) (Table 1), all variants of EBS are caused by genetic mutations in intracellular proteins, compromising their ability to provide structural support in keratinocytes of the epidermis and related tissues (Coulombe et al., 2009; Gu and Coulombe, 2007; Szeverenyi et al., 2008). The vast majority of EBS cases are due to mutations affecting either K14 or K5 which, as mentioned above, are type I and II intermediate filament (IF) genes selectively expressed in basal keratinocytes in the epidermis and related complex epithelia (Fuchs, 1995; Nelson and Sun, 1983) (Figure 1). Because of the complex substructure of keratin filaments, the perturbations engendered by mutations altering the primary structure of either K5 or K14 proteins for the structure, organization and/or regulation of filaments can range from the very subtle to the very severe, thereby accounting for a substantial fraction of the broad clinical spectra typifying EBS (see below). EBS-MD is caused by mutations in plectin (Gache et al., 1996; McLean et al., 1996), a cyto-linker protein responsible for integrating various cytoskeletal and cell adhesive elements into a functionally unified network (Wiche, 1998; see Table 1). The presence of plectin in hemidesmosomes of basal epidermal keratinocytes and at the Z-lines of skeletal myocytes account for the involvement of both skin and muscle in EBS-MD (Wiche, 1998). Mutations in type XVII alpha 1 collagen (COL17A1), a hemidesmosomal plaque protein required for tight adherence of basal keratinocytes to the basal lamina, account for a subset of patients featuring elements typical of both EBS and EB junctional (Fontao et al., 2004; Pfendner et al., 2005) (Figure 2A). Additional examples of rare forms of EBS linked to genes other than K5 and K14 are discussed in Fine et al. (Fine et al., 2008).
Early 90’s: A function for keratin filaments and a promising lead for the etiology of EBS
By the late 1980’s, several mechanisms had been proposed to account for the etiology of EBS and the fragility of basal keratinocytes that typifies this condition. These mechanisms could be assigned to either one of two broadly defined categories: a misregulated or defective enzyme, or a defect in a structural component (reviewed in Coulombe, 1993). With the notable exception of Dr. Anton-Lamprecht (Anton-Lamprecht, 1983), the literature suggests that no one was considering the possibility of defects in keratin protein or filaments in this condition. Given the heterogeneity in EBS clinical features (Table 1) and the absence of a clear understanding of keratin filament function at the time, there was no obvious reason to draw a link between the disorder and keratin filaments. In the early 90’s, however, the combination of a reverse genetic approach with the transgenic mouse technology provided key clues about the etiology of EBS (and related genodermatoses) in addition to revealing a key function for keratin filaments in their natural setting (Coulombe et al., 1991b; Fuchs et al., 1992; Vassar et al., 1991).
The cloning and sequencing of (human) keratin genes, and the resulting ability to better define the secondary structure of their protein products (e.g., Fuchs et al., 1981; Hanukoglu et al., 1982; Steinert et al., 1983), set the stage for subsequent efforts aimed at defining the key determinants in K14 and K5 co-assembly using systematic deletion mutagenesis. Initial studies of this kind revealed that K14 deletion mutants missing small portions of either end of the central rod domain had a significant, and biochemically dominant, impact on the organization of keratin filaments when expressed in cultured epithelial cells (Albers and Fuchs, 1987, 1989). Important details about the impact and mode of action of such K14 deletion mutants (Coulombe et al., 1990) and, later on, K5 deletion mutants (Wilson et al., 1992) were subsequently provided by in vitro polymerization assays making use of purified recombinant keratin proteins. Relevance to EBS, and to keratin function in vivo, was ultimately revealed when select K14 deletion mutants were tissue-specifically expressed in transgenic mice using the K14 promoter (Coulombe et al., 1991b; Vassar et al., 1991). Mutant K14-expressing transgenic mice exhibited trauma-induced blistering in the epidermis and oral mucosa, beginning at or shortly after birth, in a manner that closely approximated the key features of EBS (Figure 3). The phenotype of these mice strongly hinted that EBS could indeed arise from mutations affecting the protein-coding sequence of either K14 or K5 (Vassar et al., 1991). This possibility was further supported by publications that had described defects in keratin IF network architecture in basal keratinocytes from individuals with EBS, in the epidermis in situ (Anton-Lamprecht, 1983; Haneke and Anton-Lamprecht, 1982; Ito et al., 1991) as well as in ex vivo culture (Kitajima et al., 1989). Such mutations were indeed found shortly thereafter, initially in K14 (Bonifas et al., 1991; Coulombe et al., 1991a) and later on in K5 (Lane et al., 1992), in the genome of individuals with EBS.
In additional to providing key clue about the genetic basis of EBS, the characterization of the first cohort of transgenic mouse lines with tissue-specific expression of K14 deletion mutants (Coulombe et al., 1991b; Vassar et al., 1991) suggested a biological framework for many heretofore misunderstood features of EBS (Coulombe et al., 2009). First, mutations mapping to the α-helical rod domain of keratin can elicit the formation of aggregates of amorphous proteins in the cytoplasm in addition to dominantly disrupting filament structure (Coulombe et al., 1991b; Fuchs et al., 1992; Vassar et al., 1991). Such aggregates are diagnostic for the most severe form of EBS, EBS-DM (see Table 1), and are immunopositive for K5 and K14 (Coulombe et al., 1991a; Ishida-Yamamoto et al., 1991). Second, onset of disease occurs at birth or shortly thereafter, reflecting in part the embryonic onset of K5 and K14 gene expression (Byrne et al., 1994; Kopan and Fuchs, 1989) along with the protected environment experienced by the human fetus up until birth. Third, the broad range of clinical severity typifying EBS in humans can be mimicked in mouse skin tissue by expressing K14 mutants having either a mild, moderate, or severe impact on keratin IF assembly in vitro (Coulombe et al., 1991b). This finding helped dismiss the idea that variants such as EBS-DM and EBS-localized (Table 1) had a distinct etiology. Fourth, the ability of blisters to heal leaving no scar distinguishes EBS from the junctional and dystrophic forms of EB (Fine et al., 2008; Fine et al., 2000; Paller, 2004). From the observation that the wound-inducible keratins 6, 16, and 17 (K6, K16, K17) are induced in keratinocytes populating the margins of epidermal lesions, it has been suggested these keratins could compensate for the cellular fragility fostered by K5 or K14 mutants, allowing for normal epithelialization to occur (Coulombe et al., 1991b). Fifth, this initial set of mouse models suggested that similar defects in other keratin proteins should elicit analogous deficiencies in the epithelial cell types. This was later shown via the tissue-specific expression of a deletion mutant of K10, a keratin expressed in terminally differentiating keratinocytes of the epidermis and related cornifying epithelia (Fuchs and Green, 1980; Woodcock-Mitchell et al., 1982), in transgenic mice (Fuchs et al., 1992). These mice exhibited trauma-induced cell fragility in the suprabasal portion of epidermis, in a fashion that proved analogous to the human condition epidermolytic hyperkeratosis. Mutations in the K1 and K10 genes were later found in patients with this condition (Cheng et al., 1992; Chipev et al., 1992; Rothnagel et al., 1992).
The cell fragility that typifies EBS largely corresponds to a “loss-of-keratin-function” phenotype since mice homozygous null for K14 (Figure 3), K5, or plectin display the key features of EBS (with accompanying muscle defects in the case of plectin; Table 1). The lesser degree to which the esophagus is affected relative to epidermis in newborn K14 null mice correlates with higher levels of K15, a type I keratin related to K14, in this tissue (Lloyd et al., 1995). The presence of small amounts of K15 likely accounts for the markedly more severe blistering of skin, and earlier death, in K5 null relative to K14 null mice (Kerns et al., 2007; Lloyd et al., 1995; Peters et al., 2001). A compensatory role for K15 was also proposed based on studies of individuals with EBS whose genome bear the equivalent of a K14 null mutation (Chan et al., 1994a; Jonkman et al., 1996). Later on, the targeted expression of K16, another type I keratin homologous to K14, in basal keratinocytes of the epidermis was shown to markedly attenuate skin blistering and prevent neonatal death in K14 null mice (Paladini and Coulombe, 1999). Such findings helped foster the notion that highly homologous keratins are partially redundant in their structural support role in vivo, and paved the way for a search for small molecules that would, upon topical application, provide therapeutic relief as a result of the selective induction of a compensatory program of gene expression (see below).
