

Abstract
Hypothalamo-pituitary-adrenal (HPA) axis activity is subject to negative feedback control by glucocorticoids. Although the rapid component of this feedback is widely considered to contribute to regulation of dynamic HPA activity, few in vivo data exist on the temporal and pharmacological characteristics of this phenomenon. Thus, frequent automated blood sampling was undertaken in rats to determine the effects of acute glucocorticoid administration on basal and stress-induced corticosterone secretion. The glucocorticoid agonist methylprednisolone (5–2000 μg) or dexamethasone (5–500 μg) injected iv at the peak of the diurnal rhythm caused dose-dependent suppression of basal corticosterone secretion, which was attenuated by the glucocorticoid receptor antagonist RU38486. With 50 μg methylprednisolone, the onset of this suppression occurred at 40 min and remained significant for 120 min. However, although higher doses led to a greater and more sustained suppression of endogenous corticosterone, the response was delayed by the emergence of an initial stimulatory response that imposed a finite minimum delay. A corticosterone response to injection of CRH (1 μg, iv) during the period of maximal suppression indicated a suprapituitary site for the inhibitory effect glucocorticoid activation. This mechanism was supported by glucocorticoid injection immediately before a psychological stress (30 min, white noise); methylprednisolone caused dose-dependent attenuation of stress-induced corticosterone release and expression of the activity marker c-fos mRNA in the paraventricular nucleus but did not block the pituitary response to CRH. Thus, in rats, glucocorticoid receptor activation rapidly suppresses basal and stress-induced HPA activity that operates, at least in part, through a central mechanism of action.
The hypothalamo-pituitary-adrenal (HPA) axis is subject to negative feedback control by endogenous adrenal corticosteroids, which has been shown to act in several distinct time domains. In addition to delayed steroid feedback that regulates long-term changes in the synthesis of both CRH in the paraventricular nucleus (PVN) and ACTH in pituitary corticotrophs (1–6), more rapid effects have been suggested to contribute to the dynamic regulation of the axis (7–10). These rapid actions of corticosteroids are thought to operate either at the pituitary (11, 12) or suprapituitary levels (13, 14) and have been postulated to play important roles both in terminating the response to acute stressors (15–17) and in generating the ultradian pattern of basal HPA activity (18, 19).
Although rapid corticosteroid feedback was first proposed nearly 40 yr ago and has become widely accepted as a mechanism regulating HPA activity, data supporting the temporal dynamics and pharmacological characteristics of this phenomenon are relatively scant. The majority of studies supporting fast feedback have involved administration of corticosteroids shortly before either application of different stressors (11, 14, 20–25) or injection of CRH (11, 12, 14) to demonstrate attenuation of the secretion of ACTH. However, relatively few studies have examined the acute effects of corticosteroid feedback on basal (unstimulated) HPA activity. Early studies in rats (26) and dogs (27, 28) showed that bolus injection or infusion of cortisol caused a suppression of ACTH levels with a fixed lag of 20 min that was not reduced at higher doses. However, in these cases, the animals were both anesthetized and adrenalectomized, and no control infusions were performed. Later, Keller-Wood (21) showed that infusion of cortisol caused a significant suppression of basal ACTH levels in intact, conscious dogs, also with a delay of 40 min, although animals were restrained throughout this procedure. Studies in humans have shown that injections or infusions of corticosteroids can have a suppressive effect on basal ACTH release with onset delay of less than 1 h (10, 29–34), providing strong evidence for a rapid component of inhibition. In rodents, high doses of the corticosteroid agonist prednisolone sodium succinate (5 or 50 mg/kg iv) cause rapid decline of plasma corticosterone, reaching undetectable levels within 1 h and remaining undetectable for 4–6 h (35). More recently, using repeated blood sampling in rats, we have shown that acute iv injection of 2 mg methylprednisolone can both block the HPA response to 10 min noise stress 40 min later and suppress basal corticosterone levels when tested during the morning nadir (23) and that lower doses [500 μg (8) and 250 μg (9)] rapidly suppress basal corticosterone secretion at the diurnal acrophase.
To further characterize the pharmacology and temporal profile of rapid glucocorticoid suppression of HPA activity, we have studied the effects of acute, exogenous doses of the synthetic glucocorticoids methylprednisolone and dexamethasone on basal HPA activity measured using automated blood sampling of unhandled animals. Furthermore, to determine whether the effects occurred at a pituitary and/or central site of action, the ability of methylprednisolone to attenuate either CRH- or stress-induced HPA activity was examined.
