

Abstract
Enzyme replacement therapies for lysosomal storage disorders are often hindered by suboptimal biodistribution of recombinant enzymes after systemic injection. This is the case for Pompe disease caused by acid α-glucosidase (GAA) deficiency, leading to excess glycogen storage through the body, mainly the liver and striated muscle. Targeting intercellular adhesion molecule-1 (ICAM-1), a protein involved in inflammation and overexpressed on most cells under pathological conditions, provides broad biodistribution and lysosomal transport of therapeutic cargoes. To improve its delivery, we coupled GAA to polymer nanocarriers (~180-nm) coated with anti-ICAM. Fluorescence microscopy showed specific targeting of anti-ICAM/GAA NCs to cells, with efficient internalization and lysosomal transport, enhancing glycogen degradation over non-targeted GAA. Radioisotope tracing in mice demonstrated enhanced GAA accumulation in all organs, including Pompe targets. Along with improved delivery of Niemann-Pick and Fabry enzymes, previously described, these results indicate that ICAM-1 targeting holds promise as a broad platform for lysosomal enzyme delivery.
Keywords: ICAM-1, polymer nanocarriers, lysosomal storage disorders, enzyme replacement therapy, α-glucosidase
BACKGROUND
The lysosomal storage disorders (LSDs) are inherited diseases often due to deficiency of lysosomal enzymes.1 This is the case for Pompe disease caused by deficiency of acid α-glucosidase (GAA, an enzyme that hydrolyzes glycogen to glucose), which results in aberrant glycogen storage within lysosomes.2 Given that the liver and striated (cardiac and skeletal) muscle are more dependent on GAA activity, Pompe disease markedly affects these tissues.2–4 In addition, Pompe disease manifests in other organs, including the nervous system and the vasculature.2 Therefore, therapeutic intervention of this disease requires correction of GAA deficiency throughout the body, with emphasis on the liver and striated muscle.
Current treatment for Pompe disease utilizes enzyme replacement therapy (ERT), which substitutes deficient human GAA with a recombinant counterpart.5–8 Recombinant GAA is glycosylated and can bind to mannose-6-phosphate receptors (M6PRs) on cells, which results in clathrin-dependent endocytosis and transport to lysosomes.9,10 Systemic infusion of GAA reduces heart accumulation of glycogen, yet treatment of skeletal muscle and other tissues remain challenging.8,11 Poor access of recombinant GAA to these tissues, altered M6PR expression in LSDs, and/or antibody production against the injected enzyme reduces response to therapy.12–17 Although ERT efficacy can be enhanced by immunomodulators, this poses considerable risk,18–20 emphasizing the need for alternative therapeutic options.
An experimental strategy that enhances delivery of other lysosomal enzymes is that of coupling said enzymes to polymer nanocarriers targeted to intercellular adhesion molecule (ICAM-1).21–25 ICAM-1 is a transmembrane protein functionally involved in inflammation.26,27 It is present on endothelial and other cell types, and is over-expressed in many inflammatory pathologies, including LSDs.26,27 Binding of anti-ICAM nanocarriers to ICAM-1 results in carrier endocytosis by cells and transport to lysosomes.28,29 This strategy is expected to provide broad distribution of therapeutics through the body, most favorably to disease sites), and lysosomal delivery, amenable for LSD treatment.
Previous works have shown that polymer nanoparticles (100–200 nm in diameter) coated with anti-ICAM enhance delivery of lysosomal enzymes in cell cultures and mice, bypass clathrin-mediated endocytosis of these enzymes, and provide marked attenuation of aberrant lysosomal storage.22,24,25 Yet, the biodistribution pattern of anti-ICAM nanocarriers (anti-ICAM NCs) is slightly different depending on cargo.21,22 Also, the nature of the substrate storage of each LSD and the resulting dysfunction impacts endocytosis and intracellular trafficking,12,14,30,31 possibly affecting the delivery efficiency in particular disease settings. Whether the potential of ICAM-1-targeting strategy stands in other LSDs is an open question, e.g., accumulation of anti-ICAM NCs in skeletal muscle (a Pompe disease target) has never been tested.
In this work we have used fluorescence microscopy and radioisotope tracing to evaluate anti-ICAM/GAA NC binding, internalization, lysosomal transport, and attenuation of the excess glycogen storage in cell cultures, as well as biodistribution in mice, showing the potential of this strategy in the context of Pompe disease. We have also compared anti-ICAM/GAA NCs to formulations carrying other lysosomal enzymes to estimate the potential of the ICAM-1-targeting strategy as a broad enzyme delivery platform for LSDs.
METHODS
Antibodies and reagents
Monoclonal antibodies to human or murine ICAM-1 (anti-ICAM) were R6.526 or YN1.32 Secondary antibodies were from Jackson Immunoresearch (West Grove, PA, USA). Saccharomyces cerevisiae recombinant α-glucosidase (GAA) was from Sigma Aldrich (St. Louis, MO, USA). Polystyrene beads (100 nm-diameter) were from Polysciences (Warrington, PA, USA). Cell culture reagents were from Cellgro (Manassas, VA, USA) or Gibco BRL (Grand Island, NY, USA). Other reagents were from Sigma Aldrich.
Preparation of anti-ICAM/GAA nanocarriers and enzyme release
Model polymer nanocarriers were prepared by coating 100 nm-diameter polystyrene beads by surface adsorption with anti-ICAM (anti-ICAM NCs) or a mix of anti-ICAM and GAA (anti-ICAM/GAA NCs).22 A 95:5 antibody:enzyme mass ratio was used in vivo, as minimal amounts of GAA are needed to trace its biodistribution, and a 50:50 mass ratio was used in cell cultures to obtain measurable effects. FITC particles were used for fluorescence microscopy. Alternatively, 5% of total GAA was labeled with 125Iodine (125I-GAA). After coating, nanocarriers were washed and centrifuged to remove free anti-ICAM and GAA, resuspended in 1% bovine serum albumin (BSA) and sonicated to avoid aggregation. This protocol rendered ~180±7 nm particles (0.2 polydispersity) by dynamic light scattering (Zetasizer Nano-ZS90; Malvern Instruments Inc., Westborough, MA). This is a prototype not intended for clinical use, yet a valid model to demonstrate targeting since these particles display similar coating efficacy, targeting, and intracellular transport21,33 as anti-ICAM NCs made of poly(lactic-co-glycolic acid),34,35 a material approved by the US Food and Drug Administration. The nanocarrier concentrations used were estimated from the literature to display enough radioisotope tracing and biochemical effects. 6,7,21,22
Release of 125I-GAA from anti-ICAM NCs was determined by centrifugation to separate free enzyme from particle-bound fraction.23 Mechanical GAA release was determined after centrifugation, resuspension by pipetting, and sonication. Enzyme release was tested after incubation in storage buffer (phosphate buffer saline, PBS, supplemented with 1% bovine serum albumin, BSA) or complete cell medium (described below), at 4°C or 37°C, pH 7.4 or pH 4.5, and in the absence or presence of 50mg/mL glycogen, the enzyme substrate.