Genotype-phenotype correlations in EBS: From the bench to the bedside, and back
A comprehensive and useful database of human keratin mutations, and associated diseases, is maintained at Singapore’s Bioinformatics Institute (see www.interfil.org and (Szeverenyi et al., 2008). K5 and K14 mutations account for > 39% of all keratin disease–associated mutations characterized thus far (450 out of 1143 reports), and together they are second only to mutations in LMNA, the gene encoding lamins A and C (nuclear IF proteins), for disease-causing mutations in an IF protein–encoding gene. Mutations in K5 or K14 account for the vast majority of EBS cases, with only a small (albeit biologically significant) fraction of cases involving mutations in genes encoding IF-associated proteins (plectin, integrins, collagen XVII; see Fine et al., 2008). Most of the EBS-causing mutations occurring in K5 (>86%) and K14 (>87%) are dominantly-acting missense alleles, and they are distributed across much of the sequences of the corresponding protein products (Figure 4). This said, the majority of these mutations alter amino acid residues located within the central α-helical rod domain of K5 and K14 (Figure 4), known to play a crucial role towards the assembly and structure. of keratin filaments (Coulombe et al., 2009; Fuchs and Cleveland, 1998; Omary et al., 2004).
Figure 4. Distribution of mutations in K5 and K14 according to EBS variants.

The EBS-causing mutations that are available (September 2011) from the intermediate filament database (http://www.interfil.org; see (Szeverenyi et al., 2008)) are mapped to the secondary structure of K5 and K14 protein. Helix boundary motifs (green bars), as defined by Steinert et al. (Steinert et al., 1993), correspond to 15–20 residues segments that are very highly conserved across keratins and all other IF proteins. Trigger motifs (orange bars) are sequences capable of nucleating the formation of coiled-coil dimers (Wu et al., 2000). The Arg125 mutation hot spot in K14 (Coulombe et al., 1991a) is indicated in red. This schematic does not convey the frequency of mutations at any of the shown sites. See Table 1 for abbreviations and text for further detail.
Basic principles could readily be drawn as additional reports of EBS-causing mutations came to the fore (Coulombe, 1993; Epstein, 1992; Leigh and Lane, 1993), and are affirmed nowadays by surveying the Singapore database. For EBS-localized, EBS-generalized, and EBS-DM, a strong concordance exists between the position and nature of the mutation in the target keratin protein, the extent to which it disrupts keratin IF assembly and structure in established assays, and the severity of clinical presentation (Chan et al., 1993; Chan et al., 1994b; Gu and Coulombe, 2005; Letai et al., 1993; Morley et al., 2003; Morley et al., 1995). Corollary to this notion, the nature of the mutation affecting either K5 or K14 usually defines the sensitivity threshold that the epithelium displays toward frictional trauma, thereby contributing to whether the symptoms are “local” (e.g., EBS-localized) or “generalized” (e.g., EBS-generalized and EBS-DM). Mutations altering highly-conserved residues located within the short and highly-conserved sequence segments located at the beginning and the end of the central rod domain, in either K5 or K14, are often associated with EBS-DM (Figure 4). Further, twice as many cases of EBS-DM have been traced to mutations in K14 rather than K5 (n=106 vs. n=43, respectively), reflecting the presence of a “mutational hot spot” in codon 125 in the highly conserved segment within subdomain 1A, which normally encodes an Arg residue (Coulombe et al., 1991a; Fuchs and Coulombe, 1992; Stephens et al., 1993) (Figure 4). The very high frequency of mutations at K14’s codon 125 (~29% of all reported mutations in K14), and the corresponding (conserved) codon in other type I keratins and even non-keratin IF proteins (Omary et al., 2004; Szeverenyi et al., 2008), likely results from deamination of methylated cytosine occurring in the context of a CpG dinucleotide. The latter represents the most frequent mechanism causing mutations in the human genome (Cooper, 1988). Several lines of evidence underscore the crucial role of this specific arginine residue in keratin IF assembly and function (Coulombe et al., 1991a; Letai et al., 1993; Ma et al., 2001; Werner et al., 2004).
Conversely, the mildest variant of EBS, EBS-localized, often involve missense mutations affecting amino acids located in the non-helical “head” and “linker” domains, particularly K5 (Figure 4). The primary structure of these domains is less well-conserved than that of the helix boundary motifs and other α-helical segments of the central rod domain (Parry, 2005). Mutations in these domains are generally less consequential for IF assembly and function (Chan et al., 1993; Chan et al., 1994b; Hatzfeld and Weber, 1991; Letai et al., 1992; Letai et al., 1993). Why the analogous head and linker regions of K14 are not as frequently targeted for mutation as they are in K5 is unclear (Figure 4). One possibility is that such mutations might behave recessively (Hovnanian et al., 1993; Szeverenyi et al., 2008). Another likely factor is the presence of K15 in basal keratinocytes (Lloyd et al., 1995). Although not sufficient to prevent the consequences of severe K14 mutations, the presence of K15 might ameliorate subtle K14-generated defects in the IF network of basal keratinocytes.
EBS-generalized, which is intermediate in severity between EBS-localized and EBS-DM, is also frequently associated with mutations in the helix-initiation and helix-termination motifs but also shows a greater frequency of mutations mapping elsewhere in the rod domain, relative to EBS-DM. Another mutation bias worth mentioning is the strong association between mutations mapping to the non-helical head and tail domains of K5 protein with, respectively, the “non-classical” EBS-MP and EBS-Migr, variants (Table 1). While the analysis of phenotype-genotype correlation reported here is heavily emphasized on “mutation location” in either K5 or K14, the nature of the mutation at any given site on these two proteins also can weigh very heavily on the consequences for overall biological function and disease presentation.
The presence of cytoplasmic aggregates containing mispolymerized mutant keratin proteins, a defining characteristic of EBS-DM (Table 1), may contribute to pathophysiology at the cellular and tissue levels. Mice made to express aggregation-prone K14 mutants (Cao et al., 2001; Coulombe et al., 1991b; Vassar et al., 1991) exhibit an earlier–onset of disease, more severe blistering, and die earlier than K14 null mice (Lloyd et al., 1995). Akin to this, the EBS phenotype exhibited by individuals null for K14 is milder than autosomal-dominant cases of EBS-DM (Chan et al., 1994a; Jonkman et al., 1996; Rugg et al., 1994). Conceivably, the failure of the misfolded-protein response to resolve these aggregates may lead to cellular and tissue stress (D’Alessandro et al., 2002; Liovic et al., 2008; Loffek et al., 2010) and this could in turn impact the pathophysiology of EBS and related conditions. Such aggregates of mispolymerized IF proteins are characteristic of many additional disorders caused by mutations in genes encoding non-keratin IF proteins (Omary et al., 2004; Szeverenyi et al., 2008).
The post-EBS era: Keratin proteins fulfill multiple functions in skin epithelia
A wealth of evidence supports the view that the fragility state of basal keratinocytes accounts for the production of skin blisters in individuals with EBS, reflecting a loss of epidermal integrity following incipient mechanical trauma. This includes substantive pathophysiological findings obtained from the analysis of individuals with EBS or relevant mouse models of this condition (cf. above), along with a more detailed, and more recent, set of studies assessing the properties of mutant keratin-expressing epithelial cells in culture (Lulevich et al., 2010; Russell et al., 2004; also, see Beil et al., 2003) and mutant-keratin containing filaments in vitro (Gu et al., 2005; Ma et al., 2001). This role, referred to as structural support (Table 2), has since then been extended to virtually all keratins expressed in other epithelial settings and to all major classes of IF polymers, including the nuclear lamins (Kreplak and Fudge, 2007; Lammerding et al., 2004).
Table 2.