Materials and Methods
Animal husbandry and cannulation
All experiments were performed on virgin female Sprague Dawley rats (250–350 g) obtained from Bantin and Kingman (Hull, UK) and housed in the local animal facility at least 7 d before experimentation, initially in groups of four to six per cage. Female rats were used because the higher basal levels of corticosterone compared with males (36) enabled better resolution of the temporal profile of glucocorticoid inhibition. Animals were housed under standard environmental conditions: 14-h light, 10-h dark cycle (lights on at 0500 h) and ad libitum access to water and rat chow. The iv cannulation of the right jugular vein was performed as previously described (37). Surgery was performed at least 3 d before blood sampling to allow for postoperative recovery and adaptation to the sampling environment. Animals were housed singly after surgery, and a swivel allowed complete freedom of movement within the home cage. Cannulae were flushed with a small volume of heparinized saline (10 U/ml) daily to maintain patency. Procedures were carried out in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986 and the European Community Council Directive (86/609/EEC). Each animal underwent only one period of sampling.
Blood sampling and drug administration
Blood sampling was undertaken in a sound-isolated room using an automated sampling system (38). Animals were connected to the system through a liquid swivel and samples (37 μl whole blood diluted 1:5 in heparinized saline) automatically withdrawn every 5 or 10 min and replaced by heparinized saline. This procedure for multiple blood sampling does not cause any adverse effects (8, 9, 23, 25, 36, 37). The initial 1 h of samples was discarded to avoid any nonspecific release and measurements commenced either at 1600 h (late light phase) or 0700 h (early light phase). Injections of steroids, CRH, or vehicle were performed manually by briefly disconnecting the iv cannula from the swivel and injecting through the sampling cannula. This procedure was completed between two samples and caused minimal disturbance. Drug effects were controlled by comparison with injections of saline.
Study 1: measures of clearance rate of corticosterone and steroid cross-reaction
Because studies involved the determination of plasma corticosterone levels during injection of glucocorticoid agonists, a study was performed to examine whether measurements were affected by cross-reaction of the agonists. Seven animals underwent bilateral adrenalectomy at the same time as cannulation. In the postoperative period, they were allowed access to drinking water containing 0.9% NaCl and 0.25 mg/ml corticosterone to maintain hydromineral balance. On the fourth day at 1200 h, this solution was replaced by drinking water containing 0.9% NaCl alone to allow the corticosterone to clear from the circulation. At 1600 h, blood samples were withdrawn every 5 min for a period of 6 h. At 25, 125, and 225 min of sampling, each animal received injections of 400 μg corticosterone (Sigma-Aldrich, St. Louis, MO), 2000 μg methylprednisolone (sodium succinate, Solu-Medrone; Pharmacia & Upjohn Ltd., Milton Keynes, UK), and 500 μg dexamethasone (dexamethasone-21-phosphate; Sigma-Aldrich), respectively. These doses represented the maximum doses administered during the later pharmacological studies. Samples were assayed for corticosterone-like immunoreactivity and measured values used to calculate cross-reaction. Exponential regression was used to calculate the clearance rate (y = y0e−kt) and half-life [t½ = (ln 2)/k], where y0 is the peak corticosterone value (nanograms per milliliter), k is the rate constant, and t is time.
Study 2: effect of glucocorticoid agonists and antagonist on basal HPA activity
To examine the acute effects of glucocorticoid agonists on basal HPA activity, animals were administered injections of methylprednisolone (5 μg, n = 8; 50 μg, n = 10; 500 μg, n = 12; 2000 μg, n = 10), dexamethasone (5 μg, n = 8; 50 μg, n = 6; 500 μg, n = 7), or saline vehicle (n = 11), each in a volume of 0.1 ml. Injections were made during the late light period to coincide with the phase of the diurnal cycle when basal corticosterone secretion is near maximum (23, 37). The dynamics of the response to agonist injection were examined by rapid blood sampling every 5 min for 3 h and then every 10 min for 2 h. In all cases, samples were collected from 1600 h (3 h before lights off), and agonist injections were given at 1645 h. To determine the effect of glucocorticoid antagonism on the response to methylprednisolone, additional groups of animals were injected with either 50 μg/rat (n = 6) or 500 μg/rat (n = 6) of the antagonist RU38486 (mifepristone; gift from Dr. J. K. Belanoff, Corcept Therapeutics Inc., Menlo Park, CA) 15 min before injection of 50 μg methylprednisolone.