Cell cultures
Endothelial cells represent the first cell layer encountered by injected lysosomal enzymes and targeting these cells impacts organ accumulation. We used human umbilical vein endothelial cells (HUVECs; Clonetics, San Diego, CA, USA) maintained in M-199 supplemented as described,22 and seeded on 1% gelatin-coated coverslips. Skeletal muscle cells were isolated from C57Bl/6 mouse gastrocnemius and quadriceps by digestion with 0.2 mg/mL collagenase for 24 hours at 37°C, and seeded on matrigel (BD Biosciences, Franklin Lakes, NJ, USA). Cells were activated overnight with 10 ng/mL tumor necrosis factor-α (TNF-α; BD Biosciences, Franklin Lakes, NJ, USA), to up-regulate ICAM-1 expression as in pathology.22,26,27
Pompe disease-model cells were generated by treating cells overnight with 300 or 600 μM D(+)-turanose, a GAA competitive inhibitor.2,36 Glycogen accumulation was verified by staining fixed cells with periodic acid-Schiff (PAS).37 Semi-quantitative measurement of glycogen was assessed using an Olympus IX81 microscope (Olympus, Inc., Center Valley, PA) with a 40X objective (UPlanApo, Olympus, Inc.). Bright field micrographs were captured under blue, green and red filters (1160A-OMF, 3540B-OMF, 4040B-OMF; Semrock, Inc., Rochester, NY) with an ORCA-ER camera (Hamamatsu Corporation, Bridgewater, NJ) and combined them with SlideBook™ 4.2 (Intelligent Imaging Innovations, Inc., Denver, CO). Red glycogen was quantified with Image-Pro 6.3 (Media Cybernetics, Inc., Bethesda, MD) and normalized per cell.
Binding, internalization and lysosomal co-localization of anti-ICAM/GAA nanocarriers
Control cells or Pompe disease-model cells were incubated at 37°C with FITC anti-ICAM NCs or anti-ICAM/GAA NCs (2.5 μg GAA/mL, 7×1010 particles/mL) for 30 min to allow binding. Cells were left undisturbed or washed to remove unbound nanocarriers (pulse-chase incubation to synchronize transport) for a total time of 1 h or 3 h, to determine endocytosis. Alternatively, 3 mM amiloride (inhibitor of CAM endocytosis) or 50 μM monodansylcadaverine (MDC) (inhibitor of clathrin endocytosis) were used.28 Cells were washed, fixed and surface-bound carriers were stained using Texas-Red goat anti-mouse IgG that recognizes anti-ICAM on particles located on the cell surface (yellow color) but cannot access those endocytosed by the cell (green color).28 The nucleus was stained with blue DAPI. Fluorescence microscopy was used to determine the percentage of nanocarriers internalized per cell.28
To examine intracellular transport of nanocarriers, lysosomes were labeled by incubating cells with Texas-Red dextran (1.5 h, 37C), followed by washing and 1 h incubation with FITC anti-ICAM/GAA NCs.29 Cells were then washed and fixed, or incubated for 2 h or 4 h more to synchronize intracellular transport of carriers.29 Co-localization of green fluorescent nanocarriers with red-labeled lysosomes (yellow color) was analyzed by fluorescence microscopy.29
Glycogen degradation by anti-ICAM/GAA nanocarriers
Pompe disease-model cells were incubated at 37°C for 5 h with anti-ICAM/GAA NCs or non-targeted GAA (2.5 μg/ml GAA), in the presence of 300 μM chloroquine.22 Chloroquine is used because removal of turanose (required to avoid inhibition of the delivered GAA) may render endogenous GAA active. Chloroquine neutralizes lysosomes and inhibits endogenous lysosomal enzymes that have acidic pKa without affecting the exogenous neutral GAA. After incubation, samples were fixed, stained with PAS, and analyzed by microscopy to quantify glycogen degradation as follows:
In vivo biodistribution
Anti-ICAM/125I-GAA NCs or free 125I-GAA (51.4±3.7 μg/kg bodyweight) were injected intravenously into anesthetized C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA). Blood samples were collected at 2, 15, and 30 min. At 30 min, brain, heart, kidneys, liver, lungs, spleen, and left gastrocnemius and quadricep muscles were collected. The radioactivity and weight of organs were used to calculate: (i) percentage of injected dose per organ (%ID) to compare the biodistribution of the injected formulations, (ii) percent injected dose per gram of organ (%ID/g) to compare relative distribution amongst organs of different sizes, (iii) localization ratio (LR; organ-to-blood ratio of %ID/g) to compare tissue-to-blood distribution, and (iv) specificity index (SI, targeted-to-untargeted LR ratio in a given organ) to compare efficiency of targeted to non-targeted formulation, as described.21,22 Animal studies were performed following humane care guidelines and approved by IACUC.
Statistics
Data were calculated as mean ± standard error of the mean (SEM). Statistical significance was determined by unpaired Student’s t tests.
RESULTS
Characterization of anti-ICAM/GAA nanocarriers and enzyme release
Anti-ICAM and GAA were coated onto 100 nm nanocarriers (anti-ICAM/GAA NCs). As shown in Table 1, the 125I-GAA loading efficiency was 86.2±4.3%, rendering 277.7±1.4 GAA molecules per carrier and diameter of 180.1±0.9 nm when co-coated at a 50:50 mass ratio (selected based on the enzyme dose estimated to exert biochemical effects6,7).
Table 1.
