

Abstract
Much of the original research on desmosomes and their biochemical components was through analysis of skin and mucous membranes. The identification of desmogleins 1 and 3, desmosomal adhesion glycoproteins, as targets in pemphigus, a fatal autoimmune blistering disease of the skin and mucous membranes, provided the first link between desmosomes, desmogleins, and human diseases. The clinical and histological similarities of staphylococcal scalded skin syndrome or bullous impetigo and pemphigus foliaceus led us to identify desmoglein 1 as the proteolytic target of staphylococcal exfoliative toxins. Genetic analysis of striate palmoplantar keratoderma and hypotrichosis identified their responsible genes as desmogleins 1 and 4, respectively. More recently these fundamental findings in cutaneous biology were extended beyond the skin. Desmoglein 2, which is expressed earliest among the four isoforms of desmoglein in development and found in all desmosome-bearing epithelial cells, was found to be mutated in arrythmogenic right ventricular cardiomyopathy and has also been identified as a receptor for a subset of adenoviruses that cause respiratory and urinary tract infections. The story of desmoglein research illuminates how dermatologic research originally focused on one skin disease, pemphigus, has contributed to understanding biology and pathophysiology of many seemingly unrelated tissues and diseases.
Keywords: pemphigus, impetigo, hypotrichosis, cardiomyopathy, cadherin
Introduction
The story of the discovery of desmogleins in desmosomes, their relationship to adhesion molecules, and their targeting in diseases with loss of adhesion of cells is one of beautiful simplicity and logic, in which the research in skin has played a major role. This review will tell that story (Table 1).
Table 1.
Desmogleins targeted in human diseases
| Isoform | Type | Diseases |
|---|---|---|
| desmoglein 1 | autoimmune | pemphgius foliaceus pemphigus vulgaris (mucocutaneous type) paraneoplastic pemphigus |
| infection | staphylococcal scalded skin syndrome bullous impetigo |
|
| genetic | striate palmoplantar keratoderma | |
| desmoglein 2 | infection | respiratory and urinary tract infection (receptors for adenovirus serotypes 3, 7, 11, and 14) |
| genetic | arrythmogenic right ventricular cardiomyopathy dilated cardiomyopathy |
|
| desmoglein 3 | autoimmune | pemphigus vulgaris (mucosal dominant type, mucocutaneous type) paraneoplastic pemphigus |
| desmoglein 4 | genetic | hypotrichosis |
The story starts with the discovery of desmosomes by electron microscopy and observations that they play a role in adhesion of keratinocytes. Dermatologists already know intuitively that desmosomes are important in cell adhesion, because in spongiosis, one of the most common pathologies seen in inflammatory skin diseases such as allergic contact dermatitis, there is edema between keratinocytes separating their cell membranes, yet the cells stay attached right at the desmosomes (also called in the past “intercellular bridges”). Only when the desmosomes finally “dissolve” does a spongiotic blister form. It was in skin and stratified squamous epithelial tissue in which the presence and importance of desmosomes was discovered, but subsequently they were discovered in non-epithelial tissues such as heart (see below).
Biochemical and molecular characterization of desmogleins
Once a method of isolating desmosomes from epithelia (cow’s snout was the major source) was established, they could be biochemically characterized. The central part of the desmosome, as seen by electron microscopy as a white area between the cell membranes of adjacent cells, was thought to be the “glue” that held the desmosomes (and, thereby, the cells) together. When this area was enriched from the isolated desmosomes, its protein composition could be determined. A major protein was identified of about 160 kd, and was called desmoglein, “glein” derived from the Greek word for “glue”.
Subsequently antibodies against desmoglein were developed. These antibodies indicated that desmoglein was found in desmosomes of various tissues across various species. Desmoglein, therefore, was determined to be a widespread, and presumably important (i.e. conserved through evolution), component of the desmosome. From this work, initially in epidermis, it was found that desmoglein was also found in many other tissues such as intestine, mammary gland, trachea, bladder, liver, and, perhaps unexpectedly, heart and thymus.
Protein isolation and antibodies allowed molecular cloning of desmoglein. Surprisingly, desmoglein was found to be a group of homologous molecules encoded by a gene family. The most interesting, and scientifically satisfying, finding was that desmogleins were in a supergene family defined by classical cadherins, which were already known to be calcium-dependent adhesion proteins. Thus, desmogleins, thought the be part of the “glue” in desomomes, were found to be in a family of adhesion molecules; a very nice convergence of deductions from early morphologic findings and newer genetic cloning techniques.
The first two members of the desmoglein gene family were identified by genetic cloning as desmoglein 1 and 2, but a dermatologic disease, pemphigus vulgaris, allowed identification of a third member (see below), desmoglein 3. Analysis of the human genome database identified another desmoglein, desmoglein 4, which was also identified by mutational analysis of a genetic hair disease (localized autosomal recessive hypothrichosis) (Table 1).
Even at this early stage, characterization of a desmoglein gene family was closely intertwined with understanding skin diseases. However that interdependency reached another level when the relationship of pemphigus to desmogleins was defined.
Desmogleins identified as targets of autoantibodies in pemphigus
There are two major types of pemphigus, vulgaris and foliaceus. In both, blisters are caused from loss of cell adhesion in the deep epidermis or superficial epidermis, respectively. Pemphigus vulgaris affects mucous membranes and/or skin, whereas pemphigus foliaceus only affects skin. In both, autoantibodies are known to directly mediate the loss of cell adhesion.
The original discovery that these diseases are related to desmosomes depended on a precious commodity, time, that today (with increasing demands related to extensive grant writing; extensive regulation; the geometric progression of required educational modules for; routine clinical, laboratory and animal work; demands to earn their salaries by seeing patients; and more) is much less available. But the availability of time was the critical resource that allowed one of the authors (JRS), in early 1980, to attend a lecture by Malcolm Steinberg at Dulles Airport, 40 minutes from where I worked in Bethesda. At that time, I had been working on identifying the autoantigen in pemphigus foliaceus and knew it was an approximately 160 kd protein as identified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of epidermal extracts, followed by immunoblotting. This was precisely the same time that Malcolm Steinberg (with G. Gorbsky) was dissecting the proteins in the “desmoglea” (the electron lucent center) of the desmosome. There was no evidence then that pemphigus had anything to do with desmosomes, but since cells come apart in pemphigus I was interested in cell adhesion and, therefore, desmosomes. Since I had enough time to take several hours to attend that talk, I learned that Dr. Steinberg had identified the first desmoglein, now called desmoglein 1, as a 160 kd glycoprotein. Naively, I thought that it would make sense if desmoglein 1 was the pemphigus foliaceus antigen, because both were the same size as determined by SDS-PAGE. (Ignorance is also sometimes a great advantage to a scientist; I had no idea how many molecules extracted from epidermis might migrate in the same general area on a one dimensional SDS-PAGE gel).
In fact, the pemphigus foliaceus antigen was ultimately shown to be desmoglein 1 (Figure 1), and desmoglein 1 was shown to be in the superficial epidermis. This finding showed the first autoimmune disease of desmosomes, and had the scientifically satisfying logic that an antibody against a presumed cell adhesion molecule in a cell adhesion structure causes a disease from loss of cell adhesion and subsequent blister formation.