Study 3: CRH challenge during glucocorticoid-induced suppression
To determine whether glucocorticoid-induced suppression of HPA activity was due to suppression of pituitary-adrenal responsiveness, animals were tested for responses to CRH injection. Samples were collected every 5 min starting at 1500 h, although measurements did not start until 1600 h. Acute injection of methylprednisolone (500 μg) or saline was given at 1700 h, and CRH (1 μg/rat, iv) was injected at either 1800 or 2000 h. CRH (Bachem, Torrance, CA) was diluted in heparinized saline and stored at 4 C until use. The two time points for CRH injection (60 or 180 min after methylprednisolone) were selected to coincide with either 1) the period of maximal rate of change of corticosterone levels or 2) the period of maximal suppression of the axis. Blood sampling continued every 5 min for a minimum of 2 h after the CRH injection before changing to every 10 min.
Study 4: rapid glucocorticoid suppression of stress-induced HPA activity
To determine whether glucocorticoid feedback could rapidly suppress HPA activity induced by acute psychological stress, methylprednisolone (500 μg) or saline was injected iv 15 min before the onset of a 30-min noise (104 dB). Sampling was undertaken in the early light phase starting at 0600 h when basal corticosterone levels were low, and the noise stress commenced at 0800 h. Blood samples were collected every 5 min until 1200 h. At 15 min after the end of the noise stress, all animals received an iv injection of 1 μg CRH to determine pituitary-adrenal axis reactivity during the period of maximal poststress decline in corticosterone levels.
Finally, to demonstrate both the dose dependency and central site of action for the glucocorticoid-mediated suppression of stress-induced HPA activity, groups of animals were injected iv 15 min before the onset of a noise stress (30 min at 104 dB) with different doses of methylprednisolone (5 μg, n = 6; 50 μg, n = 5; or 500 μg, n = 5) or saline (n = 6). No automated sampling was conducted, but animals were decapitated immediately after the end of the noise, and trunk blood (∼4 ml) was collected. Six other animals injected with saline were not exposed to noise but were euthanized at the equivalent time after injection. Trunk blood samples were centrifuged at 5000 rpm for 5 min, and plasma was collected and stored at −20 C. Brains were removed and immediately stored at −80 C for later determination of c-fos mRNA expression.
Measurement of corticosterone
Total plasma corticosterone levels were measured by RIA in diluted whole blood samples (automated sampling) or in plasma (trunk blood) using a citrate buffer (pH 3.0) to denature the binding globulin, [125I]corticosterone (0.37 MBq/ml activity; ICN, High Wycombe, UK), and a specific rabbit antirat corticosterone primary antibody (gift courtesy of Professor G. Makara, Institute of Experimental Medicine, Budapest, Hungary) (see Ref. 8 for additional details). All samples from an individual animal were assayed together.
In situ hybridization histochemistry for c-fos mRNA
The hypothalamic PVN was cryosectioned at 12 μm and sections mounted on gelatin-coated slides, with the presence of the PVN confirmed by staining adjacent sections with toluidine blue. Before hybridization, sections were fixed in 4% paraformaldehyde, washed in saline/triethanolamine containing 0.25% acetic anhydride, dehydrated in ethanol and chloroform, and air dried. The c-fos mRNA levels were measured by in situ hybridization using a cDNA riboprobe as described elsewhere (39). Hybridized sections were apposed to autoradiographic film for 14 d. Relative levels of c-fos mRNA expression were calculated as OD measurements using computerized image analysis software (Image; National Institutes of Health, Bethesda, MD).
Data analysis
Hormone data are presented as the mean and sem of the absolute measures of plasma corticosterone. However, to determine the time course of the effect of acute agonist injection on plasma corticosterone levels, a normalization procedure was also applied. This normalization involved first calculating the average corticosterone levels in the preinjection period (0–45 min.) for each animal and then dividing all data for that animal by this average value. Second, to overcome the nonstationary nature of the data, the rescaled value at each time point was divided by the respective mean value for that time point in the saline control group. Thus, the mean normalized value at all time points for the saline controls was fixed at 1, and treatment groups are displayed relative to this to show the drug effect. Both raw and normalized data are presented. To determine the period of significant steroid-induced effect, t tests were performed on these normalized values at each time point to determine the first and last points that differed significantly (P < 0.05) from controls. Other comparisons were performed using t test or ANOVA, as appropriate.