Load, transport and effects of ICAM-1-targeted nanocarriers for lysosomal delivery in cell culture.
| Non- loaded | ASM | α-Gal | GAA | |
|---|---|---|---|---|
| Enzyme coating efficiency (%) | N/A | 80 | 77 | 86 |
| Enzyme molecule per NC (50:50 mass) | N/A | 230 | 530 | 278 |
| Anti-ICAM molecule per NC (50:50 mass) | 250 | 135 | 138 | 123 |
|
| ||||
| Binding (NCs per cell) | 165 | 160 | 70 | 80 |
| % Internalization (1 h) | 80 | 70 | 67 | 80 |
| % Lysosomal transport (3 h) | 75 | 70 | 70 | 65 |
| % Lysosomal storage degradation (5 h) | N/A | 98 | 70 | 75 |
| Enhanced degradation (anti-ICAM/enzyme NCs: free enzyme) | N/A | 2 | 2.5 | 3 |
The enzyme coat was stable through centrifugation, resuspension by pipetting, and sonication (11.6±0.3% enzyme release), and during storage at 4°C in buffer (0.0±0.1% to 6.1±0.5% enzyme release within 3 days; Supplementary Figure 1). Incubation of anti-ICAM/125I-GAA NCs at 37°C and neutral pH in complete cell medium also showed marked enzyme retention (e.g., 3.8±0.2% release at 3 h and 11.9±0.3% release and 72 h). Enzyme release was markedly enhanced under conditions mimicking the lysosomal environment, e.g., pH 4.5 and presence of the enzyme substrate, glycogen (3.1, 7.9, and 11.5 fold increased compared to neutral pH at 30 min, 3 h, and 8 h, respectively).
Targeting and internalization of anti-ICAM/GAA nanocarriers in Pompe disease-model cells
We generated Pompe disease-model cells by treating endothelial cells (the first layer of cells GAA encounters in the body after systemic administration) with turanose, a competitive inhibitor of GAA that induces glycogen accumulation.2 As shown in Supplementary Figure 2, cells treated overnight with 300 μM turanose accumulated increased levels of glycogen over control cells, similar to some tissues from adult-onset Pompe disease patients. 2,36
Targeting of anti-ICAM/GAA NCs in this model was assessed by fluorescence microscopy. As shown in Figure 1, A and Supplementary Figure 3, anti-ICAM/GAA NCs bound efficiently to cells: 51.3±4.7 NCs/cell by 30 min and 79.8±8.3 NCs/cell by 1 h, reaching a plateau. The level of binding of non-specific IgG NCs to control cells was 2±2 NCs/cell (data not shown), demonstrating the specificity of anti-ICAM counterparts. Importantly, anti-ICAM NCs also bound to primary skeletal muscle cells (a major target in Pompe disease) isolated from mice (Figure 1, B). In these cells carriers seemed to align in the direction of cytoskeletal fibers, a feature previously observed during internalization of anti-ICAM NCs by endothelial cells.24,28
Figure 1. Anti-ICAM/GAA NCs target and are internalized by Pompe-model cells via CAM endocytosis.

Binding of FITC-labeled anti-ICAM/GAA NCs to TNF-α activated, turanose-treated (A) HUVECs (30 min, 1 h, or 3 h, 37°C), analyzed by fluorescence microscopy, or (B) skeletal muscle cells from mice (5 h, 37°C), where arrowheads mark green carriers aligned on red-pseudocolored cytoskeletal fibers (from phase-contrast). (C–D) Micrographs (left; 1 h, 37°C) and quantification (right) of the internalization of FITC anti-ICAM/GAA NCs by HUVEC (30 min, 1 h, or 3 h, 37°C) in control media (C) or media containing amiloride to inhibit CAM endocytosis, or MDC to inhibit clathrin-coated pits. Surface bound nanocarriers were stained with Texas Red-labeled secondary antibody (double-labeled red+green yellow particles; arrows) versus internalized carriers (single-labeled green particles; arrowheads). Cell nuclei were stained with blue DAPI. Scale bar, 10 μm. Data are mean±SEM (n≥32 cells, two experiments). ***p≤0.001, by Student’s t-test (compared to control cells).
In agreement with this observation, 78.6±1.7% of all bound anti-ICAM/GAA NCs were internalized by Pompe disease-model cells within the first 30 min (Figure 1, C and Supplementary Figure 4). Particle uptake slightly increased to 85.0±1.7% and 89.4±0.4% by 1 h and 3 h. This suggests efficient intracellular transport of GAA by anti-ICAM NCs (t½ = 10 min) even in cells presenting aberrant glycogen storage, achieving almost complete uptake of nanocarriers that bind to the target.
In addition (Figure 1, D and Supplementary Figure 5) internalization of anti-ICAM/GAA NCs was inhibited by amiloride, a drug that affects cell adhesion molecule- (CAM)-mediated endocytosis (42.6±1.6% inhibition), but not MDC, an inhibitor of the clathrin-mediated pathway related to uptake of lysosomal enzymes (14.6±1.0% inhibition). This suggests that anti-ICAM NCs are capable of successfully shifting endocytosis of GAA from classical clathrin-coated pits to non-classical CAM-mediated pathway in a Pompe disease model.
Lysosomal transport of anti-ICAM/GAA nanocarriers and reduction of glycogen storage in Pompe disease-model cells
We examined the intracellular trafficking of anti-ICAM/GAA NCs labeled with green FITC in Pompe disease-model cells whose lysosomes had been pre-labeled using Texas-Red dextran.29 Fluorescence microscopy (Figure 2, A) showed a modest co-localization of green carriers with red lysosomes during the first hour (32.8±2.5%). However, delivery to lysosomes was significantly increased to 63.6±2.2% and 77.3±2.8% by 3 h and 5 h. This suggests that transport of the therapeutic cargo, GAA, to this compartment could be relatively efficient even in Pompe disease-model cells, supporting the potential of this strategy.
Figure 2. Anti-ICAM/GAA NCs traffic to lysosomes and reduce glycogen storage in Pompe-model cells.
