

Abstract
Objective and design
To determine whether Finegoldia magna protein L (PL) causes lung inflammation and, if so, whether the response is dependent on its Ig-binding B cell superantigenic property.
Material
Pulmonary inflammatory reactions were analyzed at varying time points after intratracheal administration of PL to various strains of mice.
Results
PL caused peribronchial and perivascular inflammation that peaked at 18–24 hours. Polymorphonuclear cells (PMNs) began to accumulate in bronchoalveolar lavage fluid (BALF) of PL-challenged mice by 4 hours and accounted for >90% of leukocytes by 18–24 hours. Inflammation was marked by the appearance of MIP-2, KC, TNF-α, and IL-6 in the BALF with peak levels attained 4 hours after PL administration. PL-induced pulmonary inflammation was associated with increased airway hyperreactivity following inhalation of methacholine. The inflammatory reaction was unabated in mice lacking B cells and Igs. By contrast, PL-induced inflammation was abrogated in MyD88- deficient mice. PL-induced responses required alveolar macrophages.
Conclusions
These results strongly suggest that PL-induced lung inflammation is dependent on an innate MyD88 dependent pathway rather than the Ig-binding properties of this microbial B cell superantigen. We propose that this pulmonary inflammatory reaction is caused by the interaction of PL with a Toll-like receptor expressed on alveolar macrophages.
Introduction
B cell superantigens (SAgs) are defined by their ability to: 1) stimulate a high frequency of B cells, 2) target B cells that express a particular family of VH or VL-family gene products, and 3) bind to framework domains of VH or VL expressing immunoglobulins (Igs) outside their complementarity determining regions (CDRs) [reviewed in 1, 2]. A B cell SAg can react with potentially large amounts of soluble antigen receptor molecules, i.e., immunoglobulins, in the serum, even if the host has not previously encountered the B cell SAg. Binding to a large amount of Ig molecules endows these unconventional antigens with an array of potentially dangerous biologic properties.
We previously documented that the interaction of Staphylococcus aureus protein A (SpA), the first B cell SAg to be defined, with human IgM molecules in vitro leads to activation of the classical complement cascade [3]. We subsequently reported that the interaction of SpA with human IgG in a rabbit cutaneous Arthus model leads to tissue injury characterized by features of immune complex-mediated inflammation [4]. Our studies suggested that these outcomes were imparted by the superantigenic VH3 Ig binding rather than the Fcγ binding property of this microbial protein. Given the novelty and the potential clinical relevance of B cell SAg-induced immune complex injury, we sought to elucidate cellular and molecular events initiated by the deposition of these unconventional immune complexes. Using an adaptation of a mouse peritonitis model, we identified a number of factors that contribute to this B cell SAg/IgG complex driven inflammatory process. These include mast cells, complement components, FcγRIII, TNF-α and the chemokines MIP-2 and KC [5].
In the current studies, we sought to determine the possible consequences of B cell SAg-elicited inflammation in a tissue compartment that is more relevant to human diseases, namely the lungs. The lower respiratory tree represents a compartment to which B cell SAgs might gain easy access. We used protein L (PL) as the B cell SAg rather than SpA. This product of Finegoldia magna (previously named Peptostreptococcus magnus), is a 76 to 106-kDa protein containing four or five highly homologous, consecutive extracellular Ig-binding domains [6]. It binds to the variable region framework domains [7] of human Ig κ-chains belonging to the VkI, VkIII, and VkIV-gene families regardless of the H-chain isotypes of the Igs. Since 60% of human Igs have κ-type L chains and the VκI, III, and IV families account for the majority of them, PL has the potential to interact with a significant proportion of human Igs. PL also binds avidly to mouse Ig [8]. Greater than 40% of murine peripheral IgVκ+ B cells bind this microbial protein [9, Lars Bjorck, Lund, Sweden, personal communication). Unlike SpA, PL possesses no Fcγ binding activity. Therefore, we presumed it would allow us to directly assess whether elicited pathology was caused solely by the variable region framework binding activity of this B cell SAg.