Established roles of keratin proteins in skin epithelia
| Description of functional role | Keratin(s) | Biological context | Comments # |
|---|---|---|---|
| Structural support | All | Epidermis & hair follicles | Proper structure and organization of intracellular keratin filaments is essential to withstanding frictional and mechanical stresses.1,2 |
| Cell and tissue growth | K10 | Epidermis | In mouse, loss of K10 results in enhanced proliferation in the epidermis, akin to what occurs in epidermolytic hyperkeratosis. The mechanism(s) involved remains to be defined, but well might be non-cell autonomous.3 |
| K17 | Hair follicle cycling | In mouse, loss of K17 results in premature entry into catagen, owing in part to enhanced TNF-a signaling and apoptosis. A potential mechanism is the interaction with, and negative regulation of, the adaptor protein TRADD.4,5 | |
| K17 | Wound edge | K17 participates in the increase occurring in the size of keratinocytes while in primary culture and at the wound edge in embryonic ectoderm by stimulating protein synthesis, an event that depends in part on its phosphorylation-dependent interaction with 14-3-3s (stratifin).6 | |
| Skin pigmentation | K1, K2, K5 | Epidermis | A heroic random mutagenesis screen revealed a role for K1 and K2 in skin pigmentation in mouse. Also, EBS-MP and Dowling-Degos disease (cf. Table 1) are caused by mutations at the K5 locus in humans.,7,8,9 |
| Keratinocyte migration | K6 | Wound edge | In addition to providing structural support in keratinocytes migration into wound site, K6 negatively regulates their migration speed via a direct, and inhibitory, interaction with Src kinase. The latter is believed to foster the cohesive migration of keratinocytes as a cellular sheet.10 |
| Langerhans cell density | K5 | Epidermis | In mouse, loss of K5 results in an increased Langerhans cell density in the epidermis. The same phenomenon occurs in EBS patients with K5 mutations, specifically. The mechanism is unknown, but likely involve elevated expression of select chemokines acting on DC precursors.11 |
| Immunomodulation | K17 | Skin cancer | In mouse, loss of K17 causes a delay in the onset of basaloid skin tumors and the extent of phorbol ester-induced dermatitis, correlating with reduced keratinocyte proliferation and a global switch from a Th1/Th17- dominated to a Th2-dominated immune cytokine/chemokine profile. Mechanism(s) unknown. 12 |
References: 1) Vassar, R., Coulombe, P. A., Degenstein, L., Albers, K., Fuchs, E., Cell 64:365–80 (1991). 2) Coulombe, P.A., Hutton, M. E., Vassar, R., Fuchs, E., J Cell Biol, 115:1661–74 (1991). 3) Reichelt, J. and T.M. Magin, J Cell Sci. 115:2639–50 (2002). 4) Tong, X., Coulombe, P.A. Genes & Dev. 20:1353–64 (2006). 5) Inada, H., Izawa, I., Nishizawa, M., Fujita, E., Kiyono, T., Takahashi, T., Momoi, T., Inagaki, M. J Cell Biol 155:415–26 (2001). 6) Kim, S., Wong, P., Coulombe, P.A. Nature 441:362–365 (2006). 7) Uttam, J., Hutton, E., Coulombe, P. A., Anton-Lamprecht, I., Yu, Q. C., Gedde-Dahl, T., Jr., Fine, J. D., Fuchs, E., Proc Natl Acad Sci USA 93: 9079–84 (1996). 8) Betz, R.C., Planko, L., Eigelshoven, S., Hanneken, S., Pasternack, S. M., Bussow, H., Bogaert, K. V., Wenzel, J., Braun-Falco, M., Rutten, A., Rogers, M. A., Ruzicka, T., Nothen, M. M., Magin, T. M., Kruse, R. Am J Hum Genet 78:510–9 (2006). 9) Fitch, K.R., McGowan, K. A., van Raamsdonk, C. D., Fuchs, H., Lee, D., Puech, A., Herault, Y., Threadgill, D. W., Hrabe de Angelis, M., Barsh, G. S. Genes Dev. 17:214–28 (2003). 10) Rotty, J. Coulombe, P.A., unpublished data. 11) Roth, W., Reuter, U., Wohlenberg, C., Bruckner-Tuderman, L., Magin, T. M. Hum Mutat 30:832–41 (2009). 12) DePianto, D., Kerns, M.L., Dlugosz, A.A., Coulombe, P.A. Nat. Genet. 42:910–4 (2010).
Further efforts towards the creation and characterization of transgenic mouse models alongside the study of individuals with keratin-based diseases together have (yet again) led to the discovery of several novel functions for keratin proteins in skin epithelia (Table 2). A novel function so far evidenced for K10 and K17 relates to the regulation of cell and tissue growth in the epidermis and hair follicles. Thus, dominantly-acting mutations in either K10 or its partner K1 cause epidermolytic hyperkeratosis, a condition typified by the fragility of suprabasal keratinocytes, hyperplasia in the basal compartment of epidermis, and hyperkeratosis (Omary et al., 2004; Szeverenyi et al., 2008). Nullifying the K10 gene results in hyperproliferation in the basal layer of epidermis (Reichelt and Magin, 2002) and sebaceous glands (Reichelt et al., 2004) in mouse, in the absence of any sign of fragility in suprabasal epidermal keratinocytes (Gu and Coulombe, 2007; Reichelt and Magin, 2002). Together these human and mouse findings point to a role for K10, which is expressed post-mitotically (Figure 1), in regulating the rate of proliferation in the basal layer. The mechanism underlying this remarkable phenotype is not clear at present (Chen et al., 2006; Paramio et al., 2007; Reichelt and Magin, 2002).
K17 fosters epithelial growth in multiple settings in skin tissue, whether healthy, injured, or diseased (Table 2). Mediated in part by its ability to interact with and presumably sequester 14-3-3σ (stratifin) in a phosphorylation-dependent fashion, K17 participates in the stimulation of protein synthesis and epithelial cell growth occuring in wound-proximal keratinocytes following injury to the epidermis (Kim and Coulombe, 2010; Kim et al., 2006). In anagen-stage hair follicles, K17 promotes maintenance of growth in part by antagonizing TNFα signaling towards apoptosis (Tong and Coulombe, 2006), an effect that could involve the sequestration of the adaptor protein TRADD (Inada et al., 2001). Finally, there is evidence that K17 accelerates the onset of basaloid skin tumors, in which it is faithfully expressed (Markey et al., 1992), by promoting keratinocyte proliferation correlating with the expression of pro-inflammatory and/or mitogenic cytokines and chemokines in keratinocytes (DePianto et al., 2010). The latter finding is captivating in light of the implication of specific SNPs affecting the coding sequence of K5 and enhancing the risk of developing basal cell carcinoma in human (Stacey et al., 2009). Besides, a recent genome-wide screen has implicated K6, K16, and K17 in a genetic network defining the key interrelationship between barrier function, inflammation, and tumor susceptibility in mouse skin (Quigley et al., 2009).
In keeping with the theme of chemokines, Magin and his colleagues have reported on an increased density of Langerhans cells in mice null for K5, and in individuals with EBS resulting from mutations at the K5 locus (Roth et al., 2009). This phenomenon, which does not occur in K14 null mouse skin or in K14 mutation-related cases of EBS, correlates with an upregulation in chemokines (e.g., CCl2, CCL19, CCL20) known to attract Langerhans cell precursors to the skin (Roth et al., 2009), and otherwise adds to the emerging concept that keratins may act as key immune modulators in the skin (DePianto et al., 2010).
Keratin 6, which along with K16 and K17 is rapidly induced in wound-proximal, surviving keratinocytes following injury to the skin (Paladini et al., 1996), was shown years ago to be essential to the maintenance of cell integrity in keratinocytes migrating into the wound (Wong and Coulombe, 2003). Though more fragile, K6 null mouse skin keratinocytes migrate faster than their wildtype counterpart in mechanically softer settings, such as skin explant culture and primary culture. This surprising property has been linked to K6’s ability to directly bind to, and negatively regulate, Src kinase, a key driver of cell migration (J. Rotty and P.A.C., ms. in revision). Thus K6, and keratin-associated proteins such as plakoglobin and plectin (Osmanagic-Myers et al., 2006; Yin et al., 2005), may promote the maintenance of intercellular cohesiveness in migrating sheet of keratinocytes by scaling back the extent to which a potentially oncogenic signaling protein, namely Src, is activated during tissue repair.
Pigmentation represents yet another key property of the skin potentially impacted by keratin proteins. The evidence supporting this newly defined role has been reviewed (Gu and Coulombe, 2007) so that only three elements relevant to other sections of this text will be mentioned here. First, Uttam et al. (1996) discovered that a rather peculiar missense mutation in the head domain of K5, Pro24->Leu, causes EBS with “mottled pigmentation” (EBS-MP; Table 1). Several additional cases of EBS-MP have since then been traced to the same K5 allele (Szeverenyi et al., 2008), and to distinct mutations in K5 (1649delG; Horiguchi et al., 2005) or K14 (Met119->Thr; Harel et al., 2006). The 1649delG allele of K5 can also result in EBS-Migr (Table 1), and this too is characterized by the appearance of hyper- or hypo-pigmented skin patches in adults (Gu et al., 2003). Second, a very rare condition typified by reticulate hyper-pigmentation and dark hyperkeratotic papules in skin flexural regions, Dowling-Degos disease (DDD) (Table 1), has been linked to K5 haploinsufficiency (Betz et al., 2006). Third, dominantly-acting, missense alleles in K2 and K1 elicit a dark skin phenotype, as discovered in a random-mutagenesis screen designed to identify novel determinants of coat color in mouse (Fitch et al., 2003; McGowan et al., 2006). Expression of these K2 and K1 alleles in mouse skin reproduces the keratinocyte fragility and epidermolytic hyperkeratosis-like manifestations associated with similar mutations in the human orthologs. No mechanism has been established to account for these pigmentation phenotypes, though there is preliminary evidence pointing to an interaction between K5 and components of microtubules-dependent motors, which are involved in melanin pigment transport (Betz et al., 2006; Gu et al., 2007).