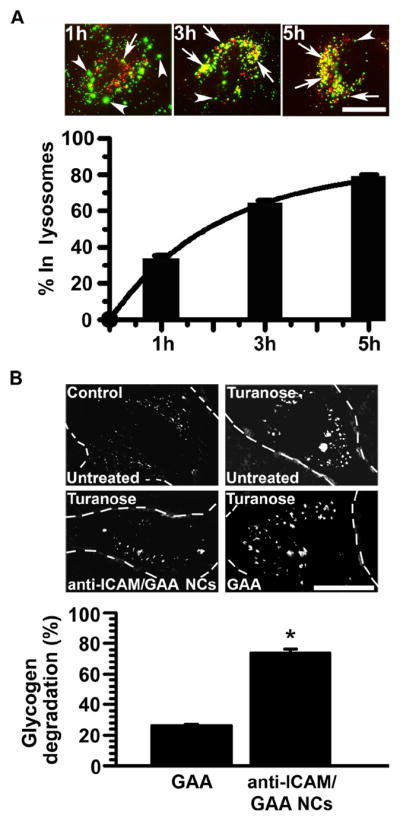
(A) FITC anti-ICAM/GAA NCs nanocarriers co-localizing with Texas-Red dextran-labeled lysosomes in Pompe disease-model cells are shown in yellow (red+green; arrowheads). Nanocarriers in other localizations are shown in green (arrows). Scale bar, 10 μm. Data are mean±SEM (n≥50 cells, two experiments). (B) Glycogen levels (determined by PAS staining) in cells treated for 5 h with a similar dose of neutral GAA coated on anti-ICAM NCs or as a free counterpart, in the presence of chloroquine, a base that inhibits the activity of endogenous acidic GAA but not exogenously administered neutral GAA. Data are mean±SEM (n≥49cells). *p≤0.05, by Student’s t-test (compared to untreated disease cells).
To confirm this, we measured glycogen reduction in Pompe disease model-cells after incubation with anti-ICAM/GAA NCs or a comparable amount of non-targeted GAA. After achieving aberrant glycogen storage in cells, turanose must be removed to avoid inhibition of the delivered enzyme. Hence, to ensure that glycogen attenuation was achieved by the exogenous neutral GAA after delivery, we inhibited the endogenous lysosomal acidic GAA by adding chloroquine to neutralize lysosomes. This strategy, used for α-Gal delivery,22 also mimics the lysosomal pH elevation observed in some LSDs,38 including severe Pompe disease in which some cells exhibit a failure to lower the lysosomal pH below 6.0.14
As shown in Figure 2, B, glycogen degradation by anti-ICAM/GAA NCs was significantly enhanced (by ~3 fold) compared to the same dose of non-targeted GAA (73.9±2.3% versus 26.0±0.9% glycogen reduction, respectively), suggesting that lysosomal delivery achieved by anti-ICAM NCs may be therapeutically relevant as compared to the free enzyme counterpart.
Biodistribution of anti-ICAM/GAA nanocarriers in mice
We then determined the in vivo biodistribution of anti-ICAM/125I-GAA NCs after intravenous administration in mice. Anti-ICAM/125I-GAA NCs rapidly disappeared from circulation, with only 2.6±0.5% of the injected dose (% ID) in blood by 1–2 min after injection, and remained low for the duration of the experiment (Supplementary Figure 6). This may be due to clearance from circulation and/or rapid targeting to ICAM-1-positive tissues. Indeed, we observed fast accumulation of anti-ICAM/125I-GAA NCs throughout the body (Figure 3, A, 30 min after injection). As expected, due to a first-pass effect of nanocarriers injected intravenously, its profuse vascularization, high ICAM-1 expression, and organ size, the lungs received the highest dose fraction (48.8±3.3 %ID). Liver accumulation followed (17.3±2.0 %ID), in accord with the size of this organ, its vascularization, high ICAM-1 expression and its role in blood clearance. Another organ of the reticulo-endothelial system, the spleen, accumulated a considerable fraction of anti-ICAM/125I-GAA NCs (2.9±0.3 %ID). Although more modest, nanocarriers were also found in the brain (0.15±0.01 %ID) and striated muscle: the heart received 0.4±0.03 %ID and the gastrocnemius and quadriceps muscles accumulated 0.07±0.01 %ID and 0.08±0.01 %ID, respectively.
Figure 3. Biodistribution of anti-ICAM/GAA NCs in mice.

Anti-ICAM/125I-GAA NCs were injected intravenously in mice, and blood and organs were collected 30 min after injection. The samples were weighed and measured for 125Iodine content. (A) Percentage of injected dose (%ID) per organ was calculated to determine 125I-GAA in circulation and tissue accumulation. (B) Percentage of injected dose per gram of tissue (%ID/g) was calculated to compare 125I-GAA delivery among organs of different size. Gastroc = gastrocnemius. Quad = quadriceps. Data are mean±SEM (n≥4 mice).
When the accumulation of anti-ICAM/125I-GAA NCs was normalized to respective tissue weight (%ID/g of organ; Figure 3, B), to compare organ specificity, the lungs remained the most heavily targeted organ with 246.8±20.4 %ID/g. This was expected as the lungs contain 20–30% endothelium in the body with high ICAM-1 expression.21 Next, high accumulation was found in the spleen (30.9±3.4%ID/g), followed by the liver (12.8±1.6%ID/g) and heart (2.9±0.2%ID/g), and finally, skeletal muscle and brain: 0.4±0.04 %ID/g and 0.3±0.04 %ID/g in gastrocnemius and quadriceps muscles, and 0.3±0.03%ID/g in brain tissue.
Comparative biodistribution of GAA delivered by anti-ICAM nanocarriers versus non-targeted GAA
We next compared the biodistribution of anti-ICAM/125I-GAA NCs to that of non-targeted 125I-GAA (Figure 4). In contrast to anti-ICAM/125I-GAA NCs, free 125I-GAA circulated significantly longer (e.g., 38.4±2.1 %ID at 30 min), suggesting lower affinity to tissues (Supplementary Figure 6). To correct for the difference in the circulating levels of 125I-GAA versus anti-ICAM/125I-GAA NCs (~10 fold higher for non-targeted GAA), we calculated the localization ratio (LR; Figure 4, A). This parameter measures %ID of a formulation per gram of organ divided by %ID/g in blood (tissue-to-blood ratio).
Figure 4. Anti-ICAM NCs enhance delivery of GAA over non-targeted enzyme in mice.

Anti-ICAM/125I-GAA NCs or a similar dose of 125I-GAA were injected intravenously in mice, and blood and organs were collected 30 min after. The samples were weighed and measured for 125Iodine content. (A) The localization ratio (LR), calculated as percent injected dose per gram of organ (%ID/g) divided by %ID/g in blood, compares tissue accumulation for these two formulations with different circulation. (B) The specificity index (SI), calculated as the LR ratio of nanocarrier-targeted versus non-targeted 125I-GAA, reflects improved enzyme delivery of anti-ICAM NCs. Gastroc=gastrocnemius. Quad=quadriceps. Data are mean±SEM (n≥3 mice). *p≤0.05, **p≤0.01, ***p≤0.001, by Student’s t-test (compared to free GAA).