In the studies reported herein, we observed that PL does indeed cause pulmonary inflammation with many histopathological features of immune complex mediated tissue injury. However, to our great surprise, we discovered that inflammation elicited by PL was not dependent on B cells or immunoglobulin. Rather the observed inflammatory reaction was dependent on an innate MyD88-dependent pathway, which we believe was likely triggered by the interaction of PL with a Toll-like receptor (TLR) expressed on alveolar macrophages.
Materials and Methods
Animals
Female C57BL/6, C3H/HeJ (TLR-4 deficient), C3H/Ouj (C3H/HeJ controls), and JHT (B cell and immunoglobulin deficient mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Myeloid differentiating factor deficient (MyD88−/−) mice were provided by S. Akira (Osaka University, Osaka, Japan). All mouse strains were used at 8–10 weeks of age. The mice were maintained under specific-pathogen free conditions at the University of Pennsylvania. The experimental protocols were approved by the University of Pennsylvania Animal Care and Use Committee (IACUC).
PL-induced lung inflammation
Mice were anesthetized with ketamine and xylazine, the trachea was exposed, and 200 μg of PL (Actigen, Oslo, Norway) was administered via intratracheal (i.t.) injection; control animals received BSA (Fraction V, Fatty Acid-Poor, Endotoxin Free) (Calbiochem, La Jolla, CA). The recombinant PL preparation was assayed by LAL test (Associates of Cape Cod, East Falmouth MA) and found to contain <0.7 ng/mg endotoxin. Mice were sacrificed at various time points (1–48 hours) after PL treatment and bronchoalveolar fluid (BALF) was analyzed for Polymorphonuclear cell (PMN) accumulation, and production of MIP-2, KC, TNF-α, IL-6 and IL-1β. Lung tissues obtained after lavage were processed for histological examination and stained with hematoxylin and eosin according to conventional procedures.
BALF collection and quantitation of PMN accumulation
Lungs were lavaged with 1 ml PBS two times at 4° after canulation of the trachea. The volume of the collected fluid was measured in each sample and the total cell count was assessed in a hemacytometer. For quantification of PMN accumulation, differential cell counts were performed on cytospins (3 min at 33 × g) and stained with HEMA 3 Solution (Fisher Scientific, Pittsburgh, PA).
ELISA determination of inflammatory cytokines/chemokines
Immunoreactive MIP-2, KC, IL6, IL-1β and TNF-α concentrations in BALFs were quantified by the proinflammatory molecule Searchlight Multiplex assay according to the manufacturer's protocol (Thermo Fisher Scientific, Needham MA) or by ELISA kits (R & D Systems, Minneapolis, MN), according to the manufacturer's instructions.
Measurements of airway hyperresponsiveness to methacholine (MCh)
In order to assess the function of the lower airways, mice were anesthetized and their tracheas were canulated and connected to a ventilator. The animals then were injected with intramuscular pancuronium bromide (3.0 mg/kg). The mice were mechanically ventilated (Harvard Apparatus Model 687) at a respiratory rate of 140 beats/min with 0.25 ml of tidal volume. Lung resistance was calculated from the measurements that were recorded by a computerized online system (Flexivent). Airway responsiveness was assessed following inhalation of nebulized saline and then increasing concentrations of MCh (3, 6, 12.5, 25 mg/ml). Groups were compared by calculating the Area Under Curve and using the Mann Whitney U test (GraphPad Prism 5.01).
Depletion of Alveolar Macrophages (AMs)
In AM depletion experiments, mice were pretreated intratracheally with liposome-encapsulated dichloromethylene diphosphonate (Lipo-clod) as previously described [10, 11]. Initial studies indicated a single dose of 100μl was sufficient to deplete macrophages by 70–80% within 24 hours. Intratracheal delivery of PBS-liposomes (lipo-PBS) did not show depletion effects and served as the control. Mice were treated with Lipo-clod or Lipo-PBS 24 hours prior to administration of PL.