Results
Study 1: measures of corticosterone clearance and steroid cross-reaction
Injection of 400 μg corticosterone into adrenalectomized rats resulted in a rapid increase of measured corticosterone levels that rose to 1326 ± 297 ng/ml within 5 min (Fig. 1). After peaking, immunoreactive steroid levels declined back to baseline with an exponential decay: rate constant 0.143 and calculated half-life 4.8 min. In contrast, injection of 2000 μg methylprednisolone caused only a very small increase in plasma steroid immunoreactivity, which peaked at the equivalent of 7.7 ± 1.4 ng/ml corticosterone and which lasted for 10 min (Fig. 1, inset). This equates to less than 0.1% of an equivalent dose of corticosterone. No measurable change in plasma corticosterone immunoreactivity was detected after injection of 500 μg dexamethasone.
Fig. 1.
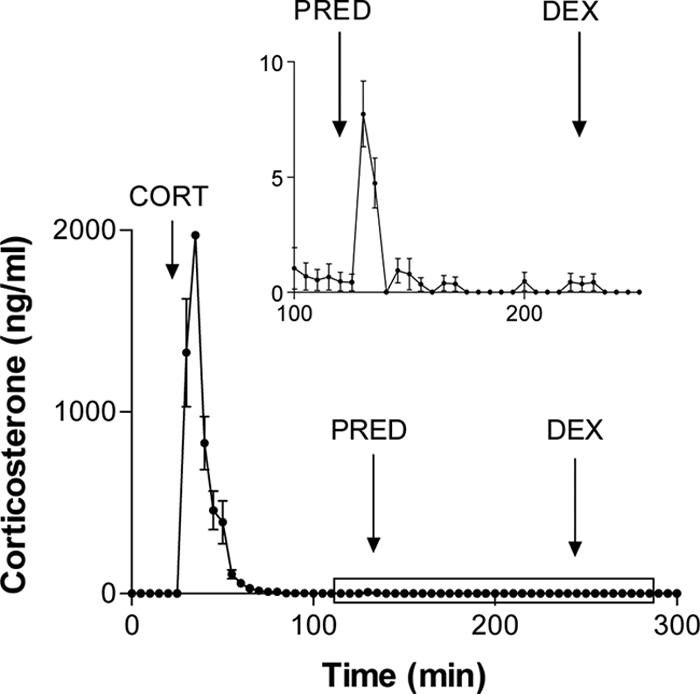
Corticosterone clearance rates and cross-reaction of glucocorticoid agonists. Adrenalectomized animals each received iv injections of corticosterone (CORT, 400 μg), methylprednisolone (PRED, 2000 μg), and dexamethasone (DEX, 500 μg). Blood samples were assayed for corticosterone-like immunoreactivity. Inset shows the period from 100–250 min on an enlarged scale. Values represent mean ± sem (n = 7).
Study 2: effect of glucocorticoid agonists and antagonist on basal HPA activity
Before injection of the glucocorticoid agonists, mean basal corticosterone levels ranged from 80–220 ng/ml, with a trend toward an increase across the time series reflecting the rising levels toward the diurnal acrophase at lights off (1900 h). These levels are within the range expected for female Sprague Dawley rats at the late light phase of the diurnal cycle (37). Injection of saline evoked no immediate change in corticosterone levels, but there was a slow decline in levels over the following 4 h in line with the normal diurnal decline that follows the acrophase to reach a stable level of 40–80 ng/ml (Fig. 2A). Injection of increasing doses of methylprednisolone caused a dose-dependent increase in the magnitude of this suppression, which at the highest dose persisted through to the end of the sampling period (Fig. 2E). The temporal profile of this steroid-induced inhibition was resolved by applying a proportional normalization procedure (Fig. 2, F–J). This revealed that injection of 5 μg methylprednisolone had little effect on corticosterone levels (Fig. 2G), but higher doses caused dose-dependent suppression with a very clear temporal profile. Fifty micrograms of methylprednisolone caused a suppression of corticosterone levels that was significant 40 min after injection and that was no longer significantly below control levels at 160 min (Fig. 2H). Notably, although a 10-fold higher dose extended the period of suppression such that recovery did not occur until 275 min after injection, the onset was delayed until 80 min (Fig. 2I). This was apparently due to a small, but not significant, stimulatory effect immediately after injection. This acute stimulatory effect was more pronounced and significant 15–25 min after injection of 2000 μg methylprednisolone (Fig. 2J). At this dose, significant suppression was detected 65 min after injection and persisted through to the end of sampling (315 min after injection).