Finally, select clinical attributes of EBS still have no sound biological basis. Topping the list of such attributes are two frequent occurrences: enhanced frequency of skin blistering during the warm season, and age-related improvement in clinical symptoms (Fine et al., 2008; Fine et al., 2000; Paller, 2004). Although experimental evidence is still lacking, one possibility is that conditions that activate the heat shock/stress response may help to resolve the aggregates of misassembled keratins (Loffek et al., 2010; Omary et al., 2004). Understanding the basis for these peculiar clinical features may lead to new insight into keratin regulation and function, epithelial biology, and innovative ideas for the treatment of EBS.
Devising an effective therapy for EB simplex: challenges and opportunities
Elucidating the etiology and gaining a better understanding of the pathophysiology of EBS has undoubtedly resulted in more extensive, and effective, means of pre- and post-natal genetic testing and counseling options (Holbrook et al., 1992; Pfendner et al., 2003; Rugg et al., 2000). Therapeutic options available to patients suffering from this chronic debilitating disorder are still lacking, however. Notwithstanding its significantly milder nature relative to the other subtypes of EB diseases, and in spite of the age-related improvement in the severity of its symptoms, EBS imposes significant restrictions and affects lifestyle well into adulthood. Yet the standard of care for EBS, i.e., minimizing trauma and preventing infections in healing blisters, remains largely preventive and palliative in spite of two decades of very substantive progress about its genetic and biological underpinnings. The broad spectrum of mutations occurring in either K5 or K14 along with their dominant mode of action, the intrinsically high cell turnover rate taking place in the epidermis, and the low incidence of EBS, an orphan disease, in the human population all contribute to the challenge of devising safe, effective, and long-lasting therapies. The relatively few formalized efforts to test therapies for EBS in human subjects have yielded inconclusive results (Langan and Williams, 2008), or need to be scaled up.
There now is compelling experimental evidence, obtained in the setting of mouse skin tissue in vivo, confirming the prediction that the impact of strong dominant-negative mutant keratin proteins can be reversed as expression of the corresponding wild-type protein (or a functional equivalent) is titrated back in epidermal basal cells (Cao et al., 2001). This is also generally consistent with a phenomenon of “revertant mosaicism” noted in the skin of individuals with EBS in which a “second hit” (e.g., a frameshift mutation) in the original mutant K14 allele generates the equivalent of a null allele (Smith et al., 2004) or a “corrected” allele (Schuilenga-Hut et al., 2002), thus negating the impact of an otherwise strong dominant-negative mutation (also, see D’Alessandro et al., 2011). The elegant study by Cao et al. (2001) also showed that wild-type keratinocytes have a growth advantage over those expressing mutant keratin in skin epithelia in vivo, a finding with obvious implications for gene therapy.
Considerable progress has been made in recent years with regards to strategies that specifically target, or interfere with, disease-causing keratin alleles in keratinocytes (Lewin et al., 2005; McLean and Moore, 2011). Various allele-specific strategies are being tested, including gene targeting (Petek et al., 2010) and trans-splicing (Wally et al., 2010; Wally et al., 2008), which are directed at the relevant genomic DNA locus and thus offer the distinct advantage of being potentially curative, and alternatively gene silencing (Atkinson et al., 2011), which targets the relevant mRNA and therefore would necessitate continuous treatment. A distinct strategy is to attempt to intervene at the protein level (Kerns et al., 2010; Kerns et al., 2007). Thus, topical application of the natural product sulforaphane significantly ameliorates the skin blistering and survival of K14 null mice, but not K5 null ones, correlating with its ability to induce the K16 and K17 expression (Kerns et al., 2007). Besides, there is evidence that stimulation of protein homeostasis pathways lessens keratin mutant protein aggregation in EBS keratinocyte cell lines (Chamcheu et al., 2011; Chamcheu et al., 2010; Lee et al., 2008; Loffek et al., 2010). Finally, there are reports showing that injection of botulinum toxin (“Botox”) in plantar skin ameliorates the blistering and diminishes pain in individuals with EBS and pachyonychia congenita (Swartling et al., 2010). These encouraging results nothwithstanding, there is much work to be done as one attempts to export and translate encouraging results obtained in virally-transformed human keratinocyte cell lines or transgenic mouse models to human skin.
Prioritizing future efforts in this field
There are multiple issues of significant interest and importance in the triad formed by keratin genes and their proteins, skin epithelia, and genodermatoses. Some stand out, however, and they are as follows. First, we sorely need a reliable, atomic-resolution model for the structure of intermediate filaments. Second, we need to better understand the mechanisms regulating keratin filament assembly in their natural setting in vivo. Third, we need to better understand the relationship between keratin filament and mechanical resilience, and whether the cell fragility that is seen in conditions such as EBS is entirely the result of defects in cellular mechanics. Related to this, we need to devise reliable biomarkers reporting on cell fragility. Fourth, we need “actionable findings” that lead to a genuine clinical improvement in patients suffering from EBS and other keratin-based genodermatoses. As a final point, perhaps, we need to understand the rationale for the remarkable diversity of keratin proteins, and the differentiation- and context-dependent nature of their expression in epithelia.
Acknowledgments
The authors thank members of the laboratory for support and Dr. Bernard Cohen for providing an illustration. Relevant efforts in the laboratory are supported by grants AR42047, AR44232 and CA160255 from the National Institutes of Health.