Figure 1.

One of the major pieces of evidence that desmoglein 1 is the pemphigus foliaceus antigen. 2-dimensional gel electrophoresis of extracts of epidermis followed by immunoblotting with pemphigus foliaceus serum and an anti-desmoglein 1 antibody shows that both identify spots with the same migration, convincing proof that both bind the same protein. (From, Koulu, L., Kusumi, A., Steinberg, M.S., Klaus Kovtun, V., and Stanley, J.R. 1984. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J. Exp. Med. 160:1509–1518).
At the time the pemphigus foliaceus antigen was shown to be desmoglein 1, the pemphigus vulgaris antigen was only known to be a glycoprotein of about 130 kd, as determined by immunoprecipitation. In addition, it was known there was some relationship of the pemphigus vulgaris antigen to desmosomes because it was shown by co-immunoprecipitation that pemphigus vulgaris antigen co-precipitated plakoglobin with the 130 kd molecule. Similarly pemphigus foliaceus sera co-precipitated plakoglobin with desmoglein 1. Plakoglobin was known to be in the plaque of the desmosome inside the cell. These studies were the first to show that the tail of desmogleins (the part inside the cell) bound a plaque protein of the desmosome. Again, a skin disease, pemphigus, was intertwined with our growing understanding of desmosomes, in this case their molecular structure.
What really brought all these observations together, in a beautiful and logical synthesis of previous findings, was the molecular cloning of pemphigus vulgaris antigen which showed it was another, previously unknown, desmoglein, now called desmoglein 3 (Figure 2). Although desmoglein 1 and 3 were both found in epidermis, they were at different levels; desmoglein 1 was superficial and desmoglein 3 was deep. All the previous observations and findings now fit together nicely: Pemphigus vulgaris and foliaceus were closely related diseases that both had loss of keratinocyte adhesion but in different tissue localizations. The autoantibodies bound closely related molecules thought to provide the “glue” in adhesion structures, the desmosomes, with resultant loss of adhesion and blisters. In pemphigus vulgaris and foliaceus the blisters were thought to occur in different tissue localizations because of the different localizations of the desmogleins. Finally, both pemphigus antigens were found to bind plakoglobin because desmogleins bind plakoglobin by their homologous tails.
Figure 2.
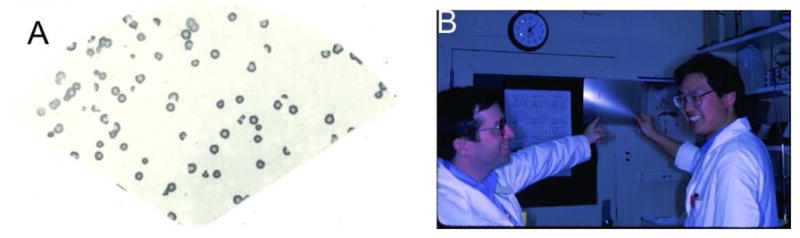
Original data from the cloning of pemphigus vulgaris antigen. A) Purified λgt11 expression phage that contain cDNA for pemphigus vulgaris antigen. A single clone multiplies in bacteria, and all its offspring were blotted to nitrocellulose. All resultant clones stain positively with pemphigus vulgaris sera. The cDNA was sequenced to show that the protein produced was desmoglein 3. B) John Stanley (left) and Masayuki Amagai on the day in 1991 when the pemphigus vulgaris antigen clone was identified. (Amagai, M., Klaus-Kovtun, V., and Stanley, J.R. 1991. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 67:869–877.)
Many more subsequent studies confirmed that the anti-desmoglein antibodies in pemphigus patients cause the disease. For example, adsorption of pemphigus sera with recombinant desmogleins resulted in loss of pathogenicity of those sera. Monoclonal anti-desmoglein antibodies cause disease when injected into neonatal mice or human skin organ culture (Figure 3). An interesting confirmation that loss of desmoglein 3 adhesion causes pemphigus is that mouse with a genetic deletion of desmoglein 3 develop oral and skin lesions with the typical histology of pemphigus vulgaris. Finally, specific proteolytic cleavage of desmoglein 1 caused lesions in epidermis histologically indistinguishable from pemphigus foliaceus (see below).
Figure 3.
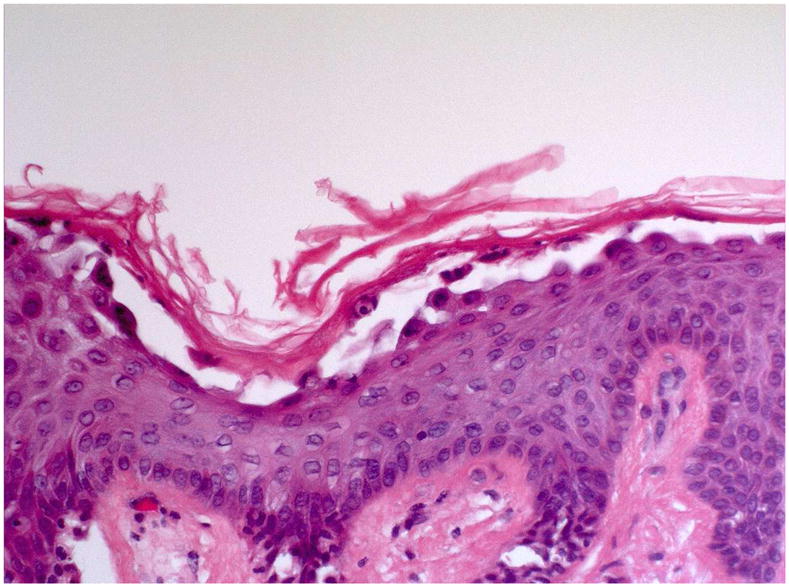
A monoclonal, monovalent anti-desmoglein 1 antibody cloned from a pemphigus foliaceus patient causes typical histology of pemphigus foliaceus when injected into normal human skin organ culture.
Desmogleins used for the diagnosis of pemphigus
cDNA isolation of desmoglein 1 and 3 allowed us to produce recombinant proteins which properly reflect their three dimensional structures by baculovirus or mammalian expression. The immunoadsorption of patients’ sera with those recombinant proteins removed their immunoreactivities on keratinocyte cell surfaces by immunofluorescence and their ability to induce blister formation in neonatal mice. Subsequently, enzyme-linked immunosorbent assay (ELISA) using recombinant desmogleins 1 and 3 were developed as a serological diagnostic tool for pemphigus. Patients with pemphigus foliaceus show only anti-desmoglein 1 IgG autoantibodies, while patients with mucosal dominant type of pemphigus vulgaris have only anti-desmoglein 3 IgG. Finally, patients with the mucocutaneous type of pemphigus vulgaris have both anti-desmogelin 3 and anti-desmoglein 1 IgG. ELISA is a powerful and objective assay that allows easy screening of large numbers of sera to characterize patients for diagnosis and research purposes.
In general, ELISA antibody titers fluctuate in parallel with the disease activity with time in any particular patient, while the titers do not necessarily reflect the disease severity among different patients. This discrepancy is because patients’ sera are polyclonal containing both pathogenic and non-pathogenic antibodies for blister formation, as demonstrated by isolation of monoclonal antibodies from mice and humans. Such pathogenicity, or the lack thereof, can be measured in neonatal mice, in skin organ culture or in keratinocyte dissociation assays of cultured human keratinocytes.