Using this parameter, accumulation of ICAM-1-targeted 125I-GAA in mouse organs significantly and consistently exceeded that of free 125I-GAA, even in tissues which showed low levels of anti-ICAM/125I-GAA NCs. For example, in cardiac and skeletal muscle, anti-ICAM/125I-GAA NCs had LRs of 1.91±0.39, 0.31±0.05 and 0.25±0.03 for heart, gastrocnemius muscle and quadriceps muscle, versus non-targeted 125I-GAA LRs of 0.17±0.01, 0.04±0.01 and 0.04±0.003. The specificity index (SI; Figure 4, B), which represents the LR ratio of targeted to non-targeted formulations, indicates that anti-ICAM NCs increased 125I-GAA delivery to all organs: 584-fold increase in lungs, 67- in spleen, 23- in liver, 10- in heart, and 6-fold in skeletal muscle and brain, indicating great potential for anti-ICAM NCs to enhance GAA delivery in vivo.
DISCUSSION
Our results indicate that ICAM-1-targeting holds potential to improve delivery of GAA for ERT of Pompe disease. Anti-ICAM/GAA NCs provided efficient targeting in Pompe disease-model cells and primary cultures of skeletal muscle, as well as internalization and lysosomal transport with attenuation of excess glycogen, surpassing the effects of free GAA. Anti-ICAM/GAA NCs also markedly enhanced enzyme delivery in vivo compared to non-targeted GAA in all tissues tested, including heart, skeletal muscle and liver, major targets in Pompe disease.
An adequate geometry of anti-ICAM carriers is crucial to modulate the efficacy of this strategy. Although cells in culture internalize anti-ICAM carriers within a wide range of geometries (~200 nm to 5 μm, spheres and elongated disks), sub-micrometer spherical carriers and micrometer elongated counterparts display more specific targeting in vivo, yet only sub-micrometer anti-ICAM NCs traffic efficiently to lysosomes.25 Hence, sub-micrometer spherical anti-CAM carriers (shown in this work) display most adequate features for lysosomal enzyme delivery.
Using such geometry parameters, coating efficacy and stability of the 125I-GAA coat on prototype anti-ICAM NCs under storage conditions and cell media were relatively high, while the enzyme was released under conditions mimicking the lysosomal microenvironment (Table 1 and Supplementary Figure 1), suitable for the intended application. Although our goal was to generate a prototype to prove targeting and biochemical effects, this configuration is still relevant since lysosomal enzymes are active at acidic lysosomal pH and act as pro-drugs prior to reaching this compartment. This loading capacity can translate into a dose of ~2 mg GAA/kg body weight, on the order of magnitude of current clinical doses of the non-targeted enzyme.7,8
Compared to nanocarriers coated at saturation with only anti-ICAM, GAA coating reduces the surface covered by antibody by ~50%. As described,39 this is expected to reduce targeting. In agreement, our result show binding of ~80 NCs/cell after 1 h incubation (Figure 1, A, and Table 1), which is a ~50% reduction compared to anti-ICAM NCs (Table 1). Nevertheless, binding was sufficient to target primary skeletal muscle cells (Figure 1, B), which express lower ICAM-1 levels than endothelial cells.27 This binding surpassed by 35-fold targeting of control IgG NCs and provided sufficient delivery of GAA as to attenuate glycogen storage (Figure 2, B).
This outcome was possible due to relatively high efficiency of uptake of anti-ICAM/GAA NCs by cells: ~80% of all cell-bound carriers at 1h (Figure 1, C, and Table 1). The internalization rate and pathway were similar to that of anti-ICAM NCs (Table 1), despite differences in the binding of these formulations. This is in agreement with previous works showing efficient uptake of anti-ICAM NCs by cells regardless of their binding level.24,28 Uptake is mediated by amiloride-sensitive CAM-mediated endocytosis, a pathway different from clathrin- and caveolar-mediated endocytosis, phagocytosis and macropinocytosis.24,28
Anti-ICAM/GAA NCs trafficked to lysosomes (e.g., 65% by 3 h), which was only minimally reduced compared to anti-ICAM NCs (75% by 3 h; Table 1). This could be due to the aberrant lysosomal storage in Pompe disease-model cells, which may affect transport, or the lower density of anti-ICAM on the carrier surface. Yet, lysosomal transport of anti-ICAM/GAA NC was sufficient to enhance glycogen degradation in Pompe-model cells by ~3 folds over non-targeted enzyme.
In vivo biodistribution of anti-ICAM/GAA NCs was also markedly enhanced in all tissues compared to non-targeted GAA (from ~5.5- to 580-fold; Figure 4, B). Main targets for Pompe disease, such as liver, heart and skeletal muscle, received ~23-, 11- and 6-fold increased GAA doses, demonstrating the efficacy of this strategy. This outcome is highly relevant since the liver represents a major storage compartment for glycogen, and the cardiac and skeletal muscles use glycogen as a source of energy.2 This is the first time that ICAM-1 targeting is demonstrated for skeletal muscle (Figures 1, B, and 4, B), which may offer an avenue for drug delivery to this tissue. Enhanced GAA delivery to the brain (~6-fold) may attenuate neuropathology and cerebral vasculopathy observed in some patients.2 Delivery to lungs, kidney, and spleen is also expected to be beneficial since these organs accumulate glycogen in GAA deficiency.2 Hence, ICAM-1 targeting could represent an alternative or complementary strategy for ERT of Pompe disease. Although previous studies indicated no apparent toxicity of anti-ICAM NCs,21 future studies will need to assess the biocompatibility of this approach.