Nonstandard abbreviations
- EB
epidermolysis bullosa
- EBS-DM
EBS Dowling-Meara
- EBS-gen
EBS generalized
- EBS-MCS
EBS with migratory circinate erythema
- EBS-MP
EBS with mottled pigmentation
- EBS-loc
EBS localized
- IF
intermediate filament
- K
keratin
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
References
- Albers K, Fuchs E. The expression of mutant epidermal keratin cDNAs transfected in simple epithelial and squamous cell carcinoma lines. J Cell Biol. 1987;105:791–806. doi: 10.1083/jcb.105.2.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers K, Fuchs E. Expression of mutant keratin cDNAs in epithelial cells reveals possible mechanisms for initiation and assembly of intermediate filaments. J Cell Biol. 1989;108:1477–93. doi: 10.1083/jcb.108.4.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton-Lamprecht I. Genetically induced abnormalities of epidermal differentiation and ultrastructure in ichthyoses and epidermolyses: pathogenesis, heterogeneity, fetal manifestation, and prenatal diagnosis. J Invest Dermatol. 1983;81:149s–53s. doi: 10.1111/1523-1747.ep12540961. [DOI] [PubMed] [Google Scholar]
- Atkinson SD, McGilligan VE, Liao H, Szeverenyi I, Smith FJ, Moore CB, et al. Development of Allele-Specific Therapeutic siRNA for Keratin 5 Mutations in Epidermolysis Bullosa Simplex. J Invest Dermatol. 2011;131:2079–86. doi: 10.1038/jid.2011.169. [DOI] [PubMed] [Google Scholar]
- Beil M, Micoulet A, von Wichert G, Paschke S, Walther P, Omary MB, Van Veldhoven PP, Gern U, Wolff-Hieber E, Eggermann J, Waltenberger J, Adler G, Spatz J, Seufferlein T. Sphingosylphosphorylcholine regulates keratin network architecture and visco-elastic properties of human cancer cells. Nat Cell Biol. 2003;5:803–11. doi: 10.1038/ncb1037. [DOI] [PubMed] [Google Scholar]
- Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, Bogaert KV, Wenzel J, Braun-Falco M, Rutten A, Rogers MA, Ruzicka T, Nothen MM, Magin TM, Kruse R. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006;78:510–9. doi: 10.1086/500850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifas JM, Rothman AL, Epstein Eh., Jr Epidermolysis bullosa simplex: evidence in two families for keratin gene abnormalities. Science. 1991;254:1202–5. doi: 10.1126/science.1720261. [DOI] [PubMed] [Google Scholar]
- Byrne C, Tainsky M, Fuchs E. Programming gene expression in developing epidermis. Development. 1994;120:2369–83. doi: 10.1242/dev.120.9.2369. [DOI] [PubMed] [Google Scholar]
- Cao T, Longley MA, Wang XJ, Roop DR. An inducible mouse model for epidermolysis bullosa simplex: implications for gene therapy. J Cell Biol. 2001;152:651–6. doi: 10.1083/jcb.152.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamcheu JC, Navsaria H, Pihl-Lundin I, Liovic M, Vahlquist A, Torma H. Chemical chaperones protect epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation: involvement of heat shock proteins and MAP kinases. J Invest Dermatol. 2011;131:1684–91. doi: 10.1038/jid.2011.93. [DOI] [PubMed] [Google Scholar]
- Chamcheu JC, Virtanen M, Navsaria H, Bowden PE, Vahlquist A, Torma H. Epidermolysis bullosa simplex due to KRT5 mutations: mutation-related differences in cellular fragility and the protective effects of trimethylamine N-oxide in cultured primary keratinocytes. Br J Dermatol. 2010;162:980–9. doi: 10.1111/j.1365-2133.2009.09615.x. [DOI] [PubMed] [Google Scholar]
- Chan Y, Anton-Lamprecht I, Yu QC, Jackel A, Zabel B, Ernst JP, et al. A human keratin 14 “knockout”: the absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev. 1994a;8:2574–87. doi: 10.1101/gad.8.21.2574. [DOI] [PubMed] [Google Scholar]
- Chan YM, Yu QC, Fine JD, Fuchs E. The genetic basis of Weber-Cockayne epidermolysis bullosa simplex. Proc Natl Acad Sci U S A. 1993;90:7414–8. doi: 10.1073/pnas.90.15.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YM, Yu QC, LeBlanc SJ, Christiano A, Pulkkinen L, Kucherlapati RS, et al. Mutations in the non-helical linker segment L1–2 of keratin 5 in patients with Weber-Cockayne epidermolysis bullosa simplex. J Cell Sci. 1994b;107:767–74. doi: 10.1242/jcs.107.4.765. [DOI] [PubMed] [Google Scholar]
- Chen J, Cheng X, Merched-Sauvage M, Caulin C, Roop DR, Koch PJ. An unexpected role for keratin 10 end domains in susceptibility to skin cancer. J Cell Sci. 2006;119:5067–76. doi: 10.1242/jcs.03298. [DOI] [PubMed] [Google Scholar]
- Cheng J, Syder AJ, Yu QC, Letai A, Paller AS, Fuchs E. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell. 1992;70:811–9. doi: 10.1016/0092-8674(92)90314-3. [DOI] [PubMed] [Google Scholar]
- Chipev CC, Korge BP, Markova N, Bale SJ, DiGiovanna JJ, Compton JG, et al. A leucine----proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell. 1992;70:821–8. doi: 10.1016/0092-8674(92)90315-4. [DOI] [PubMed] [Google Scholar]
- Collin C, Moll R, Kubicka S, Ouhayoun JP, Franke WW. Characterization of human cytokeratin 2, an epidermal cytoskeletal protein synthesized late during differentiation. Exp Cell Res. 1992;202:132–41. doi: 10.1016/0014-4827(92)90412-2. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Youssouffian H. The CpG dinucleotide and human genetic diseases. Hum Genet. 1988;78:151–5. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Coulombe PA, Bernot KM. Keratins and the Skin. In: Lane MD, Lennarz W, editors. Encyclopedia of Biological Chemistry. New York: Elsevier Science; 2004. [Google Scholar]
- Coulombe PA, Chan YM, Albers K, Fuchs E. Deletions in epidermal keratins leading to alterations in filament organization in vivo and in intermediate filament assembly in vitro. J Cell Biol. 1990;111:3049–64. doi: 10.1083/jcb.111.6.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe PA, Fuchs E. Epidermolysis bullosa simplex. Semin Dermatol. 1993;12:173–90. [PubMed] [Google Scholar]
- Coulombe PA, Hutton ME, Letai A, Hebert A, Paller AS, Fuchs E. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: genetic and functional analyses. Cell. 1991a;66:1301–11. doi: 10.1016/0092-8674(91)90051-y. [DOI] [PubMed] [Google Scholar]
- Coulombe PA, Hutton ME, Vassar R, Fuchs E. A function for keratins and a common thread among different types of epidermolysis bullosa simplex diseases. J Cell Biol. 1991b;115:1661–74. doi: 10.1083/jcb.115.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe PA, Kerns ML, Fuchs E. Epidermolysis bullosa simplex: a paradigm for disorders of tissue fragility. J Clin Invest. 2009;119:1784–93. doi: 10.1172/JCI38177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessandro M, Coats SE, Jonkmann MF, Leigh IM, Lane EB. Keratin 14-null cells as a model to test the efficacy of gene therapy approaches in epithelial cells. J Invest Dermatol. 2011;131:1412–9. doi: 10.1038/jid.2011.19. [DOI] [PubMed] [Google Scholar]
- D’Alessandro M, Russell D, Morley SM, Davies AM, Lane EB. Keratin mutations of epidermolysis bullosa simplex alter the kinetics of stress response to osmotic shock. J Cell Sci. 2002;115:4341–51. doi: 10.1242/jcs.00120. [DOI] [PubMed] [Google Scholar]
- DePianto D, Kerns ML, Dlugosz AA, Coulombe PA. Keratin 17 promotes epithelial proliferation and tumor growth by polarizing the immune response in skin. Nat Genet. 2010;42:910–4. doi: 10.1038/ng.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein EH., Jr Molecular genetics of epidermolysis bullosa. Science. 1992;256:799–804. doi: 10.1126/science.1375393. [DOI] [PubMed] [Google Scholar]
- Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Fine JD, Eady RA, Bauer EA, Briggaman RA, Bruckner-Tuderman L, Christiano A, et al. Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. J Am Acad Dermatol. 2000;42:1051–66. [PubMed] [Google Scholar]
- Fitch KR, McGowan KA, van Raamsdonk CD, Fuchs H, Lee D, Puech A, Herault Y, Threadgill DW, Hrabe de Angelis M, Barsh GS. Genetics of dark skin in mice. Genes Dev. 2003;17:214–28. doi: 10.1101/gad.1023703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontao L, Tasanen K, Huber M, Hohl D, Koster J, Bruckner-Tuderman L, et al. Molecular consequences of deletion of the cytoplasmic domain of bullous pemphigoid 180 in a patient with predominant features of epidermolysis bullosa simplex. J Invest Dermatol. 2004;122:65–72. doi: 10.1046/j.0022-202X.2003.22125.x. [DOI] [PubMed] [Google Scholar]