Pathophysiology of blister formation in pemphigus
There are two major, not necessarily exclusive, theories of how pemphigus antibodies cause blisters. One theory is that antibodies cause steric hindrance of the desmoglein adhesion site thus interfering directly with adhesion. The other theory is that antibodies cause intracellular signaling that leads to loss of adhesion.
Pemphigus antibodies cause steric hindrance
Epitope mapping of pemphigus autoantibodies and monoclonal antibodies from mice and humans has demonstrated that pathogenic antibodies bind to the amino-terminal extracellular domain of desmogleins that is predicted to form the trans-adhesive interface between cells, based on the crystal structures of classic cadherins, molecules in the same gene family as desmogleins. In addition, pathogenic pemphigus foliaceus monoclonal antibodies cloned from patients bind to the mature desmoglein 1 which reveals the active adhesion site only after the proprotein is cleaved, but not to the proprotein which does not form the adhesion site. So-called “knockout mice” lacking desmoglein 3 show similar, if not identical, acantholytic lesions mimicking the phenotype of mucosal dominant type of pemphigus vulgaris. These mice show that lack of function (i.e. adhesion) of desmoglein 3, without antibody-induced signaling (e.g., by crosslinking a cell surface receptor), show the typical histology of pemphigus vulgaris. Desmoglein compensation theory logically explains the site of blister formation in the skin and mucous membranes in patients with pemphigus, suggesting blisters are formed as a result of the loss of adhesive function of each desmoglein isoform. If antibody signaling caused loss of adhesion, then one would expect that whenever antibodies bound desmoglein 3 a blister should result because of the resultant signaling, yet in fact this is not the case wherever desmoglein 1 is also present. These observations suggest that pemphigus blisters are initially caused by steric hindrance and not by the activation of a signaling pathway that causes the initial loss of adhesion, although such signaling may occur after loss of adhesion and may amplify the initial loss of cell adhesion. Furthermore, these data do not negate that modulation of signaling pathways that control desmosome stability could be useful in counteracting pemphigus antibody-induced loss of adhesion.
Pemphigus antibodies cause intracellular signaling
On the other hand, when pathogenic pemphigus antibodies bind to the keratinocyte cell surface, several signaling events have been shown to take place. The most widely studied are those pathways that involve p38 mitogen-activated protein kinase (p38MAP kinase) and plakoglobin. The most general indication of signaling is that pemphigus antibodies cause protein phosphorylation changes in keratinocytes. Studies of these changes indicated that p38MAP kinase was a major pathway stimulated by both pemphigus vulgairs and pemphigus foliaceus autoantibodies. These pathways may be activated only after cells lose their adhesion, but, even so, they probably increase the acantholytic effects of pemphigus antibodies, and blocking them may improve disease (see below). Another observations regarding signaling in pemphigus was that keratinocytes genetically engineered to lack plakoglobin were not as susceptible to pemphigus antibodies as were wild type cells. This observation led to a pathway involving c-myc, which was shown to be elevated in pemphigus antibody-stimulated keratinocytes.
Some of these signaling pathways are probably involved in normal homeostasis of desmosomes and their components, such as transport to the cell surface, into the desmosome, and internalization. Pemphigus antibodies may act through modulation of these physiologic pathways. For example, as a result of signals induced by pemphigus antibodies that cause loss desmoglein adhesion as desmogleins reach the cell membrane, desmogleins may be depleted first from the cell membrane thereby decreasing or eliminating the pool of desmogleins for their incorporation into desmosomes, ultimating resulting in depletion of desmogleins in desmosomes leading to desmosome dysfunction (i.e. acantholysis). Although it is difficult at this time to detemine which are the most critical signaling pathways contributing to pathogenicity, signaling pathways may be an important target in designing therapy for pemphigus in the future (see below).
Immunology of development of anti-desmoglein antibodies
Why do patients with pemphigus produce harmful autoantibodies against desmogleins? This is an ultimate and fundamental question that we need to answer in the future. To begin to address this question, upstream events of pathogenic antibody production have been investigated. It is probable that both anti-desmoglein B cells and desmoglein-peptide specific T cells that provide help to those B cells are necessary for anti-desmoglein antibody formation. In support of this model, circulating B cells producing anti-desmoglein 3 antibodies were detected by enzyme-linked immunospot (ELISPOT) assay in patients. Circulating T cells reacting with desmoglein 3 were detected in patents, but perhaps surprisingly, were also detected in normal people. To investigate the formation of anti-desmoglein antibodies in an animal model, an active disease mouse model for pemphigus vulgaris was developed by adoptive transfer of lymphocytes from desmoglein 3 deficient mice to immune-deficient desmoglein 3-expressing mice. This model was used to isolate anti-desmoglein 3 pathogenic monoclonal antibodies as well as desmoglein 3-specific T cells that help B cells to produce pathogenic antibodies. Both the T and B cells were needed for antibody formation. Aire (autoimmune regulator, a transcription factor) was also shown to play a role in regulating desmoglein 3 expression in the medullary thymic epithelial cells and in selection of T cells in thymus. Presumably expression of desmoglein 3 in thymus helps provide tolerance to that antigen in most people.
To further clarify the mechanisms for central and peripheral tolerance to desmoglein 3-reacting T cells and B cells will be an important step to develop antigen-specific immunosuppressive therapies.
Desmogleins are targets in infectious diseases
The story of the identification of desmoglein 1 as the target of staphylococcus exfoliative toxin (Table 1) illustrates the importance of having physicians as scientists.
The blisters in bullous impetigo and staphylococcal scalded skin disease have been known for many years to be caused by a toxin made and secreted by staphylococcus aureus, called exfoliative toxin. In bullous impetigo, the toxin is produced locally, causing blisters at sites of infection. In staphylococcal scalded skin syndrome, which usually is seen in infants or very young children, the toxin circulates and causes blisters distant from sites of infection. When injected into mice this toxin causes blisters. However, for many years after is discovery, it was not clear how this toxin caused blisters in the epidermis. Even after the toxin was molecularly cloned and shown to have the structure of a serine protease (i.e. a protease with a serine in its active site) that seemed to need a specific substrate in order to orient its catalytic site to be active, that substrate could not be identified. Ultimately, it was dermatologists, with their knowledge of skin diseases, who were able to solve this longstanding puzzle.
Even as early as the 18th century, astute clinicians who were superb morphologists (probably because they had little technology beyond their eyes and ears) recognized that staphylococcal scalded skin syndrome resembled pemphigus. Because of this observation, staphylococcal scalded skin syndrome was sometimes called “pemphigus neonatorum”. In addition, it was known that injection of exfoliative toxin into neonatal mice caused blisters whose histology was identical to that resulting from injection of pemphigus foliaceus antibodies into mice. In fact, any practicing dermatologists knows that whenever a biopsy of a patient with pemphigus foliaceus is taken, the report always indicates that although the histology is consistent with pemphigus foliaceus it is also consistent with bullous impetigo or staphylococcal scalded skin syndrome. Once it was learned that pemphigus foliaceus was caused by antibodies against desmoglein 1, it was a small step in logic to hypothesize that exfoliative toxin’s protein substrate might be desmoglein 1. In both cases the function of desmoglein 1 would be impaired with similar resultant histology.