Fig. 2.

Acute iv injection of methylprednisolone (PRED) causes dose-dependent suppression of basal corticosterone levels. Data show the mean ± sem of the absolute hormone levels (left panels) and the data after normalization to the saline controls (right panels): A and F, Saline controls, n = 11; B and G, 5 μg, n = 8; C and H, 50 μg, n = 12; D and I, 500 μg, n = 12; E and J, 2000 μg, n = 10 (see text for normalization procedure). All injections (indicated by the broken line in each panel) were given at 1645 h when diurnal levels of circulating plasma corticosterone are high. The hatched bar in each panel indicates the dark period commencing at 1900 h. The asterisks in H–J show the first and last time points that are significantly different from saline-injected animals (P < 0.05). Note that the onset of feedback inhibition does not occur until after 40 min after methylprednisolone injection. The dagger in J indicates three successive samples that are significantly greater than the saline controls (P < 0.05). The horizontal lines in the right panels show the line of equality with saline values (i.e. normalized values equal 1).
Similar temporal profiles were observed after injection of dexamethasone but at lower doses than those of methylprednisolone: injection of 5 μg caused significant suppression of plasma corticosterone between 95 and 205 min after injection (Fig. 3F), 50 μg suppressed levels between 50 and 195 min after injection (Fig. 3G), whereas 500 μg caused a significant stimulatory effect 15–20 min after injection followed by a persistent suppression that commenced 55 min after injection (Fig. 3H).
Fig. 3.

Effects of acute iv injection of dexamethasone (DEX) on basal corticosterone levels. Data show the mean ± sem of the absolute hormone levels (left panels) and the data after normalization to the saline controls (right panels): A and E, saline controls, n = 11; B and F, 5 μg, n = 8; C and G, 50 μg, n = 6; D and H, 500 μg, n = 7. All injections (indicated by the broken line in each panel) were given at 1645 h when diurnal levels of circulating plasma corticosterone are high. The hatched bar in each panel indicates the dark period. The asterisks in F–H show the first and last time points that are significantly different from saline-injected animals (P < 0.05). Note that the onset of feedback inhibition occurs only after 50 min after DEX injection. The dagger in H indicates three successive samples that are significantly greater than the saline controls (P < 0.05). The horizontal lines in the right panels show the line of equality with saline values (i.e. normalized values equal 1). Saline control data are the same as in Fig. 2, A and E.
Administration of the glucocorticoid receptor (GR) antagonist RU38486 15 min before injection of 50 μg methylprednisolone modified the temporal profile of the steroid-induced suppression. When the antagonist was administered at an equivalent dose (50 μg) the onset of the suppression was unaffected, but recovery occurred earlier, with significant differences between groups at 150–155 and 170–180 min after injection (Fig. 4A). A higher antagonist dose (500 μg) caused a variable effect, either blocking the inhibition or, in some animals, evoking a significant stimulation of corticosterone levels (Fig. 4B).
Fig. 4.