Results
Kinetics of PL-mediated lung inflammation
The lung inflammatory response was determined by analyzing the kinetics of appearance of inflammatory cells in the lung tissue and BALF. Histological analysis of lungs obtained 1–48 hours following i.t. administration of C57BL/6 mice with PL revealed an inflammatory response characterized by a peribronchial and perivascular infiltrate of inflammatory cells (Figure 1). The inflammatory cell influx developed within the first 4 hours after PL instillation at which time PMNs represented the majority of the infiltrating cells. The reaction was most prominent from 18 to 24 hours with a subsequent decline by 48 hours, as assessed by hematoxylin/eosin staining of the lung tissue. The lungs of mice that received BSA i.t. did not show evidence of inflammation.
Figure 1. PL-induced lung histopathology in C57BL/6 mice.

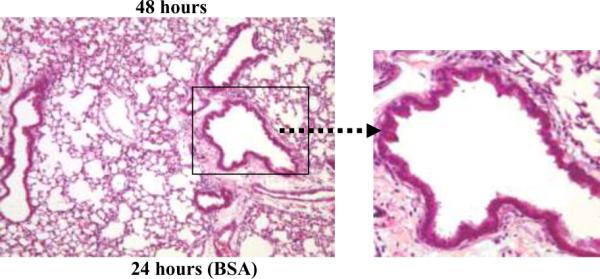
Representative hematoxalin and eosin-stained sections of lavaged and paraffin-embedded lung tissue from C57BL/6 mice at 1–48 hours post PL challenge (n>5 at each time point) and 24 hours post BSA challenge (bottom left). PL induced peribronchial and perivascular inflammation. Original magnification, × 100; insets magnification × 400. The peak inflammatory response occurred 18–24 hours after i.t. administration of PL. Solid arrows indicate peribronchial inflammation.
Inflammatory cells appearing in the BALF of C57BL/6 mice treated with PL were markedly enriched for PMNs. Accumulation of PMNs was not apparent until 4 h with numbers peaking between 18–24 hours (Figure 2). PMNs consistently accounted for >90% of the leukocytes recovered from the BALF during this time period. PMN recruitment was not observed in the BALF of mice treated with BSA.
Figure 2. Kinetics of PMN infiltration in BALF.
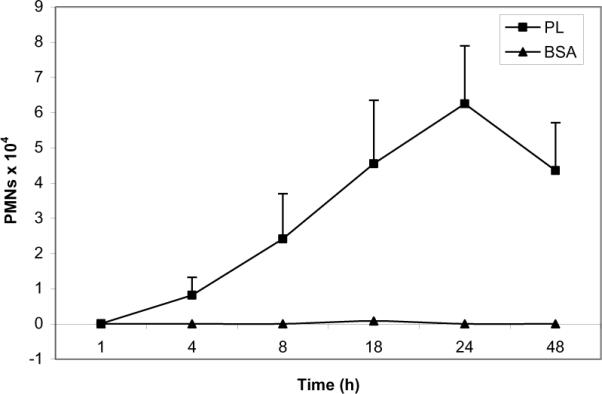
C57BL/6 mice (n>5) were administered PL i.t. At varying time points thereafter, animals were sacrificed and their lungs were lavaged. The total number of PMNs was determined and expressed as means ± SD. PMNs began to appear in the BALF of PL-treated animals at 4 hours, peaking at 18–24 hrs.
The results indicate that PL elicits an inflammatory response in the lungs characterized by a peribronchial and perivascular inflammatory cell infiltrate along with accumulation of PMNS in the BALF. The latter observation indicates that the inflammation elicited by PL induced a rapid chemotaxis of PMNs which are typically not present in this pulmonary compartment.
Kinetics of Cytokine/CXC Chemokine Production
To further dissect mechanisms responsible for the PL-induced inflammatory response, we analyzed the recovered BALF for TNF-α, MIP-2, KC, IL6 and IL1β (Figure 3). Local production of these factors has previously been associated with other causes of neutrophil-enriched inflammatory reactions in the lung [12]. MIP-2 and KC, two functionally related CXC chemokines that are homologues of human IL-8, accounted for the highest concentrations of analytes with peaks observed at 4 hours post challenge, falloff by eight hours and return to baseline by 24 hours. Peak levels of TNF-α and IL-6 were observed between 4–8 hours post challenge. IL-1β was not detected. BSA treated control mice showed <10pg/ml for all analytes tested (data not shown). Taken together, these data indicate that PL-elicited lung inflammation results in enhanced production of TNF-α, MIP-2, KC, and IL-6 but not IL-1β.