- Freedberg IM, Tomic-Canic M, Komine M, Blumenberg M. Keratins and the keratinocyte activation cycle. J Invest Dermatol. 2001;116:633–40. doi: 10.1046/j.1523-1747.2001.01327.x. [DOI] [PubMed] [Google Scholar]
- Fuchs E. Keratins and the skin. Ann Rev Cell Dev Biol. 1995;11:123–53. doi: 10.1146/annurev.cb.11.110195.001011. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–19. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Coulombe PA. Of mice and men: genetic skin diseases of keratin. Cell. 1992;69:899–902. doi: 10.1016/0092-8674(92)90607-e. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Esteves RA, Coulombe PA. Transgenic mice expressing a mutant keratin 10 gene reveal the likely genetic basis for epidermolytic hyperkeratosis. Proc Natl Acad Sci U S A. 1992;89:6906–10. doi: 10.1073/pnas.89.15.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs E, Green H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 1980;19:1033–42. doi: 10.1016/0092-8674(80)90094-x. [DOI] [PubMed] [Google Scholar]
- Fuchs EV, Coppock SM, Green H, Cleveland DW. Two distinct classes of keratin genes and their evolutionary significance. Cell. 1981 doi: 10.1016/0092-8674(81)90362-7. [DOI] [PubMed] [Google Scholar]
- Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, et al. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;97:2289–98. doi: 10.1172/JCI118671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu LH, Coulombe PA. Keratin function in skin epithelia: a broadening palette with surprising shades. Curr Opin Cell Biol. 2007;19:13–23. doi: 10.1016/j.ceb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Gu LH, Coulombe PA. Defining the properties of the nonhelical tail domain in type II keratin 5: insight from a bullous disease-causing mutation. Mol Biol Cell. 2005;16:1427–38. doi: 10.1091/mbc.E04-06-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu LH, Coulombe PA. Keratin function in skin epithelia: a broadening palette with surprising shades. Curr Opin Cell Biol. 2007;19:13–23. doi: 10.1016/j.ceb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Gu LH, Kim SC, Ichiki Y, Park J, Nagai M, Kitajima Y. A usual frameshift and delayed termination codon mutation in keratin 5 causes a novel type of epidermolysis bullosa simplex with migratory circinate erythema. J Invest Dermatol. 2003;121:482–5. doi: 10.1046/j.1523-1747.2003.12424.x. [DOI] [PubMed] [Google Scholar]
- Haneke E, Anton-Lamprecht I. Ultrastructure of blister formation in epidermolysis bullosa hereditaria: V. Epidermolysis bullosa simplex localisata type Weber-Cockayne. J Invest Dermatol. 1982;78:219–23. doi: 10.1111/1523-1747.ep12506502. [DOI] [PubMed] [Google Scholar]
- Hanukoglu I, Fuchs E. The cDNA sequence of a human epidermal keratin: divergence of sequence but conservation of structure among intermediate filament proteins. Cell. 1982;31:243–52. doi: 10.1016/0092-8674(82)90424-x. [DOI] [PubMed] [Google Scholar]
- Harel A, Bergman R, Indelman M, Sprecher E. Epidermolysis bullosa simplex with mottled pigmentation resulting from a recurrent mutation in KRT14. J Invest Dermatol. 2006;126:1654–7. doi: 10.1038/sj.jid.5700296. [DOI] [PubMed] [Google Scholar]
- Hatzfeld M, Franke WW. Pair formation and promiscuity of cytokeratins: formation in vitro of heterotypic complexes and intermediate-sized filaments by homologous and heterologous recombinations of purified polypeptides. J Cell Biol. 1985;101:1826–41. doi: 10.1083/jcb.101.5.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M, Weber K. Modulation of keratin intermediate filament assembly by single amino acid exchanges in the consensus sequence at the C-terminal end of the rod domain. J Cell Sci. 1991;99:351–62. doi: 10.1242/jcs.99.2.351. [DOI] [PubMed] [Google Scholar]
- Hesse M, Zimek A, Weber K, Magin TM. Comprehensive analysis of keratin gene clusters in humans and rodents. Eur J Cell Biol. 2004;83:19–26. doi: 10.1078/0171-9335-00354. [DOI] [PubMed] [Google Scholar]
- Holbrook KA, Wapner R, Jackson L, Zaeri N. Diagnosis and prenatal diagnosis of epidermolysis bullosa herpetiformis (Dowling-Meara) in a mother, two affected children, and an affected fetus. Prenat Diagn. 1992;12:725–39. doi: 10.1002/pd.1970120906. [DOI] [PubMed] [Google Scholar]
- Horiguchi Y, Sawamura D, Mori R, Nakamura H, Takahashi K, Shimizu H. Clinical heterogeneity of 1649d elG mutation in the tail domain of keratin 5: a Japanese family with epidermolysis bullosa simplex with mottled pigmentation. J Invest Dermatol. 2005;125:83–5. doi: 10.1111/j.0022-202X.2005.23790.x. [DOI] [PubMed] [Google Scholar]
- Horn HM, Tidman MJ. The clinical spectrum of epidermolysis bullosa simplex. Br J Dermatol. 2000;142:468–72. doi: 10.1046/j.1365-2133.2000.03358.x. [DOI] [PubMed] [Google Scholar]
- Hovnanian A, Pollack E, Hilal L, Rochat A, Prost C, Barrandon Y, et al. A missense mutation in the rod domain of keratin 14 associated with recessive epidermolysis bullosa simplex. Nat Genet. 1993;3:327–32. doi: 10.1038/ng0493-327. [DOI] [PubMed] [Google Scholar]
- Inada H, Izawa I, Nishizawa M, Fujita E, Kiyono T, Takahashi T, Momoi T, Inagaki M. Keratin attenuates tumor necrosis factor-induced cytotoxicity through association with TRADD. J Cell Biol. 2001;155:415–26. doi: 10.1083/jcb.200103078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida-Yamamoto A, McGrath JA, Chapman SJ, Leigh IM, Lane EB, Eady RA. Epidermolysis bullosa simplex (Dowling-Meara type) is a genetic disease characterized by an abnormal keratin-filament network involving keratins K5 and K14. J Invest Dermatol. 1991;97:959–68. doi: 10.1111/1523-1747.ep12491885. [DOI] [PubMed] [Google Scholar]
- Ito M, Okuda C, Shimizu N, Tazawa T, Sato Y. Epidermolysis bullosa simplex (Koebner) is a keratin disorder. Ultrastructural and immunohistochemical study. Arch Dermatol. 1991;127:367–72. [PubMed] [Google Scholar]
- Jonkman MF, Heeres K, Pas HH, van Luyn MJ, Elema JD, Corden LD, et al. Effects of keratin 14 ablation on the clinical and cellular phenotype in a kindred with recessive epidermolysis bullosa simplex. J Invest Dermatol. 1996;107:764–9. doi: 10.1111/1523-1747.ep12365805. [DOI] [PubMed] [Google Scholar]
- Kerns M, DePianto D, Yamamoto M, Coulombe PA. Differential modulation of keratin expression by sulforaphane occurs via Nrf2-dependent and -independent pathways in skin epithelia. Mol Biol Cell. 2010;21:4068–75. doi: 10.1091/mbc.E10-02-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns ML, DePianto D, Dinkova-Kostova AT, Talalay P, Coulombe PA. Reprogramming of keratin biosynthesis by sulforaphane restores skin integrity in epidermolysis bullosa simplex. Proc Natl Acad Sci U S A. 2007;104:14460–5. doi: 10.1073/pnas.0706486104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol. 2010;11:75–81. doi: 10.1038/nrm2818. [DOI] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21:1581–97. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–5. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- Kitajima Y, Inoue S, Yaoita H. Abnormal organization of keratin intermediate filaments in cultured keratinocytes of epidermolysis bullosa simplex. Arch Dermatol Res. 1989;281:5–10. doi: 10.1007/BF00424265. [DOI] [PubMed] [Google Scholar]
- Kopan R, Fuchs E. A new look into an old problem: keratins as tools to investigate determination, morphogenesis, and differentiation in skin. Genes Dev. 1989;3:1–15. doi: 10.1101/gad.3.1.1. [DOI] [PubMed] [Google Scholar]
- Kreplak L, Fudge D. Biomechanical properties of intermediate filaments: from tissues to single filaments and back. Bioessays. 2007;29:26–35. doi: 10.1002/bies.20514. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–8. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane EB, Rugg EL, Navsaria H, Leigh IM, Heagerty AH, Ishida YA, et al. A mutation in the conserved helix termination peptide of keratin 5 in hereditary skin blistering. Nature. 1992;356:244–6. doi: 10.1038/356244a0. [DOI] [PubMed] [Google Scholar]