In fact, that simple but eloquent hypothesis was shown to be correct. Exfoliative toxin was shown to cleave one peptide bound in one substrate, desmoglein 1 (Figure 4). The use of this toxin to enhance staphylococcus pathogenicity and infectivity is beautiful in its simplicity. Essentially, staphylococcus has evolved to produce a toxin that targets one peptide bond of one molecule to enable it to grow under the stratum corneum (the barrier of the skin), but enables it to be superficial enough so that when one child touches another the bacteria can be passed on.
Figure 4.

Coomassie blue gel showing cleavage of the 84-kD extracellular domain of desmoglein 1 to a 50 kD and 34 kD fragment, increasing with time of incubation with exfoliative toxin A. (Hanakawa, Y., Schechter, N.M., Lin, C., Nishifuji, K., Amagai, M., and Stanley, J.R. 2004. Enzymatic and molecular characteristics of the efficiency and specificity of exfoliative toxin cleavage of desmoglein 1. J. Biol. Chem. 279:5268–5277.)
Adenovirus provides another example of desmoglein involvement in infection. Most of human adenoviruses (serotypes A to F) use the coxsackie adenovirus receptor as a primary attachment receptor. However, serotype B adenovirus, which cause respiratory and urinary tract infections, does not use this receptor, but has recently been found to bind to desmoglein 2 as the primary high-affinity receptor (Table 1). Adenovirus binding of desmoglein 2 triggers an epithelial-to-mesenchymal-like transition, leading to transient opening of intercellular junctions and penetration of virus into the subepithelial cell layers and the blood stream, allowing the virus to spread.
Desmogleins are targets in genetic diseases
Mutations in genes encoding desmogleins have also been described in skin, hair, as well as heart diseases (Table 1). Striate palmoplantar keratoderma, an autosomal dominant disorder, is characterized by longitudinal hyperkeratotic lesions on the palms associated with focal or diffuse thickening of the plantar skin. Haploinsufficiency mutations in the gene for desmoglein 1, desmocollin 2, or desmoplakin, have been shown to underlie this skin disorder. Mutations in the gene encoding desmoglein 4 cause localized autosomal recessive hypothrichosis in humans and the lanceolate hair phenotype in mice. More recently, it has been discovered that arrhythmogenic right ventricular cardiomyopathy, which is clinically characterized by right ventricular enlargement and dysfunction, fibrofatty replacement of myocytes in the right ventricle, characteristic electrocardiographic abnormalities, ventricular arrhythmia, and sudden death, is caused by mutations in the genes for desmoglein 2, desmocollin 2, plakoglobin, plakophilin 2, or desmoplakin. These genetic findings underscore the importance of desmoglein (and other desmosomal molecules) mediated adhesive function in tissue integrity and function.
Desmogleins do more than provide adhesion of cells
Experiments showing that misexpression of desmoglein isoforms where they are not normally present causes differentiation defects indicate that desmogleins may do more than simply provide adhesion. For example, desmoglein 3 expression in the superficial epidermis, where it is not normally present, cause abnormal epidermal differentiation and statum corneum formation with increased transepidermal water loss. In mice with a genetic deletion of desmoglein 2 there is a defect in blastocyst proliferation resulting in failure of implantation. Conversely, forced expression of desmoglein 2 in the superficial epidermis causes increased proliferation. The above findings indicate profound effects of desmogleins on keratinocyte proliferation and differentiation. Presently, it is thought that these effects are produced through signal transduction pathways, although the exact pathways have not been elucidated. One major contributor to signaling through desmosomes may be plakoglobin. As discussed above, desmogleins bind to plakoglobin, which is related to β-catenin, a well characterized molecule important in the Wnt signaling pathway. This pathway is important in proliferation and differentiation as well as embryological development. Amount and isoforms of desmoglein may control amount of free plakoglobin that could contribute to signaling. However, much work needs to be done in the future to link signaling to specific molecules and pathways.
Further evidence indicating that desmogleins do more than just provide cell adhesion are the observations that demonstrate that desmoglein 1 is critical for proper stratification and differentiation of epidermis. Interestingly, this effect, which is associated with down regulations of the EGFR-Erk1/2 signaling pathway, requires neither extracellular adhesion nor intracellular plakoglobin binding. These results implicate pathways independent of plakoglobin in desmoglein signaling.
The future: therapy of pemphigus based on the understanding of its pathophysiology and immunology
Anti-idiotypic therapy
Recent genetic analysis of monoclonal antibodies cloned from pemphigus vulgaris and foliaceus patients indicates that both pathogenic and non-pathogenic monoclonal antibodies are found, and that there are a limited number of parenteral B cell clones for these antibodies. A limited set, but several different, immunoglobulin variable heavy chain genes are used in patients to make pathogenic antibodies, therefore, it probably is not possible to target variable heavy chains to treat disease. However, when the complementarity determining region 3 (CDR3) of the antibodies was analyzed, pathogenic antibodies shared a common sequences. (The CDR3 region of antibodies is thought to be the major region that binds the corresponding antigen). Randomization and site-directed mutagenesis of the heavy chain CDR3 sequences in some of these antibodies illustrated the importance of only a few, or even one, amino acids for binding and/or pathogenicity. Such data suggest that it might be possible to target common sequences in the CDR3 regions of pathogenic pemphigus antibodies to prevent disease. Reagents derived from phage display cloning of monoclonal antibodies from patients could be used to screen for such blockers.
Rituximab
From knowledge that pemphigus is an autoantibody-mediated disease with a limited set of anti-desmoglein non-tolerant B cells, as discussed above, it makes sense that eliminating B cells might improve disease, especially if these non-tolerant set of B cells cannot come back after B cell depletion. In fact, the success of rituximab for therapy tends to validate this idea. Rituximab is an anti-CD20 antibody. CD20 is found on all mature B cells, but not the stem B cells. B cell pools are re-populated from stem B cells (i.e. B cells that have not formed immunoglobulins against specific antigens). Rituximab should, therefore, in theory, eliminate anti-desmoglein B cell clones.
In fact, rituximab has shown excellent results in treating pemphigus patients refractory to standard therapy (e.g. prednisone plus immunosuppressives like azathioprine or mycophenolate mofetil). A single cycle (2 injections over 2 weeks or 4 injections over a month) can lead to complete remission is over 80% of patients, although some relapse and some may need low dose prednisone therapy. Rituxan causes a profound depletion of circulating B cells, but presumably does not deplete plasma cells, which do not have CD20 on their surface. Since in most patients anti-desmoglein antibodies go down after rituximab therapy, the anti-desmoglein antibody-producing plasma cells may be short-lived and need frequent replenishment by B cells.
In any case, elimination of circulating B cells has proven to be excellent therapy for patients with refractory pemphigus.
Signaling
As discussed above, pemphigus antibody binding to desmoglein on the keratinocyte cell surface causes signal transduction which can exacerbate acantholysis. Therefore, if the right signaling pathways can be identified then pharmacologically interrupting them might be beneficial in patients. For instance, blocking the p38MAP kinase pathway in a mouse model of pemphigus has been shown to modulate disease.