Figure 3. BALF Cytokine/Chemokine Assay.
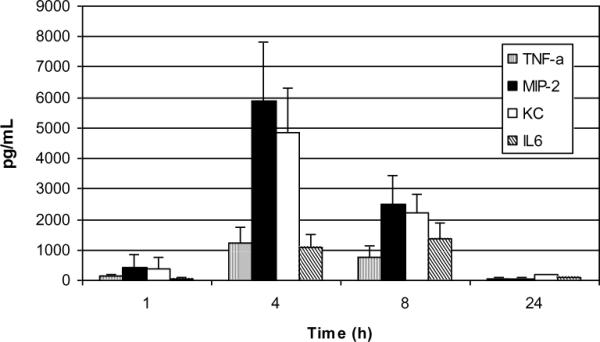
Concentrations of MIP-2, KC, TNF, IL-6 and IL-1β were analyzed in the BALF of PL-challenged C57BL/6 mice (n>4 per group) and expressed as means ± SD. Peak levels of analytes were apparent at 4–8 hours with obvious falloff at 24 hours. MIP-2 and KC levels were significantly higher at 4 hours vs. 8 hours P= 0.010 and 0.007 (respectively); Student's t-test. Levels of each analyte were < 10 pg/ml in BSA treated animals.
PL-induced airway hyperreactivity
Given the elicitation of peribronchial inflammation by PL, we sought to determine if this pathologic picture is accompanied by functional changes. Airway hyperresponsiveness was assessed in a group of mice at the height of the inflammatory response, 24 hours after intratracheal treatment, and prior to performing the bronchoalveolar lavage. We measured airway resistance to inhaled MCh in PL treated and control animals using the Flexivent system as previously described in our laboratory [13]. Figure 4 shows that PL-treated mice (n=6) had an enhanced airway resistance to inhaled MCh (3–25.0 mg/ml) at 24 hours compared to BSA controls (n=4; p<0.05, Area Under Curve, compared with the Mann-Whitney U test).
Figure 4. PL-induced airway hyperreactivity.

Lung function measurements were performed on a subset of PL-treated (n=6) and BSA-treated mice (n=4) 24 hours after treatment. Lung resistance was assessed using the Flexivent system in anesthetized, canulated and ventilated mice following inhalation of increasing doses of MCh as indicated. Data are expressed as the percent change over baseline. The baseline values for the PL-treated and BSA-treated groups were 0.97±0.08 and 0.96±0.09 cmH2O.s/min. The Area Under Curve values (expressed as means ± SD) were significantly different between the PL and BSA-treated groups (p<0.05; Mann-Whitney U test).
Lack of role for B cells and Ig in the PL –induced Inflammatory Reaction
In order to determine whether the PL-mediated inflammatory response is due to the B cell superantigenic Ig-binding property of PL, we administered it to JHT mice (kindly provided by Dr. Robert Eisenberg, University of Pennsylvania). These mice lack peripheral B cells and serum Igs due to a targeted lesion in the Jh locus [14]). Surprisingly, we observed no diminution in the intensity of the inflammatory response as gauged by histopathology (Figure 5a and 5b). These results excluded Ig-binding as a mechanism accounting for PL-induced inflammation.
Figure 5. PL-induced lung response in JHT and MyD88−/− mice.


Eighteen hours following i.t. administration of PL to JHT (a) and MyD88−/− (c) mice and their respective wild-type controls, lungs were examined for histopathology. Shown are representative (n=3) hematoxalin and eosin stained lung sections (100× magnification) revealing the presence of an intact pulmonary inflammatory reaction in JHT mice (a) compared to similarly stained sections from PL-challenged wild-type mice (b). By contrast, histopathological abnormalities are markedly attenuated in MyD88−/− (c) vs (d) wild-type control mice. Solid arrows indicate peribronchial inflammation and stippled arrow indicates perivascular inflammation..