This is the third example where ICAM-1-targeting shows a marked improvement, in cell culture and in vivo, in the context of LSD enzymes. Other examples include acid sphingomyelinase (ASM) deficient in types A-B Niemann-Pick disease40 and α-galactosidase (α-Gal) deficient in Fabry disease.41 The loading efficiency of these enzymes on anti-ICAM NCs was similar: 77 to 86% for anti-ICAM:enzyme 50:50 mass ratio formulations (Table 1). In all cases, this diminishes the density of anti-ICAM on the carrier by ~40–50% (Table 1). Although this results in a ~50% reduction in NC targeting for α-Gal and GAA (in cell cultures), this effect is not observed for anti-ICAM/ASM NCs (Table 1). ASM on the NC surface may provide affinity to the cell surface, through binding to its substrate, sphingomyelin, abundantly present at the plasmalemma.30
In all cases, anti-ICAM/enzyme NC uptake was efficient: ~70–80% of cell bound NCs (Table 1), which occurred by CAM-mediated endocytosis (Figure 1, D, and 22,25). This provided similarly efficient lysosomal delivery of ASM, α-Gal and GAA (65–70%; Table 1), resulting in a substantial reduction of lysosomal storage of sphingomyelin, globotriaosylceramide and glycogen, substrates of these enzymes, respectively (Table 1). While 70–75% substrate reduction was observed for α-Gal and GAA, ASM delivered by anti-ICAM NCs reduced substrate levels to control values, which may be due to experimental differences: an acidic enzyme (ASM) was tested in genetic models24,25 versus neutral α-Gal22 and GAA tested in pharmacological models. In any instance, the enzymes delivered by anti-ICAM NCs improved substrate reduction by 2- to 3-fold compared to non-targeted counterparts (Table 1). These differences may be more acute in vivo, where binding of non-targeted enzymes may be further hindered due to blood flow.
Indeed, non-targeted ASM, α-Gal and GAA accumulated more poorly than their anti-ICAM NC counterparts in all tissues in mice, including the brain and peripheral organs (SI, fold increase in the tissue-to-blood enzyme level when administered on nanocarriers; Table 2). In all cases, although brain accumulation was significantly improved by anti-ICAM NCs (~4–7 fold increase), this organ received the lowest dose compared to the lung and liver, which received the highest dose. First pass effect after intravenous injection and high ICAM-1 expression associated to the lung and liver may account for this pattern.
Table 2.
Biodistribution of ICAM-1-targeted nanocarriers for lysosomal enzyme delivery in mice.
| Blood | Brain
|
Heart
|
Kidney
|
Liver
|
Lung
|
Spleen
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| %ID | %ID | SI | %ID | SI | %ID | SI | %ID | SI | %ID | SI | %ID | SI | |
| Anti-ICAM NC | 4.71 ± 0.94 | 0.19 ± 0.03 | 0.34 ± 0.04 | 1.34 ± 0.07 | 31.90 ± 3.58 | 26.35 ± 3.22 | 4.08 ± 0.48 | ||||||
| Anti-ICAM/ASM NC | 6.94 ± 2.96 | 0.27 ± 0.05 | 7.24 | 0.35 ± 0.02 | 9.25 | 1.37 ± 0.05 | 5.06 | 39.60 ± 2.81 | 14.54 | 21.51 ± 2.73 | 195.18 | 4.08 ± 0.37 | 50.00 |
| Anti-ICAM/αGal NC | 5.59 ± 0.33 | 0.13 ± 0.01 | 3.76 | 0.33 ± 0.04 | 5.06 | 1.89 ± 0.14 | 1.40 | 18.41 ± 1.22 | 21.22 | 36.24 ± 3.47 | 351.44 | 3.11 ± 0.43 | 52.94 |
| Anti-ICAM/GAA NC | 3.32 ± 0.46 | 0.15 ± 0.01 | 5.56 | 0.41 ± 0.03 | 10.97 | 2.08 ± 0.18 | 8.74 | 17.32 ± 2.05 | 23.07 | 48.75 ± 3.25 | 584.15 | 2.91 ± 0.27 | 66.55 |
We observed some differences in tissue distribution of anti-ICAM NCs loaded with ASM, α-Gal or GAA (Table 2). Anti-ICAM/ASM NCs provided the highest enhancement in brain delivery (7-fold), which is relevant since this organ is a more important target for type A Niemann-Pick disease.2,40,41 Furthermore, despite lower density of targeting antibodies (Table 1), anti-ICAM/ASM NCs displayed greater brain accumulation than anti-ICAM NCs, perhaps due to ASM binding to abundant sphingomyelin in this organ. For an unknown reason, anti-ICAM/GAA provided the greatest affinity to the lungs (585 fold over GAA). This formulation also displayed higher accumulation in the heart (11 fold over GAA), which constitutes a very prevalent target for Pompe disease. This formulation also targeted the kidneys, liver and spleen at higher specificity compared to its non-targeted counterpart (9-, 23- and 67-fold over GAA).
Given that ultimately all tissues are affected at some degree by LSDs, and since anti-ICAM NCs enhances enzyme delivery to main targets of intervention in all three cases, ICAM-1-targeting strategy holds considerable promise as a general platform to improve ERT delivery for LSDs.
Supplementary Material
(A) Anti-ICAM/125I-GAA NCs were incubated in either storage buffer (1% BSA-PBS, pH 7.4) at 4°C or in a physiological-like fluid (cell medium supplemented with serum, pH 7.4) at 37°C. Samples were collected at the indicated time points to determine the release of 125I-GAA from carrier particles after separation from 125I-GAA stably bound to the nanocarrie r coat by centrifugation. (B) Release of 125I-GAA from anti-ICAM/125I-GAA NCs at 37°C in a lysosomal-like environment (e.g., serum-supplemented cell medium, pH 4.5, presence of glycogen) was compared to that of similar counterparts incubated at neutral pH and in the absence of the enzyme substrate (fold increase is represented). Data are mean±SEM (n=6). **p≤0.01, ***p≤0.001, by Student’s t-test (GAA release in lysosomal-like environment compared to GAA release in saline).
TNF-α-activated HUVECs were incubated overnight in control media versus media containing 300 μM or 600 μM turanose (to inhibit GAA), then washed and fixed, followed by Periodic acid-Schiff (PAS) staining to label glycogen (upper panels). Red color images were then isolated from full color micrographs to quantify glycogen staining (arrows), shown in arbitrary units (A.U., bottom graph). Scale bar, 10 μm. Data are mean±SEM (n≥47 cells, two experiments). ***p≤0.001, compares turanose-treated cells to control cells by Student’s t-test.
Fluorescence microscopy of TNF-α activated, turanose-treated HUVECs incubated at 37°C with FITC-labeled anti-ICAM/GAA NCs for 30 min, 1 h, or 3 h. Cell nuclei were stained with blue DAPI. Scale bar, 10 μm.
TNF-α activated, turanose-treated HUVECs were incubated at 37°C with FITC-labeled anti-ICAM/GAA NCs for 30 min, 1 h, or 3 h. After cell fixation, surface-bound carriers were stained with a Texas-Red labeled secondary antibody as in Figure 1B, yielding yellow-colored particles (arrows), versus internalized counterparts that appear green (arrowheads). Cell nuclei were stained with blue DAPI. Scale bar, 10 μm.