In addition, studies, discussed above, have shown that the normal system of cycling of desmosomal components may be interrupted in pemphigus, causing depletion of desmoglein in desmosomes with subsequent acantholysis. Tools might be found to perturb signaling to affect this physiologic process in order to increase desmoglein synthesis and subsequent incorporation into desmosomes, thereby, stabilizing them and counteracting the depletion induced by pemphigus antibodies. It may be that corticosteroids, which are already known to be very effective in therapy, act by this precise mechanism, in that they increase desmoglein synthesis through signaling.
Acknowledgments
Work supported by grants from the National Institututes of Arthritis, Musculoskeletal, and Skin Diseases of the National Institutes of Health (JRS) and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MA)
Important JID References
Introduction
- Multiple authors. Milestones Cutaneous Biology: Desmosome. J Invest Dermatol. 2007 Jan;:E1–E16. (published online) [Google Scholar]
- Multiple authors. Milestones Cutaneous Biology: Autoimmne bullous diseases. J Invest Dermatol. 2008 Oct;:E15–E32. (published online) [Google Scholar]
- Matoltsy AG. Desmosomes, filaments, and keratohyaline granules: their role in the stabilization and keratinization of the epidermis. J Invest Dermatol. 1975;65:127–142. doi: 10.1111/1523-1747.ep12598093. (Describes the importance of desmosomes for stability of epidermis and a method for isolating desmosomes from epithelium later widely used to biochemically characterize desmosomes. This method allowed identification of desmosomal proteins on which all further biochemical and molecular work was based.) [DOI] [PubMed] [Google Scholar]
Biochemical and molecular characterization of desmogleins
- Konohana A, Konohana I, Roberts GP, Marks R. Biochemical changes in desmosomes of bovine muzzle epidermis during differentiation. J Invest Dermatol. 1987;89:353–357. doi: 10.1111/1523-1747.ep12471752. (Shows changes in desmoglein amounts as epidermis differentiates. This paper presaged the finding that a different isoform of desmoglein was produced in more differentiated epidermis and that desmoglein undergoes biochemical changes in the stratum corneum that may be related to desquamation) [DOI] [PubMed] [Google Scholar]
- Lundstrom A, Egelrud T. Evidence that cell shedding from plantar stratum corneum in vitro involves endogenous proteolysis of the desmosomal protein desmoglein I. J Invest Dermatol. 1990;94:216–220. doi: 10.1111/1523-1747.ep12874531. (More evidence that degradation of desmoglein 1 occurs during desquamation) [DOI] [PubMed] [Google Scholar]
- Roh JY, Stanley JR. Plakoglobin binding by human Dsg3 (pemphigus vulgaris antigen) in keratinocytes requires the cadherin-like intracytoplasmic segment. J Invest Dermatol. 1995;104:720–724. doi: 10.1111/1523-1747.ep12606963. (This report determines which part of the desmoglein 3 tail binds to plakoglobin) [DOI] [PubMed] [Google Scholar]
- Whittock NV, Bower C. Genetic evidence for a novel human desmosomal cadherin, desmoglein 4. J Invest Dermatol. 2003;120:523–530. doi: 10.1046/j.1523-1747.2003.12113.x. [DOI] [PubMed] [Google Scholar]
Desmogleins identified as targets of autoantibodies in pemphigus
- Stanley JR, Hawley Nelson P, Poirier M, Katz SI, Yuspa SH. Detection of pemphigoid antigen, pemphigus antigen, and keratin filaments by indirect immunofluorescence in cultured human epidermal cells. J Invest Dermatol. 1980;75:183–186. doi: 10.1111/1523-1747.ep12522615. (Presence of pemphigus antigen in keratinocyte culture indicated that such cells could be used for immumoprecipation and immunoblotting to biochemically characterize the antigens) [DOI] [PubMed] [Google Scholar]
- Stanley JR, Klaus Kovtun V, Sampaio SA. Antigenic specificity of fogo selvagem autoantibodies is similar to North American pemphigus foliaceus and distinct from pemphigus vulgaris autoantibodies. J Invest Dermatol. 1986;87:197–201. doi: 10.1111/1523-1747.ep12695334. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Ogawa MM, Konohana A, Nishikawa T. Detection of pemphigus vulgaris and pemphigus foliaceus antigens by immunoblot analysis using different antigen sources. J Invest Dermatol. 1990;94:327–331. doi: 10.1111/1523-1747.ep12874456. (Immunoblotting has become an important approach to characterizing autoimmune blistering skin diseases, including those with anti-desmoglein antibodies) [DOI] [PubMed] [Google Scholar]
- Korman NJ, Eyre RW, Zone J, Stanley JR. Drug-induced pemphigus: autoantibodies directed against the pemphigus antigen complexes are present in penicillamine and captopril-induced pemphigus. J Invest Dermatol. 1991;96:273–276. doi: 10.1111/1523-1747.ep12464471. (Same desmoglein antigens are found in these special types of pemphigus as in spontaneously occurring pemphigus) [DOI] [PubMed] [Google Scholar]
- Rappersberger K, Roos N, Stanley JR. Immunomorphological and biochemical identification of the pemphigus foliaceus autoantigen within desmosomes. J Invest Dermatol. 1992;99:323–330. doi: 10.1111/1523-1747.ep12616659. [DOI] [PubMed] [Google Scholar]
- Olague-Alcala M, Giudice GJ, Diaz LA. Pemphigus foliaceus sera recognize an N-terminal fragment of bovine desmoglein 1. J Invest Dermatol. 1994;102:882–885. doi: 10.1111/1523-1747.ep12382794. [DOI] [PubMed] [Google Scholar]
- Emery DJ, Diaz LA, Fairley JA, Lopez A, Taylor AF, Giudice GJ. Pemphigus foliaceus and pemphigus vulgaris autoantibodies react with the extracellular domain of desmoglein-1. J Invest Dermatol. 1995;104:323–328. doi: 10.1111/1523-1747.ep12665364. [DOI] [PubMed] [Google Scholar]
- Kowalczyk AP, Anderson JE, Borgwardt JE, Hashimoto T, Stanley JR, Green KJ. Pemphigus sera recognize conformationally sensitive epitopes in the amino-terminal region of desmoglein-1 (Dsg1) J Invest Dermatol. 1995;105:147–152. doi: 10.1111/1523-1747.ep12316680. (Many of the pemphigus antibody-defined epitopes on desmogleins are conformational. This type of data led to the findings that pathogenic anti-desmoglein antibodies tend to bind calcium-stabilized conformationally sensitive epitopes) [DOI] [PubMed] [Google Scholar]
- Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, Nishikawa T. Pemphigus vulgaris and pemphigus foliaceus sera show an inversely graded binding pattern to extracellular regions of desmosomes in different layers of human epidermis. J Invest Dermatol. 1995;105:153–159. doi: 10.1111/1523-1747.ep12316695. [DOI] [PubMed] [Google Scholar]
- Amagai M, Koch PJ, Nishikawa T, Stanley JR. Pemphigus vulgaris antigen (Desmoglein 3) is localized in the lower epidermis, the site of blister formation in patients. J Invest Dermatol. 1996;106:351–355. doi: 10.1111/1523-1747.ep12343081. [DOI] [PubMed] [Google Scholar]