Dependence of PL-induced inflammatory reaction on MyD88
The above findings indicated that the PL-induced inflammatory lung reaction was not dependent upon the Ig binding activity of PL. We therefore focused on two innate immune mechanisms that would not require binding of PL to Igs and presensitization of the recipient mice. Initial studies conducted in C3 deficient mice (kindly provided by Dr. John Lambris, University of Pennsylvania) excluded the involvement of complement pathway activation, since lung histology was not altered when PL was administered to the mice i.t. (data not shown). Since many microbial proteins possess pathogen associated molecular patterns that stimulate TLRs [15], we addressed the involvement of these innate immunity receptors by conducting studies in MyD88−/− knockout mice. MyD88 is an adaptor protein necessary for signaling of most TLRs [15] as well as IL-1β, IL-18 [16, 17] and IL-33 receptors [17, 18]. MyD88 knockout mice demonstrated an absence of the histopathological abnormalities seen in wild-type controls (Figure 5c and 5d, representative of three mice), a marked reduction in BALF PMNS (Figure 6a; n=5; p<0.005, Students t-test) vs wild-type mice and markedly reduced cytokines/chemokines in BALF vs. wild-type mice (Figure 6b; n=5; p<0.001, Student's t-test).at 18, 18, and 4 hours after i.t. administration of PL.
Figure 6. PL-induced responses in MyD88 −/− mice.
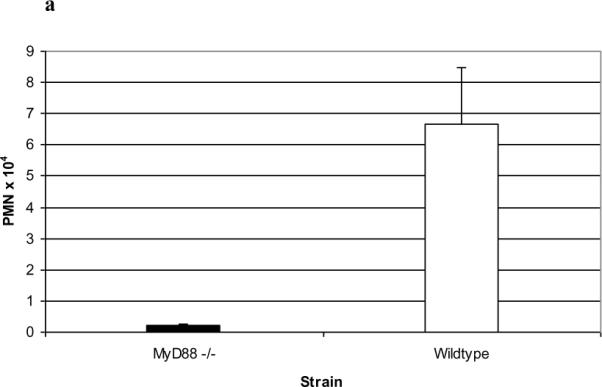
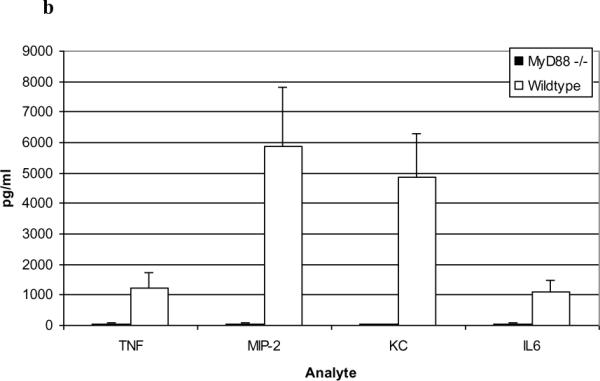
PL-treated MyD88−/− mice demonstrated a marked reduction in BALF PMN numbers vs. wild-type mice (a; n=3; P<0.005, Students t-test) and markedly reduced cytokines/chemokines in BALF vs. wild-type mice in the BALF at 4 hours (b; n=5; p<0.001, Student's t-test). Data are expressed as means ± SD.
To begin to address the possibility that MyD88 was recruited following the interaction of PL with a TLR, we repeated our studies in TLR-4 deficient C3H/HeJ mice and C3H/OUj control mice. The PL-induced histopathological and BALF PMN responses were intact in the C3H/HeJ mice (Figure 7a, b).
Figure 7. PL-induced histopathology in C3H/Hej (TLR-4 deficient) mice).

Representative hematoxalin and eosin-stained sections of lavaged and paraffin-embedded lung tissue from TLR-4 deficient C3H/Hej mice (a) and their respective wild-type controls (b) prepared 18 hours after PL challenge. Original magnification × 100. Arrows indicate peribronchial inflammation.