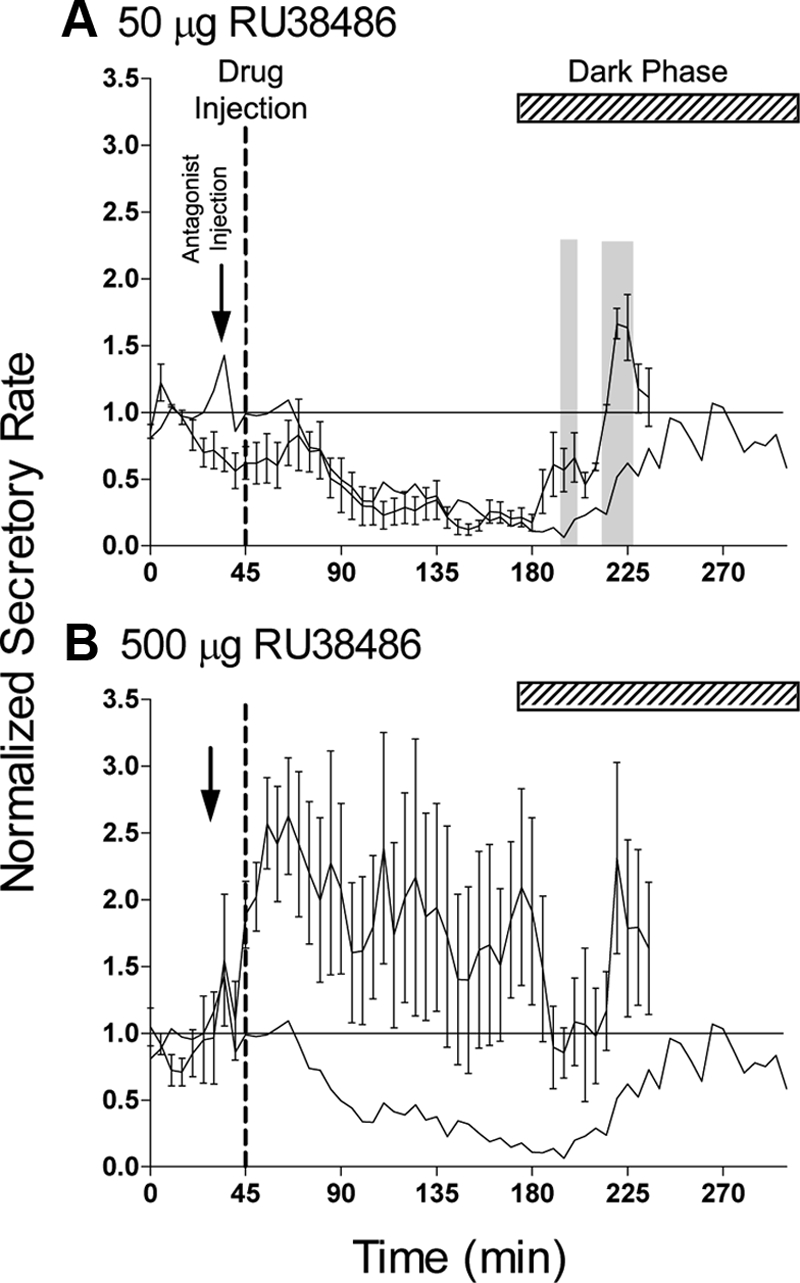
Effect of the GR antagonist RU38486 on the rapid inhibitory effect of iv injection of 50 μg methylprednisolone. Injection of methylprednisolone (indicated by the broken line) was preceded by injection of either 50 μg (A, n = 6) or 500 μg (B, n = 6) RU38486 (indicated by arrow). Data are the values after the normalization procedure and are mean ± sem. The solid line in each panel shows the mean response to 50 μg methylprednisolone in the absence of antagonist (data from Fig. 2H). The shaded areas in A indicate points at which the antagonist-treated data differ significantly from methylprednisolone alone (P < 0.05, repeated-measures ANOVA). The horizontal lines show the line of equality with saline values (i.e. normalized values equal 1), and the hatched bar in each panel indicates the dark period.
Study 3: CRH challenge during glucocorticoid-induced suppression
To test pituitary-adrenal responsiveness during steroid-induced suppression of corticosterone secretion, CRH injections were performed after acute administration of methylprednisolone. Injection of CRH (1 μg, iv) at the time of the diurnal acrophase induced no further increase in plasma corticosterone levels in saline-pretreated animals (Fig. 5A). However, when endogenous steroid secretion was suppressed by injection of 500 μg methylprednisolone, CRH was able to reinstate corticosterone secretion, irrespective of whether it was administered during the period of maximal rate of change (Fig. 5C) or the period of maximal inhibition (Fig. 5D). Interestingly, this dose of CRH produced a persistent increase in pituitary-adrenal activity. Saline injection during the period of suppression had no effect on corticosterone levels (Fig. 5B), indicating that the stimulatory effect was not due to nonspecific HPA activation.
Fig. 5.