- Langan SM, Williams HC. A systematic review of randomized controlled trials of treatments for inherited forms of epidermolysis bullosa. Clin Exp Dermatol. 2008 doi: 10.1111/j.1365-2230.2008.02789.x. [DOI] [PubMed] [Google Scholar]
- Lee D, Santos D, Al-Rawi H, McNeill AM, Rugg EL. The chemical chaperone trimethylamine N-oxide ameliorates the effects of mutant keratins in cultured cells. Br J Dermatol. 2008;159:252–5. doi: 10.1111/j.1365-2133.2008.08596.x. [DOI] [PubMed] [Google Scholar]
- Leigh IM, Lane EB. Mutations in the genes for epidermal keratins in epidermolysis bullosa and epidermolytic hyperkeratosis. [Review] Arch Dermatol. 1993;129:1571–7. [PubMed] [Google Scholar]
- Letai A, Coulombe PA, Fuchs E. Do the ends justify the mean? Proline mutations at the ends of the keratin coiled-coil rod segment are more disruptive than internal mutations. J Cell Biol. 1992;116:1181–95. doi: 10.1083/jcb.116.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Coulombe PA, McCormick MB, Yu QC, Hutton E, Fuchs E. Disease severity correlates with position of keratin point mutations in patients with epidermolysis bullosa simplex. Proc Natl Acad Sci U S A. 1993;90:3197–201. doi: 10.1073/pnas.90.8.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin AS, Glazer PM, Milstone LM. Gene therapy for autosomal dominant disorders of keratin. J Investig Dermatol Symp Proc. 2005;10:47–61. doi: 10.1111/j.1087-0024.2005.10207.x. [DOI] [PubMed] [Google Scholar]
- Liovic M, Lee B, Tomic-Canic M, D’Alessandro M, Bolshakov VN, Lane EB. Dual-specificity phosphatases in the hypo-osmotic stress response of keratin-defective epithelial cell lines. Exp Cell Res. 2008;314:2066–75. doi: 10.1016/j.yexcr.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Lloyd C, Yu QC, Cheng J, Turksen K, Degenstein L, Hutton E, et al. The basal keratin network of stratified squamous epithelia: defining K15 function in the absence of K14. J Cell Biol. 1995;129:1329–44. doi: 10.1083/jcb.129.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffek S, Woll S, Hohfeld J, Leube RE, Has C, Bruckner-Tuderman L, et al. The ubiquitin ligase CHIP/STUB1 targets mutant keratins for degradation. Hum Mutat. 2010;31:466–76. doi: 10.1002/humu.21222. [DOI] [PubMed] [Google Scholar]
- Lu H, Chen J, Planko L, Zigrino P, Klein-Hitpass L, Magin TM. Induction of Inflammatory Cytokines by a Keratin Mutation and their Repression by a Small Molecule in a Mouse Model for EBS. J Invest Dermatol. 2007 doi: 10.1038/sj.jid.5700918. [DOI] [PubMed] [Google Scholar]
- Lulevich V, Yang HY, Isseroff RR, Liu GY. Single cell mechanics of keratinocyte cells. Ultramicroscopy. 2010;110:1435–42. doi: 10.1016/j.ultramic.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Ma L, Yamada S, Wirtz D, Coulombe PA. A ‘hot-spot’ mutation alters the mechanical properties of keratin filament networks. Nat Cell Biol. 2001;3:503–6. doi: 10.1038/35074576. [DOI] [PubMed] [Google Scholar]
- Markey AC, Lane EB, Macdonald DM, Leigh IM. Keratin expression in basal cell carcinomas. Br J Dermatol. 1992;126:154–60. doi: 10.1111/j.1365-2133.1992.tb07813.x. [DOI] [PubMed] [Google Scholar]
- McGowan KA, Aradhya S, Fuchs H, de Angelis MH, Barsh GS. A mouse keratin 1 mutation causes dark skin and epidermolytic hyperkeratosis. J Invest Dermatol. 2006;126:1013–6. doi: 10.1038/sj.jid.5700241. [DOI] [PubMed] [Google Scholar]
- McGowan KM, Coulombe PA. The wound repair associated keratins 6, 16, and 17: Insights into the role of intermediate filaments in specifying cytoarchitecture. In: Harris JR, Herrmann H, editors. Subcellular Biochemistry: Intermediate Filaments. London: Plenum Publishing Co; 1998. pp. 141–65. [PubMed] [Google Scholar]
- McLean WH, Moore CB. Keratin disorders: from gene to therapy. Hum Mol Genet. 2011;20:R189–97. doi: 10.1093/hmg/ddr379. [DOI] [PubMed] [Google Scholar]
- McLean WH, Pulkkinen L, Smith FJ, Rugg EL, Lane EB, Bullrich F, et al. Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev. 1996;10:1724–35. doi: 10.1101/gad.10.14.1724. [DOI] [PubMed] [Google Scholar]
- MIller SJ, Sun TT, Coulombe PA. Epidermal growth and differentiation. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Lefell DJ, editors. Fitzpatrick’s Dermatology in General Medicine. 7. New York: McGraw Hill; 2008. pp. 375–82. [Google Scholar]
- Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. [Review] Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- Morley SM, D’Alessandro M, Sexton C, Rugg EL, Navsaria H, Shemanko CS, et al. Generation and characterization of epidermolysis bullosa simplex cell lines: scratch assays show faster migration with disruptive keratin mutations. Br J Dermatol. 2003;149:46–58. doi: 10.1046/j.1365-2133.2003.05493.x. [DOI] [PubMed] [Google Scholar]
- Morley SM, Dundas SR, James JL, Gupta T, Brown RA, Sexton CJ, Navsaria HA, Leigh IM, Lane EB. Temperature sensitivity of the keratin cytoskeleton and delayed spreading of keratinocyte lines derived from EBS patients. J Cell Sci. 1995;108:3463–71. doi: 10.1242/jcs.108.11.3463. [DOI] [PubMed] [Google Scholar]
- Nelson WG, Sun TT. The 50- and 58-kdalton keratin classes as molecular markers for stratified squamous epithelia: cell culture studies. J Cell Biol. 1983;97:244–51. doi: 10.1083/jcb.97.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Guin WM, Schermer A, Lynch M, Sun T-T. Differentiation-specific expression of keratin pairs. In: Goldman RD, Steinert PM, editors. Cellular and molecular biology of intermediate filaments. Plenum Publishing Corp; 1990. pp. 301–34. [Google Scholar]
- Omary MB, Coulombe PA, McLean WHI. Intermediate filament proteins and their associated diseases. N Engl J Med. 2004;351:2087–100. doi: 10.1056/NEJMra040319. [DOI] [PubMed] [Google Scholar]
- Osmanagic-Myers S, Gregor M, Walko G, Burgstaller G, Reipert S, Wiche G. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J Cell Biol. 2006;174:557–68. doi: 10.1083/jcb.200605172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini RD, Coulombe PA. The functional diversity of epidermal keratins revealed by the partial rescue of the keratin 14 null phenotype by keratin 16. J Cell Biol. 1999;146:1185–201. doi: 10.1083/jcb.146.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini RD, Takahashi K, Bravo NS, Coulombe PA. Onset of re-epithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: defining a potential role for keratin 16. J Cell Biol. 1996;132:381–97. doi: 10.1083/jcb.132.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paller AS. The complexities of epidermolysis bullosa “simplex”. J Invest Dermatol. 2004;122:vi–vii. doi: 10.1046/j.1523-1747.2003.22143.x. [DOI] [PubMed] [Google Scholar]
- Paramio JM, Santos M, Jorcano JL. The ends of a conundrum? J Cell Sci. 2007;120:1145–7. doi: 10.1242/jcs.005348. author reply 7–8. [DOI] [PubMed] [Google Scholar]
- Parry DA. Microdissection of the sequence and structure of intermediate filament chains. Adv Protein Chem. 2005;70:113–42. doi: 10.1016/S0065-3233(05)70005-X. [DOI] [PubMed] [Google Scholar]
- Petek LM, Fleckman P, Miller DG. Efficient KRT14 targeting and functional characterization of transplanted human keratinocytes for the treatment of epidermolysis bullosa simplex. Mol Ther. 2010;18:1624–32. doi: 10.1038/mt.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B, Kirfel J, Bussow H, Vidal M, Magin TM. Complete cytolysis and neonatal lethality in keratin 5 knockout mice reveal its fundamental role in skin integrity and in epidermolysis bullosa simplex. Mol Biol Cell. 2001;12:1775–89. doi: 10.1091/mbc.12.6.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfendner E, Rouan F, Uitto J. Progress in epidermolysis bullosa: the phenotypic spectrum of plectin mutations. Exp Dermatol. 2005;14:241–9. doi: 10.1111/j.0906-6705.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- Pfendner EG, Nakano A, Pulkkinen L, Christiano AM, Uitto J. Prenatal diagnosis for epidermolysis bullosa: a study of 144 consecutive pregnancies at risk. Prenat Diagn. 2003;23:447–56. doi: 10.1002/pd.619. [DOI] [PubMed] [Google Scholar]