Cellular basis of PL-induced lung inflammation: interrogation of AMs
Having obtained data suggesting TLRs mediate PL-induced-inflammation in the lungs, we turned our attention to a resident effector cell population that expresses these innate immune receptors, namely AMs. The AM is known to promote the migration of neutrophils into this compartment by producing a variety of mediators, including TNF-α, MIP-2, and KC [12]. Therefore, we investigated the involvement of AMs in the PL-induced inflammatory lung reaction in mice depleted of these cells by treatment with Lipo-clod [10,11] prior to i.t. challenge with PL. Lipo-clod treatment resulted in approximately 80% AM depletion within 24 hours compared to control mice receiving Lipo-PBS (data not shown). PL-induced PMN influx was significantly lower in Lipo-clod treated mice than in Lipo-PBS control animals (Figure 8a; n=3; P<0.001 Student's t test) and the histopathological alterations at 24 hours were much less apparent (Figure 8b, representative of 3 experiments).
Figures 8. Role of alveolar macrophages in PL-induced inflammatory reactions.

C57BL/6 mice were treated i.t. with Cl2MDP-encapsulated liposomes (Lipo-clod) or Lipo-PBS as a control; PL was administered i.t. 24 hours later and BALF PMN numbers (a) and histopathological reactions (b) were then assessed 24 hours thereafter as described in the standard protocol. AM depletion by Lipo-clod decreased PL-induced BALF PMN numbers expressed as means ± SD (n=3 Lipo-PBS/PL and 4 Lipo-clod/PL-treated mice; P<0.001, Student's t-test) and histopathological reactions (representative experiment).
Discussion
These studies were originally intended to extend our previous studies of B cell SAg mediated immune complex inflammation [3–5]. We turned our attention to PL because we believed it to be a “cleaner” B cell SAg than SpA since it possessed V-region binding activity outside the CDRs but lacked the Fcγ binding activity of SpA. Moreover, the B cell superantigenic Ig-binding properties of PL are thought to be an important virulence factor of the F. magna strains that produce it [19, 20].
The results of the studies reported herein indicate that the PL cell wall component of F. magna induces an inflammatory reaction characterized by the rapid accumulation of PMNs in the BALF and a peribronchial and perivascular infiltrate of inflammatory cells peaking between 18 and 24 hours. The cellular changes were associated with the appearance of elevated levels of MIP-2, KC, TNF-α, and IL6 in BALF, peaking at 4–8 hours, but not IL-1β. The temporal pattern of appearance of these mediators suggests that one or more may contribute to the PMN infiltration. Although our studies do not allow for a conclusion to be drawn about which of these mediators is responsible for the attraction of PMNs, the CXC chemokines MIP-2 and KC are likely to be involved given their robust chemoattractant properties. Of note, the same profile of BALF chemokines and cytokines we observed in the PL-induced reaction was observed in models of lung inflammation caused by the deposition of conventional immune complexes (12) Thus, our findings were consistent with the hypothesis that PL-induced inflammation was elicited by PL/IgG containing immune complexes. Such a mechanism would not be expected to require sensitization of the mice to PL, since reactive Ig Vk-binding molecules would be present in the endogenous murine Ig repertoire [8, 9]. Indeed, the kinetics of the PL-induced pulmonary changes in the naïve mice were consistent with this supposition. Our findings are physiologically relevant since they were associated with a significant airway hyperresponsiveness to MCh 24 hours after PL exposure of the mice. Further, the kinetics of the PL-induced airway changes in the naïve mice strongly suggest that PL activated the innate, rather than the adaptive immune response in the lung.
Our results are highlighted by the finding that the Ig-binding property of PL is not responsible for its proinflammatory action in the lung. This unexpected result was revealed in studies of the JHT mice, which lack both B cells and Ig molecules [13]. We therefore shifted our focus to potential innate immune mechanisms that would not require binding of PL to Igs and presensitization of the recipient mice. The preservation of lung inflammation in C3 deficient mice ruled out the recruitment of two potent complement pathway activation by-products, C3a and C5a. However, the abrogation of all components of the inflammatory responses in MyD88 knockout mice suggested a requirement for one or more TLRs, IL-1β, IL-18 or IL-33 since all signal through a MyD88-dependent pathway. We think that binding of PL to a TLR is the most likely explanation for the elicitation of the observed inflammatory reactions. It is unlikely that IL-1β is an important contributor to the pulmonary inflammation since it was not detected in the BALF of PL-challenged animals. Although we did not measure IL-18 or IL-33 levels in BALF, we think these IL-1 family cytokines are not key players since they tend to elicit TH2-cytokines and typically are not associated with PMN-rich inflammatory responses when linked to either innate or adaptive immune pathway-induced airway hyperreactivity [17, 18, 21–23].