Internalization of FITC-labeled anti-ICAM/GAA NCs by TNF-α activated, turanose-treated HUVECs was tested (as described in Figure 1B) after 1 h incubation at 37°C in either control cell medium (Ctr) or medium containing amiloride (Amil, to inhibit CAM-mediated endocytosis) or monodansyl cadaverine (MDC, to inhibit clathrin-mediated endocytosis). Arrows mark surface-bound yellow nanocarriers. Arrowheads mark internalized green nanocarriers. Cell nuclei were stained with blue DAPI. Scale bar, 10 μm.
Anesthetized mice were intravenously injected with either anti-ICAM/125I-GAA NCs or a similar dose of non-targeted 125I-GAA. Blood samples were taken from retro-orbital sinus 2 min, 15 min, and 30 min after injection, from which percent of injected dose (%ID) was calculated. Data are mean±SEM (n≥4 mice). ***p≤0.001, compares ICAM-1-targeted GAA to non-targeted GAA by Student’s t-test.
Acknowledgments
This work was supported by the NSF Research Experience for Undergraduates program of the Bioengineering Department in the University of Maryland (LN), Nanobiotechnology Program of the Maryland Department of Business and Economic Development, Minta Martin Foundation, AHA 09BGIA2450014, and R01HL098416 (SM).
The authors thank Edward Lim and Dave Dolak (Malvern Instruments Inc., Worcestershire, UK) for providing DLS size measurement of anti-ICAM/GAA NCs.
Footnotes
The authors do not have any competing interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–65. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 2.Hirschhorn R, Reuser AJJ. Glycogen storage Disease Type II: Acid a-Glucosidase (Acid Maltase) Deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8. Vol. 3. New York: McGraw-Hill; 2001. pp. 3389–420. [Google Scholar]
- 3.Malicdan MC, Noguchi S, Nonaka I, Saftig P, Nishino I. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromuscul Disord. 2008;18:521–9. doi: 10.1016/j.nmd.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Shea L, Raben N. Autophagy in skeletal muscle: implications for Pompe disease. Int J Clin Pharmacol Ther. 2009;47 (Suppl 1):S42–7. doi: 10.5414/cpp47042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrow TA, Hopkin RJ, Leslie ND, Tinkle BT, Grabowski GA. Enzyme reconstitution/replacement therapy for lysosomal storage diseases. Curr Opin Pediatr. 2007;19:628–35. doi: 10.1097/MOP.0b013e3282f161f2. [DOI] [PubMed] [Google Scholar]
- 6.Martiniuk F, Chen A, Donnabella V, Arvanitopoulos E, Slonim AE, Raben N, et al. Correction of glycogen storage disease type II by enzyme replacement with a recombinant human acid maltase produced by over-expression in a CHO-DHFR(neg) cell line. Biochem Biophys Res Commun. 2000;276:917–23. doi: 10.1006/bbrc.2000.3555. [DOI] [PubMed] [Google Scholar]
- 7.Merk T, Wibmer T, Schumann C, Kruger S. Glycogen storage disease type II (Pompe disease)--influence of enzyme replacement therapy in adults. Eur J Neurol. 2009;16:274–7. doi: 10.1111/j.1468-1331.2008.02377.x. [DOI] [PubMed] [Google Scholar]
- 8.Schoser B, Hill V, Raben N. Therapeutic approaches in glycogen storage disease type II/Pompe Disease. Neurotherapeutics. 2008;5:569–78. doi: 10.1016/j.nurt.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth Defects Orig Artic Ser. 1980;16:77–84. [PubMed] [Google Scholar]
- 10.Sly WS, Kaplan A, Achord DT, Brot FE, Bell CE. Receptor-mediated uptake of lysosomal enzymes. Prog Clin Biol Res. 1978;23:547–51. [PubMed] [Google Scholar]
- 11.Fukuda T, Roberts A, Plotz PH, Raben N. Acid alpha-glucosidase deficiency (Pompe disease) Curr Neurol Neurosci Rep. 2007;7:71–7. doi: 10.1007/s11910-007-0024-4. [DOI] [PubMed] [Google Scholar]
- 12.Cardone M, Porto C, Tarallo A, Vicinanza M, Rossi B, Polishchuk E, et al. Abnormal mannose-6-phosphate receptor trafficking impairs recombinant alpha-glucosidase uptake in Pompe disease fibroblasts. Pathogenetics. 2008;1:6. doi: 10.1186/1755-8417-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Vries JM, van der Beek NA, Kroos MA, Ozkan L, van Doorn PA, Richards SM, et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genet Metab. 2010;101:338–45. doi: 10.1016/j.ymgme.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–8. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 15.Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99:26–33. doi: 10.1016/j.ymgme.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koeberl DD, Luo X, Sun B, McVie-Wylie A, Dai J, Li S, et al. Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol Genet Metab. 2011 doi: 10.1016/j.ymgme.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun B, Li S, Bird A, Yi H, Kemper A, Thurberg BL, et al. Antibody formation and mannose-6-phosphate receptor expression impact the efficacy of muscle-specific transgene expression in murine Pompe disease. J Gene Med. 2010;12:881–91. doi: 10.1002/jgm.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joseph A, Munroe K, Housman M, Garman R, Richards S. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clin Exp Immunol. 2008;152:138–46. doi: 10.1111/j.1365-2249.2008.03602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–9. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun B, Kulis MD, Young SP, Hobeika AC, Li S, Bird A, et al. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther. 2010;18:353–60. doi: 10.1038/mt.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garnacho C, Dhami R, Simone E, Dziubla T, Leferovich J, Schuchman EH, et al. Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J Pharmacol Exp Ther. 2008;325:400–8. doi: 10.1124/jpet.107.133298. [DOI] [PubMed] [Google Scholar]
- 22.Hsu J, Serrano D, Bhowmick T, Kumar K, Shen Y, Kuo YC, et al. Enhanced endothelial delivery and biochemical effects of alpha-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release. 2011;149:323–31. doi: 10.1016/j.jconrel.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muro S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:189–204. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, et al. Control of endothelial targeting and intracellular delivery of therapeutic enzymes by modulating the size and shape of ICAM-1-targeted carriers. Mol Ther. 2008;16:1450–8. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muro S, Schuchman EH, Muzykantov VR. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther. 2006;13:135–41. doi: 10.1016/j.ymthe.2005.07.687. [DOI] [PubMed] [Google Scholar]