- Porter RM, Lunny DP, Ogden PH, Morley SM, McLean WH, Evans A, Harrison DL, Rugg EL, Lane EB. K15 expression implies lateral differentiation within stratified epithelial basal cells. Lab Invest. 2000;80:1701–10. doi: 10.1038/labinvest.3780180. [DOI] [PubMed] [Google Scholar]
- Quigley DA, To MD, Perez-Losada J, Pelorosso FG, Mao JH, Nagase H, et al. Genetic architecture of mouse skin inflammation and tumour susceptibility. Nature. 2009;458:505–8. doi: 10.1038/nature07683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt J, Furstenberger G, Magin TM. Loss of Keratin 10 Leads to Mitogen-activated Protein Kinase (MAPK) Activation, Increased Keratinocyte Turnover, and Decreased Tumor Formation in Mice. J Invest Dermatol. 2004;123:973–81. doi: 10.1111/j.0022-202X.2004.23426.x. [DOI] [PubMed] [Google Scholar]
- Reichelt J, Magin TM. Hyperproliferation, induction of c-Myc and 14-3-3sigma, but no cell fragility in keratin-10-null mice. J Cell Sci. 2002;115:2639–50. doi: 10.1242/jcs.115.13.2639. [DOI] [PubMed] [Google Scholar]
- Roth W, Reuter U, Wohlenberg C, Bruckner-Tuderman L, Magin TM. Cytokines as genetic modifiers in K5−/− mice and in human epidermolysis bullosa simplex. Hum Mutat. 2009;30:832–41. doi: 10.1002/humu.20981. [DOI] [PubMed] [Google Scholar]
- Rothnagel JA, Dominey AM, Dempsey LD, Longley MA, Greenhalgh DA, Gagne TA, et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science. 1992;257:1128–30. doi: 10.1126/science.257.5073.1128. [DOI] [PubMed] [Google Scholar]
- Rugg EL, Baty D, Shemanko CS, Magee G, Polak S, Bergman R, et al. DNA based prenatal testing for the skin blistering disorder epidermolysis bullosa simplex. Prenat Diagn. 2000;20:371–7. doi: 10.1002/(sici)1097-0223(200005)20:5<371::aid-pd818>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Rugg EL, McLean IWH, Lane EB, Pitera R, McMillan JR, Dopping-Hepenstal PJC, Navsaria HA, Leigh IM, Eady RAJ. A functional “knockout” of human keratin 14. Genes Dev. 1994;8:2563–73. doi: 10.1101/gad.8.21.2563. [DOI] [PubMed] [Google Scholar]
- Russell D, Andrews PD, James J, Lane EB. Mechanical stress induces profound remodelling of keratin filaments and cell junctions in epidermolysis bullosa simplex keratinocytes. J Cell Sci. 2004;117:5233–43. doi: 10.1242/jcs.01407. [DOI] [PubMed] [Google Scholar]
- Schuilenga-Hut PH, Scheffer H, Pas HH, Nijenhuis M, Buys CH, Jonkman MF. Partial revertant mosaicism of keratin 14 in a patient with recessive epidermolysis bullosa simplex. J Invest Dermatol. 2002;118:626–30. doi: 10.1046/j.1523-1747.2002.01715.x. [DOI] [PubMed] [Google Scholar]
- Schweizer J, Bowden PE, Coulombe PA, Langbein L, Lane EB, Magin TM, Maltais L, Omary MB, Parry DA, Rogers MA, Wright MW. New consensus nomenclature for mammalian keratins. J Cell Biol. 2006;174:169–74. doi: 10.1083/jcb.200603161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FJ, Morley SM, McLean WH. Novel mechanism of revertant mosaicism in Dowling-Meara epidermolysis bullosa simplex. J Invest Dermatol. 2004;122:73–7. doi: 10.1046/j.0022-202X.2003.22129.x. [DOI] [PubMed] [Google Scholar]
- Stacey SN, Sulem P, Masson G, Gudjonsson SA, Thorleifsson G, Jakobsdottir M, et al. New common variants affecting susceptibility to basal cell carcinoma. Nat Genet. 2009;41:909–14. doi: 10.1038/ng.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert PM, Idler WW, Zimmerman SB. Self-assembly of bovine epidermal keratin filaments in vitro. J Mol Biol. 1976;108:547–67. doi: 10.1016/s0022-2836(76)80136-2. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Marekov LN, Fraser RD, Parry DA. Keratin intermediate filament structure. Crosslinking studies yield quantitative information on molecular dimensions and mechanism of assembly. Journal Of Molecular Biology. 1993;230:436–52. doi: 10.1006/jmbi.1993.1161. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Rice RH, Roop DR, Trus BL, Steven AC. Complete amino acid sequence of a mouse epidermal keratin subunit and implications for the structure of intermediate filaments. Nature. 1983;302:794–800. doi: 10.1038/302794a0. [DOI] [PubMed] [Google Scholar]
- Stephens K, Sybert VP, Wijsman EM, Ehrlich P, Spencer A. A keratin 14 mutational hot spot for epidermolysis bullosa simplex, Dowling-Meara: implications for diagnosis. J Invest Dermatol. 1993;101:240–3. doi: 10.1111/1523-1747.ep12365079. [DOI] [PubMed] [Google Scholar]
- Swartling C, Karlqvist M, Hymnelius K, Weis J, Vahlquist A. Botulinum toxin in the treatment of sweat-worsened foot problems in patients with epidermolysis bullosa simplex and pachyonychia congenita. Br J Dermatol. 2010;163:1072–6. doi: 10.1111/j.1365-2133.2010.09927.x. [DOI] [PubMed] [Google Scholar]
- Swensson O, Langbein L, McMillan JR, Stevens HP, Leigh IM, McLean WH, et al. Specialized keratin expression pattern in human ridged skin as an adaptation to high physical stress. Br J Dermatol. 1998;139:767–75. doi: 10.1046/j.1365-2133.1998.02499.x. [DOI] [PubMed] [Google Scholar]
- Szeverenyi I, Cassidy AJ, Chung CW, Lee BT, Common JE, Ogg SC, et al. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29:351–60. doi: 10.1002/humu.20652. [DOI] [PubMed] [Google Scholar]
- Tong X, Coulombe PA. Keratin 17 modulates hair follicle cycling in a TNFalpha-dependent fashion. Genes Dev. 2006;20:1353–64. doi: 10.1101/gad.1387406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttam J, Hutton E, Coulombe PA, Anton-Lamprecht I, Yu QC, Gedde-Dahl T, Jr, Fine JD, Fuchs E. The genetic basis of epidermolysis bullosa simplex with mottled pigmentation. Proc Natl Acad Sci U S A. 1996;93:9079–84. doi: 10.1073/pnas.93.17.9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Coulombe PA, Degenstein L, Albers K, Fuchs E. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell. 1991;64:365–80. doi: 10.1016/0092-8674(91)90645-f. [DOI] [PubMed] [Google Scholar]
- Wally V, Brunner M, Lettner T, Wagner M, Koller U, Trost A, et al. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum Mol Genet. 2010;19:4715–25. doi: 10.1093/hmg/ddq405. [DOI] [PubMed] [Google Scholar]
- Wally V, Klausegger A, Koller U, Lochmuller H, Krause S, Wiche G, et al. 5′ trans-splicing repair of the PLEC1 gene. J Invest Dermatol. 2008;128:568–74. doi: 10.1038/sj.jid.5701152. [DOI] [PubMed] [Google Scholar]
- Werner NS, Windoffer R, Strnad P, Grund C, Leube RE, Magin TM. Epidermolysis bullosa simplex-type mutations alter the dynamics of the keratin cytoskeleton and reveal a contribution of actin to the transport of keratin subunits. Mol Biol Cell. 2004;15:990–1002. doi: 10.1091/mbc.E03-09-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci. 1998;111:2477–86. doi: 10.1242/jcs.111.17.2477. [DOI] [PubMed] [Google Scholar]
- Wilson AK, Coulombe PA, Fuchs E. The roles of K5 and K14 head, tail, and R/K L L E G E domains in keratin filament assembly in vitro. J Cell Biol. 1992;119:401–14. doi: 10.1083/jcb.119.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P, Coulombe PA. Loss of keratin 6 (K6) proteins reveals a function for intermediate filaments during wound repair. J Cell Biol. 2003;163:327–37. doi: 10.1083/jcb.200305032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock-Mitchell J, Eichner R, Nelson WG, Sun TT. Immunolocalization of keratin polypeptides in human epidermis using monoclonal antibodies. J Cell Biol. 1982;95:580–8. doi: 10.1083/jcb.95.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KC, Bryan JT, Morasso MI, Jang SI, Lee JH, Yang JM, et al. Coiled-coil trigger motifs in the 1B and 2B rod domain segments are required for the stability of keratin intermediate filaments [In Process Citation] Mol Biol Cell. 2000;11:3539–58. doi: 10.1091/mbc.11.10.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin T, Getsios S, Caldelari R, Kowalczyk AP, Muller EJ, Jones JC, et al. Plakoglobin suppresses keratinocyte motility through both cell-cell adhesion-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2005;102:5420–5. doi: 10.1073/pnas.0501676102. [DOI] [PMC free article] [PubMed] [Google Scholar]