To date, thirteen types of TLRs have been described in mammals and all but TLR-3 have been associated with MyD88 signaling [reviewed in 15]. We can exclude TLR-4 as a PL target. since the pulmonary reactions were unaltered in the TLR-4 defective C3H/HeJ mouse strain. In addition, the latter observation implies that the PL-induced effects cannot be caused by the negligible amount of endotoxin in the recombinant PL preparation under study since endotoxin signals via TLR-4. Studies are currently underway in mouse strains deficient in other TLRs to indentify the putative PL-binding receptor.
Having obtained data strongly suggesting that TLR mediates PL-induced-inflammation in the lungs, we turned our attention to a resident effector cell population that expresses these innate immune receptors, namely AMs. The PL-induced reactions were markedly diminished in AM-depleted mice, suggesting an important role for AMs in their pathogenesis. However, the responses were not completely abrogated. This finding suggests that the macrophages that escaped depletion or perhaps another cell type that expresses TLRs, e.g., epithelial cells, dendritic cells and/or mast cells, accounted for the residual response.
The interaction of PL with another component of the innate immune system has recently been reported [24]. PL was demonstrated to bind to the antibacterial proteins S100A8/A9 via a site not involved with Ig binding. It was suggested that this interaction protected PL-secreting strains from the bactericidal actions of these antibacterial proteins and thereby promoted inflammation.
In summary, our results reveal a novel pro-inflammatory mechanism initiated by a microbial protein with known B cell superantigenic properties. Whereas the virulence of F. magna has been formerly associated with the superantigenic Ig binding property of PL, our findings indicate that this property does not account for PL-induced pulmonary inflammation. Rather, the experimental results strongly suggest pulmonary inflammation is mediated by a MyD88-dependent activity of this microbial protein, which we believe is most likely initiated by its binding to a TLR expressed by pulmonary macrophages. Accordingly, our results add to the growing list of mechanisms by which this bacterial protein may exploit components of the adaptive and innate immune system to elicit inflammation.
Acknowledgements
This work was supported by National Institutes of Health Grants AI065582 (A.I.L.) and P30ES013508; R01AI072197; 1RC1ES018505 (A.H.).
References
- 1.Levinson AI, Kozlowski L, Zheng Y, Wheatley L. B-cell superantigens: definition and potential impact on the immune response. J Clin Immunol. 1995;15:26S–36S. doi: 10.1007/BF01540891. [DOI] [PubMed] [Google Scholar]
- 2.Silverman GJ. B-cell superantigens. Immunol Today. 1997;18:379–386. doi: 10.1016/s0167-5699(97)01101-8. [DOI] [PubMed] [Google Scholar]
- 3.Kozlowski LM, Silverman G, Lambris JD, Levinson AI. Complement activation by a B cell superantigen. J Immunol. 1996;157:1200–12006. 1996. [PubMed] [Google Scholar]
- 4.Kozlowski LM, Li W, Goldschmidt M, Levinson AI. In vivo inflammatory response to a prototypic B cell superantigen: elicitation of an Arthus reaction by staphylococcal protein A. J Immunol. 1998;160:5246–5252. 1998. [PubMed] [Google Scholar]
- 5.Anderson AL, Sporici R, Lambris J, Larosa D, Levinson AI. Pathogenesis of B-cell superantigen-induced immune complex-mediated inflammation. Infect Iimmun. 2006;74:1196–1203. doi: 10.1128/IAI.74.2.1196-1203.2006. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kastern W, Sjobring U, Bjorck L. Structure of peptostreptococcal protein L and identification of a repeated immunoglobulin light chain-binding domain. J Biol Chem. 1992;267:12820–12825. [PubMed] [Google Scholar]