Effect of CRH on corticosterone secretion during methylprednisolone-induced negative feedback. A, Injection of CRH (1 μg, iv) 60 min after an injection of saline has little effect on basal HPA activity during the late light phase of the diurnal cycle (n = 4). B, Injection of methylprednisolone (PRED, 500 μg, iv) caused rapid and sustained suppression of corticosterone levels that was unaffected by subsequent injection of saline (n = 3). CRH injection was able to cause a rapid increase in corticosterone levels when given either 60 min after methylprednisolone during the maximal rate of suppression (C, n = 12) or at 240 min when corticosterone levels had reached a nadir (D, n = 4). Values are mean ± sem. Hatched bar shows the dark period. Note that for injections performed during the dark period (B and D), animals were exposed to a brief period of light.
Study 4: rapid glucocorticoid suppression of stress-induced HPA activity
To determine whether methylprednisolone caused acute suppression of centrally driven HPA activation, steroid injections were performed before a psychological stressor (30 min, 104 dB white noise). Preceding and immediately after the injection of 500 μg methylprednisolone, corticosterone levels did not differ between animals receiving steroid or saline (Fig. 6). In saline-treated animals, noise stress evoked an increase in corticosterone that was detectable after 10 min and that reached a peak at 15 min from the onset. Methylprednisolone-treated animals showed no difference from saline controls at 5 and 10 min after the onset of noise, but thereafter, corticosterone levels were significantly suppressed, both during the stress and in the 10 min after the end of the stimulus. Despite the marked suppression of corticosterone levels at this time, injection of 1 μg CRH was able to evoke renewed secretory activity.
Fig. 6.

Effect of acute glucocorticoid injection on noise stress-induced corticosterone secretion during the diurnal nadir. Single injection of methylprednisolone (PRED, 500 μg, n = 12, ▴) given 15 min before the onset of a 30-min white noise stress (104 dB, shaded area) caused a significant attenuation of the corticosterone response compared with saline-injected controls (n = 10, ■). Asterisks indicate time points that significantly differ between groups before injection of CRH (P < 0.05, repeated-measures ANOVA). Injection of CRH (1 μg, iv) at 100 min confirmed pituitary-adrenal responsiveness. Data are mean ± sem.
The suppression of noise-induced corticosterone levels by methylprednisolone was confirmed in trunk blood samples obtained from animals pretreated with different steroid doses (Fig. 7A): 500 μg, but not 5 or 50 μg, methylprednisolone reduced the levels of corticosterone at the end of a 30-min noise stress. This dose also attenuated the levels of noise-induced c-fos mRNA expression in the PVN (Fig. 7B).
Fig. 7.
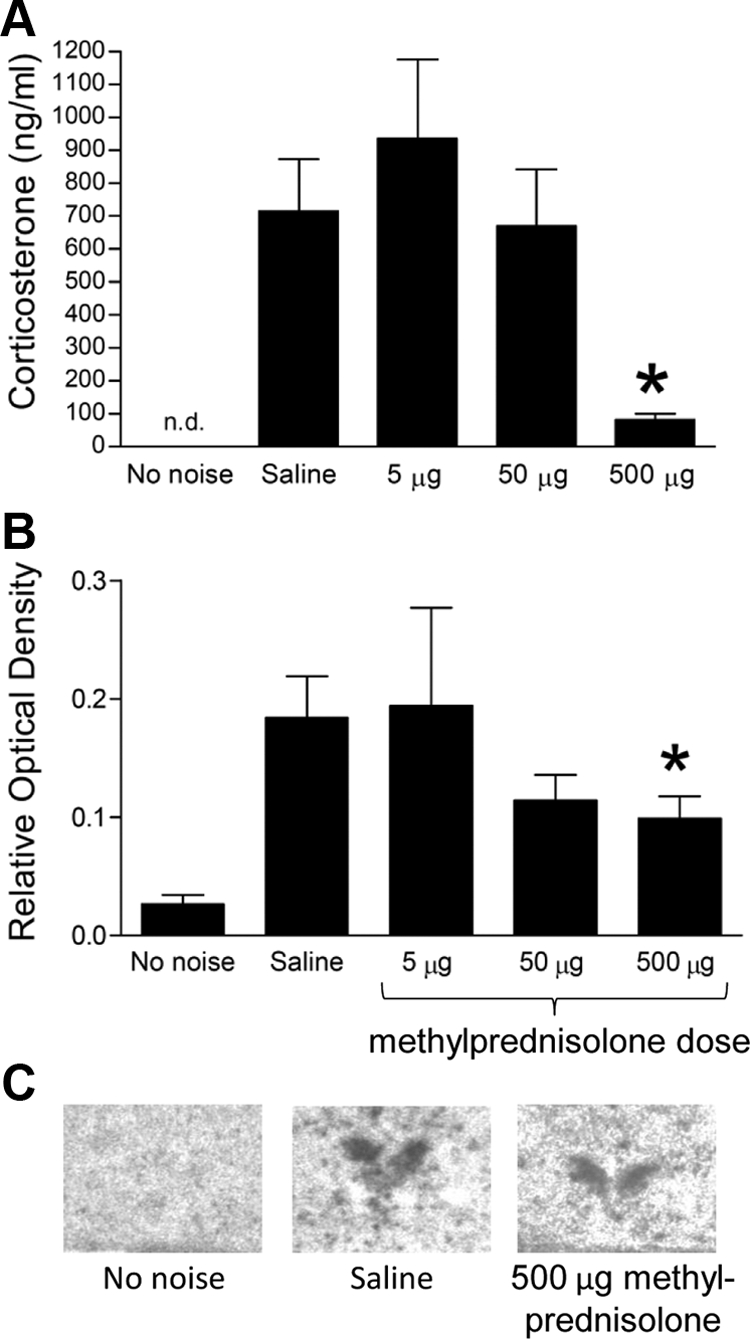