- Memar OM, Rajaraman S, Thotakura R, Tyring SK, Fan JL, Seetharamaiah GS, Lopez A, Jordon RE, Prabhakar BS. Recombinant desmoglein 3 has the necessary epitopes to adsorb and induce blister-causing antibodies. J Invest Dermatol. 1996;106:261–268. doi: 10.1111/1523-1747.ep12340663. [DOI] [PubMed] [Google Scholar]
- Ding X, Diaz LA, Fairley JA, Giudice GJ, Liu Z. The anti-desmoglein 1 autoantibodies in pemphigus vulgaris sera are pathogenic. J Invest Dermatol. 1999;112:739–743. doi: 10.1046/j.1523-1747.1999.00585.x. [DOI] [PubMed] [Google Scholar]
- Andl CD, Stanley JR. Central role of the plakoglobin-binding domain for desmoglein 3 incorporation into desmosomes. J Invest Dermatol. 2001;117:1068–1074. doi: 10.1046/j.0022-202x.2001.01528.x. [DOI] [PubMed] [Google Scholar]
- Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128:939–948. doi: 10.1038/sj.jid.5701132. (Monoclonal anti-desmoglein 1 antibodies cloned from a pemphigus foliaceus patient cause blisters with typical histology in mice and human skin organ culture) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokouchi M, Saleh MA, Kuroda K, Hachiya T, Stanley JR, Amagai M, Ishii K. Pathogenic Epitopes of Autoantibodies in Pemphigus Reside in the Amino-Terminal Adhesive Region of Desmogleins Which Are Unmasked by Proteolytic Processing of Prosequence. J Invest Dermatol. 2009;129:2156–2166. doi: 10.1038/jid.2009.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Desmogleins used for the diagnosis of pemphigus
- Amagai M, Hashimoto T, Green KJ, Shimizu N, Nishikawa T. Antigen-specific immunoabsorption of pathogenic autoantibodies in pemphigus foliaceus. J Invest Dermatol. 1995;104:895–901. doi: 10.1111/1523-1747.ep12606168. (Proof that anti-desmoglein 1 antibodies in sera are pathogenic) [DOI] [PubMed] [Google Scholar]
- Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Invest Dermatol. 2005;124:939–946. doi: 10.1111/j.0022-202X.2005.23714.x. (This paper describes an assay, that has become widely used, to measure pemphigus antibody pathogenicity) [DOI] [PubMed] [Google Scholar]
Pathophysiology of blister formation in pemphigus
- Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. 1999;112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x. [DOI] [PubMed] [Google Scholar]
- Muller E, Caldelari R, de BA, Baumann D, Bierkamp C, Balmer V, Suter VMM. Pathogenesis in pemphigus vulgaris: A central role for the armadillo protein plakoglobin. J Invest Dermatol. 2000;115:332. doi: 10.1046/j.1523-1747.2000.00abs-2.x. [DOI] [PubMed] [Google Scholar]
- Williamson L, Hunziker T, Suter MM, Muller EJ. Nuclear c-Myc: a molecular marker for early stage pemphigus vulgaris. J Invest Dermatol. 2007;127:1549–1555. doi: 10.1038/sj.jid.5700735. [DOI] [PubMed] [Google Scholar]
- Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. 2008;128:738–740. doi: 10.1038/sj.jid.5701080. [DOI] [PubMed] [Google Scholar]
- Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol. 2009;129:908–918. doi: 10.1038/jid.2008.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, Amagai M, Kowalczyk AP. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol. 2011;131:706–718. doi: 10.1038/jid.2010.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Immunology of development of anti-desmoglein antibodies
- Hertl M, Karr RW, Amagai M, Katz SI. Heterogeneous MHC II restriction pattern of autoreactive desmoglein 3 specific T cell responses in pemphigus vulgaris patients and normals. J Invest Dermatol. 1998;110:388–392. doi: 10.1046/j.1523-1747.1998.00156.x. [DOI] [PubMed] [Google Scholar]
- Nishifuji K, Amagai M, Kuwana M, Iwasaki T, Nishikawa T. Detection of antigen-specific B cells in patients with pemphigus vulgaris by enzyme-linked immunospot assay: requirement of T cell collaboration for autoantibody production. J Invest Dermatol. 2000;114:88–94. doi: 10.1046/j.1523-1747.2000.00840.x. [DOI] [PubMed] [Google Scholar]
- Wada N, Nishifuji K, Yamada T, Kudoh J, Shimizu N, Matsumoto M, Peltonen L, Nagafuchi S, Amagai M. Aire-dependent thymic expression of desmoglein 3, the autoantigen in pemphigus vulgaris, and its role in T-cell tolerance. J Invest Dermatol. 2011;131:410–417. doi: 10.1038/jid.2010.330. [DOI] [PubMed] [Google Scholar]
Desmogleins are targets in infectious diseases
- Elias PM, Fritsch P, Dahl MV, Wolff K. Staphylococcal toxic epidermal necrolysis: Pathogenesis and studies on the subcellular site of action of exfoliatin. J Invest Dermatol. 1975;65:501–512. doi: 10.1111/1523-1747.ep12610196. (Exfoliative toxin causes acantholysis similar to pemphigus antibodies) [DOI] [PubMed] [Google Scholar]
- Amagai M, Yamaguchi T, Hanakawa Y, Nishifuji K, Sugai M, Stanley JR. Staphylococcal exfoliative toxin B specifically cleaves desmoglein 1. J Invest Dermatol. 2002;118:845–850. doi: 10.1046/j.1523-1747.2002.01751.x. [DOI] [PubMed] [Google Scholar]
Desmogleins are targets in genetic diseases
- Whittock NV, Smith FJ, Wan H, Mallipeddi R, Griffiths WA, Dopping-Hepenstal P, Ashton GH, Eady RA, McLean WH, McGrath JA. Frameshift mutation in the V2 domain of human keratin 1 results in striate palmoplantar keratoderma. J Invest Dermatol. 2002;118:838–844. doi: 10.1046/j.1523-1747.2002.01750.x. [DOI] [PubMed] [Google Scholar]
- Moss C, Martinez-Mir A, Lam H, Tadin-Strapps M, Kljuic A, Christiano AM. A recurrent intragenic deletion in the desmoglein 4 gene underlies localized autosomal recessive hypotrichosis. J Invest Dermatol. 2004;123:607–610. doi: 10.1111/j.0022-202X.2004.23311.x. [DOI] [PubMed] [Google Scholar]
Desmogleins do more than provide adhesion of cells
- Green KJ, Simpson CL. Desmosomes: new perspectives on a classic. J Invest Dermatol. 2007;127:2499–2515. doi: 10.1038/sj.jid.5701015. [DOI] [PubMed] [Google Scholar]
The future: therapy of pemphigus based on the understanding of its pathophysiology and immunology (Anti-idiotypic therapy)
Anti-idiotypic therapy
- Payne AS, Siegel DL, Stanley JR. Targeting pemphigus autoantibodies through their heavy-chain variable region genes. J Invest Dermatol. 2007;127:1681–1691. doi: 10.1038/sj.jid.5700790. [DOI] [PubMed] [Google Scholar]
- Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128:939–948. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rituximab
- Mouquet H, Musette P, Gougeon ML, Jacquot S, Lemercier B, Lim A, Gilbert D, Dutot I, Roujeau JC, D’Incan M, et al. B-cell depletion immunotherapy in pemphigus: effects on cellular and humoral immune responses. J Invest Dermatol. 2008;128:2859–2869. doi: 10.1038/jid.2008.178. [DOI] [PubMed] [Google Scholar]
- Eming R, Nagel A, Wolff-Franke S, Podstawa E, Debus D, Hertl M. Rituximab exerts a dual effect in pemphigus vulgaris. J Invest Dermatol. 2008;128:2850–2858. doi: 10.1038/jid.2008.172. [DOI] [PubMed] [Google Scholar]