- 7.Graille M, Stura EA, Housden NG, Beckingham JA, Bottomley SP, Beale D, Taussig MJ, Sutton BJ, Gore MG, Charbonnier JB. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure. 2001;9:679–687. doi: 10.1016/s0969-2126(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 8.De Chateau M, Nilson BH, Erntell M, Myhre E, Magnusson CG, Akerstrom B, Bjorck L. On the interaction between protein L and immunoglobulins of various mammalian species. Scandinavian J Immunol. 1993;37:399–405. doi: 10.1111/j.1365-3083.1993.tb03310.x. [DOI] [PubMed] [Google Scholar]
- 9.Viau M, Longo NS, Lipsky PE, Bjorck L, Zouali M. Specific in vivo deletion of B-cell subpopulations expressing human immunoglobulins by the B-cell superantigen protein L. Infection Immun. 2004;72:3515–3523. doi: 10.1128/IAI.72.6.3515-3523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skokowa J, Ali SR, Felda O, Kumar V, Konrad S, Shushakova N, Schmidt RE, Piekorz RP, Nurnberg B, Spicher K, Birnbaumer L, Zwirner J, Claassens JW, Verbeek JS, van Rooijen N, Kohl J, Gessner JE. Macrophages induce the inflammatory response in the pulmonary Arthus reaction through G alpha i2 activation that controls C5aR and Fc receptor cooperation. J Immunol. 2005;174:3041–3050. doi: 10.4049/jimmunol.174.5.3041. [DOI] [PubMed] [Google Scholar]
- 11.van Rooijen N, van Kesteren-Hendrikx E. In vivo” depletion of macrophages by liposome-mediated “suicide. Meth Enzymol. 2003;373:3–16. doi: 10.1016/s0076-6879(03)73001-8. [DOI] [PubMed] [Google Scholar]
- 12.Chouchakova N, Skokowa J, Baumann U, Tschernig T, Philippens KM, Nieswandt B, Schmidt RE, Gessner JE. Fc gamma RIII-mediated production of TNF-alpha induces immune complex alveolitis independently of CXC chemokine generation. J Immunol. 2001;166:5193–5200. doi: 10.4049/jimmunol.166.8.5193. [DOI] [PubMed] [Google Scholar]
- 13.Jain D, Keslacy S, Tliba O, Cao Y, Kierstein S, Amin K, Panettieri RA, Jr., Haczku A, Amrani Y. Essential role of IFNbeta and CD38 in TNFalpha-induced airway smooth muscle hyper-responsiveness. Immunobiol. 2008;213:499–509. doi: 10.1016/j.imbio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, Huszar D. Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int Immunol. 1993;5:647–656. doi: 10.1093/intimm/5.6.647. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 16.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 17.Kroeger KM, Sullivan BM, Locksley RM. IL-18 and IL-33 elicit Th2 cytokines from basophils via a MyD88- and p38alpha-dependent pathway. J. Leuk. Biol. 2009;86:769–778. doi: 10.1189/jlb.0708452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, Hoshino T, Fujimoto J, Nakanishi K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 19.Kastern W, Holst E, Nielsen E, Sjobring U, Bjorck L. Protein L, a bacterial immunoglobulin-binding protein and possible virulence determinant. Infect Immun. 1990;58:1217–1222. [Google Scholar]
- 20.Ricci S, Medaglini D, Marcotte H, Olsen A, Pozzi G, Bjorck L. Immunoglobulin-binding domains of peptostreptococcal protein L enhance vaginal colonization of mice by Streptococcus gordonii. Microb Pathogenesis. 2001;30:229–235. doi: 10.1006/mpat.2000.0427. [DOI] [PubMed] [Google Scholar]
- 21.Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, Komai-Koma M, Pitman N, Li Y, Niedbala W, McKenzie AN, Teixeira MM, Liew FY, Xu D. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–4790. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 22.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, Pitman N, Mirchandani A, Rana B, van Rooijen N, Shepherd M, McSharry C, McInnes IB, Xu D, Liew FY. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–6477. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- 23.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 24.Akerstrom B, Bjorck L. Bacterial Surface Protein L Binds and Inactivates Neutrophil Proteins S100A8/A9. J Immunol. 2009;183:4583–4592. doi: 10.4049/jimmunol.0901487. [DOI] [PubMed] [Google Scholar]