- 26.Marlin SD, Springer TA. Purified intercellular adhesion molecule-1 (ICAM-1) is a ligand for lymphocyte function-associated antigen 1 (LFA-1) Cell. 1987;51:813–9. doi: 10.1016/0092-8674(87)90104-8. [DOI] [PubMed] [Google Scholar]
- 27.Muro S. Intercellular Adhesion Molecule-I and Vascular Cell Adhesion Molecule-I. In: Aird WC, editor. Endothelial Biomedicine. Cambridge University Press; 2007. pp. 1058–70. [Google Scholar]
- 28.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, et al. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci. 2003;116:1599–609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 29.Muro S, Cui X, Gajewski C, Murciano JC, Muzykantov VR, Koval M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am J Physiol Cell Physiol. 2003;285:C1339–47. doi: 10.1152/ajpcell.00099.2003. [DOI] [PubMed] [Google Scholar]
- 30.Pagano RE, Puri V, Dominguez M, Marks DL. Membrane traffic in sphingolipid storage diseases. Traffic. 2000;1:807–15. doi: 10.1034/j.1600-0854.2000.011101.x. [DOI] [PubMed] [Google Scholar]
- 31.Parenti G, Andria G. Pompe Disease: from New Views on Pathophysiology to Innovative Therapeutic Strategies. Curr Pharm Biotechnol. 2011 doi: 10.2174/138920111795542606. [DOI] [PubMed] [Google Scholar]
- 32.Jevnikar AM, Wuthrich RP, Takei F, Xu HW, Brennan DC, Glimcher LH, et al. Differing regulation and function of ICAM-1 and class II antigens on renal tubular cells. Kidney Int. 1990;38:417–25. doi: 10.1038/ki.1990.221. [DOI] [PubMed] [Google Scholar]
- 33.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, et al. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J Pharmacol Exp Ther. 2006;317:1161–9. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 34.Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D, L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125:193–209. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–47. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 36.Broadhead DM, Butterworth J. alpha-Glucosidase in Pompe’s disease. J Inherit Metab Dis. 1978;1:153–4. doi: 10.1007/BF01805584. [DOI] [PubMed] [Google Scholar]
- 37.Hagemans ML, Stigter RL, van Capelle CI, van der Beek NA, Winkel LP, van Vliet L, et al. PAS-positive lymphocyte vacuoles can be used as diagnostic screening test for Pompe disease. J Inherit Metab Dis. 2010;33:133–9. doi: 10.1007/s10545-009-9027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bach G, Chen CS, Pagano RE. Elevated lysosomal pH in Mucolipidosis type IV cells. Clin Chim Acta. 1999;280:173–9. doi: 10.1016/s0009-8981(98)00183-1. [DOI] [PubMed] [Google Scholar]
- 39.Calderon AJ, Bhowmick T, Leferovich J, Burman B, Pichette B, Muzykantov V, et al. Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J Control Release. 2011;150:37–44. doi: 10.1016/j.jconrel.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuchman EH, Desnick RJ. Niemann-Pick Disease Types A and B: Acid Sphingomyelinase Deficiencies. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8. Vol. 3. New York: McGraw-Hill; 2001. pp. 3589–610. [Google Scholar]
- 41.Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A Deficiency: Fabry Disease. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8. Vol. 3. New York: McGraw-Hill; 2001. pp. 3733–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Anti-ICAM/125I-GAA NCs were incubated in either storage buffer (1% BSA-PBS, pH 7.4) at 4°C or in a physiological-like fluid (cell medium supplemented with serum, pH 7.4) at 37°C. Samples were collected at the indicated time points to determine the release of 125I-GAA from carrier particles after separation from 125I-GAA stably bound to the nanocarrie r coat by centrifugation. (B) Release of 125I-GAA from anti-ICAM/125I-GAA NCs at 37°C in a lysosomal-like environment (e.g., serum-supplemented cell medium, pH 4.5, presence of glycogen) was compared to that of similar counterparts incubated at neutral pH and in the absence of the enzyme substrate (fold increase is represented). Data are mean±SEM (n=6). **p≤0.01, ***p≤0.001, by Student’s t-test (GAA release in lysosomal-like environment compared to GAA release in saline).
TNF-α-activated HUVECs were incubated overnight in control media versus media containing 300 μM or 600 μM turanose (to inhibit GAA), then washed and fixed, followed by Periodic acid-Schiff (PAS) staining to label glycogen (upper panels). Red color images were then isolated from full color micrographs to quantify glycogen staining (arrows), shown in arbitrary units (A.U., bottom graph). Scale bar, 10 μm. Data are mean±SEM (n≥47 cells, two experiments). ***p≤0.001, compares turanose-treated cells to control cells by Student’s t-test.
Fluorescence microscopy of TNF-α activated, turanose-treated HUVECs incubated at 37°C with FITC-labeled anti-ICAM/GAA NCs for 30 min, 1 h, or 3 h. Cell nuclei were stained with blue DAPI. Scale bar, 10 μm.
TNF-α activated, turanose-treated HUVECs were incubated at 37°C with FITC-labeled anti-ICAM/GAA NCs for 30 min, 1 h, or 3 h. After cell fixation, surface-bound carriers were stained with a Texas-Red labeled secondary antibody as in Figure 1B, yielding yellow-colored particles (arrows), versus internalized counterparts that appear green (arrowheads). Cell nuclei were stained with blue DAPI. Scale bar, 10 μm.
Internalization of FITC-labeled anti-ICAM/GAA NCs by TNF-α activated, turanose-treated HUVECs was tested (as described in Figure 1B) after 1 h incubation at 37°C in either control cell medium (Ctr) or medium containing amiloride (Amil, to inhibit CAM-mediated endocytosis) or monodansyl cadaverine (MDC, to inhibit clathrin-mediated endocytosis). Arrows mark surface-bound yellow nanocarriers. Arrowheads mark internalized green nanocarriers. Cell nuclei were stained with blue DAPI. Scale bar, 10 μm.
Anesthetized mice were intravenously injected with either anti-ICAM/125I-GAA NCs or a similar dose of non-targeted 125I-GAA. Blood samples were taken from retro-orbital sinus 2 min, 15 min, and 30 min after injection, from which percent of injected dose (%ID) was calculated. Data are mean±SEM (n≥4 mice). ***p≤0.001, compares ICAM-1-targeted GAA to non-targeted GAA by Student’s t-test.