- Zambruno G, Borradori L. Rituximab immunotherapy in pemphigus: therapeutic effects beyond B-cell depletion. J Invest Dermatol. 2008;128:2745–2747. doi: 10.1038/jid.2008.330. [DOI] [PubMed] [Google Scholar]
Signaling
- Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, Amagai M, Kowalczyk AP. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol. 2011;131:706–718. doi: 10.1038/jid.2010.389. (Basis for idea that using methods, e.g. signaling, to increase desmoglein 3 sythesis might be effective therapy for pemphigus) [DOI] [PMC free article] [PubMed] [Google Scholar]
Selected non-JID References
Introduction
- Chambers R, de Renyi GS. The structure of the cells in tissues as revealed by microdissection. I. The physical relationships of cells in epithelia. Am J Anat. 1925;35:385–402. (Cell bridges, now known as desmosomes, hold epithelial cells together) [Google Scholar]
- Porter R. Observations on the submicroscopic structure of animal epidermis. Anat Rec. 1954;118:433, Abstr. (Given credit for first description of desmosomes in epithelia by electron microscopy in Staehelin, L.A. 1974. Structure and function of intercellular junctions. Int. Rev. Cytol. 39:191–283.) [Google Scholar]
- Skerrow CJ, Matoltsy AG. Isolation of epidermal desmosomes. J Cell Biol. 1974;63:515–523. doi: 10.1083/jcb.63.2.515. (Preparation of desmosomes from cow snout) [DOI] [PMC free article] [PubMed] [Google Scholar]
Biochemical and molecular characterization of desmogleins
- Gorbsky G, Steinberg MS. Isolation of the intercellular glycoproteins of desmosomes. J Cell Biol. 1981;90:243–248. doi: 10.1083/jcb.90.1.243. (Isolation of “desmoglea” central part of desmosome and identification of desmoglein glycoprotein) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelz M, Duden R, Cowin P, Franke WW. A constitutive transmembrane glycoprotein of Mr 165000 (desmoglein) in epidermal and non-epidermal desmosomes. I. Biochemical identification of the polypeptide. Eur J Cell Biol. 1986;42:177–183. (Anti-desmoglein antibodies and distribution of desmogleins) [PubMed] [Google Scholar]
- Cowin P, Garrod DR. Antibodies to epithelial desmosomes show wide tissue and species cross-reactivity. Nature. 1983;302:148–150. doi: 10.1038/302148a0. [DOI] [PubMed] [Google Scholar]
(Next 4 references: Molecular cloning of desmogleins 1 and 2 and homology to classical cadherins)
- Koch PJ, Walsh MJ, Schmelz M, Goldschmidt MD, Zimbelmann R, Franke WW. Identification of desmoglein, a constitutive desmosomal glycoprotein, as a member of the cadherin family of cell adhesion molecules. Eur J Cell Biol. 1990;53:1–12. [PubMed] [Google Scholar]
- Nilles LA, Parry DAD, Powers EE, Angst BD, Wagner RM, Green KJ. Structural analysis and expression of human desmoglein: a cadherin-like component of the desmosome. J Cell Sci. 1991;99:809–821. doi: 10.1242/jcs.99.4.809. [DOI] [PubMed] [Google Scholar]
- Wheeler GN, Parker AE, Thomas CL, Ataliotis P, Poynter D, Arnemann J, Rutman AJ, Pidsley SC, Watt FM, Rees DA, et al. Desmosomal glycoprotein DGI, a component of intercellular desmosome junctions, is related to the cadherin family of cell adhesion molecules. Proc Natl Acad Sci USA. 1991;88:4796–4800. doi: 10.1073/pnas.88.11.4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer S, Koch PJ, Franke WW. Identification of the ubiquitous human desmoglein, Dsg2, and the expression catalogue of a subfamily of desmosomal cadherins. Exp Cell Res. 1994;211:391–399. doi: 10.1006/excr.1994.1103. [DOI] [PubMed] [Google Scholar]
Desmogleins identified as targets of autoantibodies in pemphigus
- Koulu L, Kusumi A, Steinberg MS, Klaus Kovtun V, Stanley JR. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med. 1984;160:1509–1518. doi: 10.1084/jem.160.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korman NJ, Eyre RW, Klaus-Kovtun V, Stanley JR. Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N Engl J Med. 1989;321:631–635. doi: 10.1056/NEJM198909073211002. [DOI] [PubMed] [Google Scholar]
- Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–877. doi: 10.1016/0092-8674(91)90360-b. (Cloning of pemphigus vulgaris antigen indicates it is desmoglein 3.) [DOI] [PubMed] [Google Scholar]
- Amagai M, Hashimoto T, Shimizu N, Nishikawa T. Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest. 1994;94:59–67. doi: 10.1172/JCI117349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–1102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–2178. doi: 10.4049/jimmunol.170.4.2170. (The famous mouse monoclonal antibody, AK23, directed against desmoglein 3 causes pemphigus vulgaris lesions in mice) [DOI] [PubMed] [Google Scholar]
- Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. (Anti-desmoglein 3 monoclonal antibodies from a pemphigus vulgaris patient cause typical pemphigus vulgaris histology when injected in neonatal mice or human skin organ culture.) [DOI] [PMC free article] [PubMed] [Google Scholar]
Desmogleins used for diagnosis of pemphigus
- Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, Gamou S, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010–2017. [PubMed] [Google Scholar]
Pathophysiology of blister formation in pemphigus
Pemphigus antibodies cause steric hindrance
- Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–1102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461–468. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol. 2001;167:5439–5448. doi: 10.4049/jimmunol.167.9.5439. [DOI] [PubMed] [Google Scholar]
- Li N, Aoki V, Hans-Filho G, Rivitti EA, Diaz LA. The role of intramolecular epitope spreading in the pathogenesis of endemic pemphigus foliaceus (fogo selvagem) J Exp Med. 2003;197:1501–1510. doi: 10.1084/jem.20022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pemphigus antibodies cause intracellular signaling
- Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, Müller E. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. 2001;153:823–834. doi: 10.1083/jcb.153.4.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, Rubenstein DS. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 2005;280:23778–23784. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- Rubenstein DS, Diaz LA. Pemphigus antibody induced phosphorylation of keratinocyte proteins. Autoimmunity. 2006;39:577–586. doi: 10.1080/08916930600971885. [DOI] [PubMed] [Google Scholar]
- Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem. 2007;282:13804–13812. doi: 10.1074/jbc.M611365200. [DOI] [PubMed] [Google Scholar]
- Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. 2008;173:1628–1636. doi: 10.2353/ajpath.2008.080391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly PS, Berkowitz P, Bektas M, Lee HE, Chua M, Diaz LA, Rubenstein DS. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. 2010;285:8936–8941. doi: 10.1074/jbc.M109.087999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem. 2011;286:1283–1291. doi: 10.1074/jbc.M110.172874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson L, Raess NA, Caldelari R, Zakher A, de BA, Posthaus H, Bolli R, Hunziker T, Suter MM, Muller EJ. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006;25:3298–3309. doi: 10.1038/sj.emboj.7601224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP. Pemphigus vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism. J Biol Chem. 2008;283:18303–18313. doi: 10.1074/jbc.M710046200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Immunology of development of anti-desmoglein antibodies
- Wucherpfennig KW, Yu B, Bhol K, Monos DS, Argyris E, Karr RW, Ahmed AR, Strominger JL. Structural basis for major histocompatibility complex (MHC)-linked susceptibility to autoimmunity: charged residues of a single MHC binding pocket confer selective presentation of self-peptides in pemphigus vulgaris. Proc Natl Acad Sci U S A. 1995;92:11935–11939. doi: 10.1073/pnas.92.25.11935. (First identification of desmoglein 3-specific T cells in human) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MS, Swartz SJ, Lopez A, Ding X, Fernandez-Vina MA, Stastny P, Fairley JA, Diaz LA. Development and characterization of desmoglein-3 specific T cells from patients with pemphigus vulgaris. J Clin Invest. 1997;99:31–40. doi: 10.1172/JCI119130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. 2000;105:625–631. doi: 10.1172/JCI8748. (Development of active disease mouse model for pemphigus vulgaris) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–2178. doi: 10.4049/jimmunol.170.4.2170. (Pathogenic monoclonal antibodies against desmoglein 3 was isolated) [DOI] [PubMed] [Google Scholar]
- Takahashi H, Amagai M, Nishikawa T, Fujii Y, Kawakami Y, Kuwana M. Novel system evaluating in vivo pathogenicity of desmoglein 3-reactive T cell clones using murine pemphigus vulgaris. J Immunol. 2008;181:1526–1535. doi: 10.4049/jimmunol.181.2.1526. [DOI] [PubMed] [Google Scholar]
Desmogleins are targets in infectious diseases
- Melish ME, Glasgow LA. The staphylococcal scalded-skin syndrome. Development of an experimental model. N Engl J Med. 1970;282:1114–1119. doi: 10.1056/NEJM197005142822002. (Exfoliative toxin causes a blister in neonatal mice) [DOI] [PubMed] [Google Scholar]
- Dancer SJ, Garratt R, Saldanha J, Jhoti H, Evans R. The epidermolytic toxins are serine proteases. FEBS Letters. 1990;268:129–132. doi: 10.1016/0014-5793(90)80990-z. [DOI] [PubMed] [Google Scholar]
- Vath GM, Earhart CA, Rago JV, Kim MH, Bohach GA, Schlievert PM, Ohlendorf DH. The structure of the superantigen exfoliative toxin A suggests a novel regulation as a serine protease. Biochemistry. 1997;36:1559–1566. doi: 10.1021/bi962614f. (Exfoliative toxin may cleave a specific protein receptor.) [DOI] [PubMed] [Google Scholar]
- Cavarelli J, Prevost G, Bourguet W, Moulinier L, Chevrier B, Delagoutte B, Bilwes A, Mourey L, Rifai S, Piemont Y, et al. The structure of Staphylococcus aureus epidermolytic toxin A, an atypic serine protease, at 1.7A resolution. Structure. 1997;5:813–824. doi: 10.1016/s0969-2126(97)00235-9. [DOI] [PubMed] [Google Scholar]
- Amagai M, Matsuyoshi N, Wang ZH, Andl C, Stanley JR. Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nature Med. 2000;6:1275–1277. doi: 10.1038/81385. [DOI] [PubMed] [Google Scholar]
- Hanakawa Y, Schechter NM, Lin C, Nishifuji K, Amagai M, Stanley JR. Enzymatic and molecular characteristics of the efficiency and specificity of exfoliative toxin cleavage of desmoglein 1. J Biol Chem. 2004;279:5268–5277. doi: 10.1074/jbc.M311087200. [DOI] [PubMed] [Google Scholar]
- Wang H, Li ZY, Liu Y, Persson J, Beyer I, Moller T, Koyuncu D, Drescher MR, Strauss R, Zhang XB, Wahl JK, 3rd, Urban N, Drescher C, Hemminki A, Fender P, Lieber A. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med. 2011;17:96–104. doi: 10.1038/nm.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Desmogleins are targets in genetic diseases
- Kljuic A, Bazzi H, Sundberg JP, Martinez-Mir A, O’Shaughnessy R, Mahoney MG, Levy M, Montagutelli X, Ahmad W, Aita VM, et al. Desmoglein 4 in hair follicle differentiation and epidermal adhesion: evidence from inherited hypotrichosis and acquired pemphigus vulgaris. Cell. 2003;113:249–260. doi: 10.1016/s0092-8674(03)00273-3. (Desmoglein 4 associated with localized autosomal recessive hypothrichosis) [DOI] [PubMed] [Google Scholar]
- Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–1179. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
Desmogleins do more than provide adhesion of cells
- Eshkind L, Tian Q, Schmidt A, Franke WW, Windoffer R, Leube RE. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol. 2002;81:592–598. doi: 10.1078/0171-9335-00278. [DOI] [PubMed] [Google Scholar]
- Brennan D, Hu Y, Joubeh S, Choi YW, Whitaker-Menezes D, O’Brien T, Uitto J, Rodeck U, Mahoney MG. Suprabasal Dsg2 expression in transgenic mouse skin confers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Sci. 2007;120:758–771. doi: 10.1242/jcs.03392. [DOI] [PubMed] [Google Scholar]
- Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, Cornwell M, Green KJ. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol. 2009;185:1243–1258. doi: 10.1083/jcb.200809044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta. 2008;1778:572–587. doi: 10.1016/j.bbamem.2007.07.014. [DOI] [PubMed] [Google Scholar]
The future: therapy of pemphigus based on the understanding of its pathophysiology and immunology
Anti-idiotypic therapy
- Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J, Kacir S, Ishii K, Payne AS, Siegel DL, Stanley JR. Antibodies to the desmoglein 1 precursor proprotein but not to the mature cell surface protein cloned from individuals without pemphigus. J Immunol. 2009;183:5615–5621. doi: 10.4049/jimmunol.0901691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Invest. 2010;120:4111–4117. doi: 10.1172/JCI44425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rituximab
- Joly P, Mouquet H, Roujeau JC, D’Incan M, Gilbert D, Jacquot S, Gougeon ML, Bedane C, Muller R, Dreno B, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–552. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
Signaling
- Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci U S A. 2006;103:12855–12860. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Arredondo J, Chernyavsky AI, Kitajima Y, Pittelkow M, Grando SA. Pemphigus vulgaris IgG and methylprednisolone exhibit reciprocal effects on keratinocytes. J Biol Chem. 2004;279:2135–2146. doi: 10.1074/jbc.M309000200. (Corticosteroids may by therapeutic in pemphigus by increasing synthesis of desmogleins) [DOI] [PubMed] [Google Scholar]