Glucocorticoid-induced suppression of HPA and c-fos mRNA response to noise stress. A, Injection of methylprednisolone (5–500 μg) 15 min before a 30-min noise stress significantly attenuated corticosterone levels measured from trunk blood samples obtained immediately after the end of the noise. *, P < 0.001, 500-μg dose vs. either saline or 5-μg dose (one-way ANOVA followed by post hoc Tukey's test). Corticosterone levels in animals receiving no noise were not determined (n.d.). B, Expression of c-fos mRNA in the hypothalamic PVN of animals receiving either saline or an acute dose of methylprednisolone (5–500 mg) 15 min before a 30-min noise stress. A second control group (no noise) is also included. *, P < 0.05, saline vs. 5 mg (one-way ANOVA followed by post hoc Tukey's test). C, Example autoradiograms of the PVN region in three treatment groups.
Discussion
These data provide a detailed characterization of glucocorticoid-induced rapid suppression of basal (nonstimulated) HPA activity in the freely behaving rat. They build on our initial observations of rapid HPA suppression with methylprednisolone (8, 9, 23) and show that this phenomenon involves GR activation and is mediated, at least in part, through a central site of action, because pituitary-adrenal responses can be reactivated by exogenous CRH when both spontaneous (basal) and stress-induced HPA activity are suppressed. The data support the evidence of corticosteroid-induced rapid suppression of HPA activity in humans (10, 29–34) and suggest that observations of a finite temporal delay between corticosteroid administration and onset of the suppressive response may arise from an initial increase in corticosterone secretion induced by high glucocorticoid concentrations.
Dynamics of rapid HPA responses to corticosteroids
Steroid negative feedback of the HPA axis is thought to occur as a result of plasma glucocorticoid concentrations rising to levels that exceed the range that is considered physiologically homeostatic. The present data demonstrate that an increase in GR activation (either by methylprednisolone or dexamethasone) leads to dose-dependent suppression of basal HPA axis activity. The effect lasts up to several hours and is reversible at lower doses but is subject to a finite delay, with the earliest time point when suppression is detected being 40 min after steroid injection.
Although rapid, rate-sensitive corticosteroid negative feedback was first reported during the late 1960s and 1970s (40–42; reviewed in Ref. 7), most studies involved indirect measures or examination of the effect of steroids on stimulated release. In respect of basal (unstimulated) HPA activity, bolus injection or infusion of a corticosteroid causes suppression of ACTH levels in anesthetized and adrenalectomized rats (2.56 or 5.12 μg corticosterone/100 g·min) (26) or dogs (bolus 292–317 μg/kg cortisol) (28), and similar effects have been seen in conscious and restrained intact dogs (3–22.5 μg cortisol/kg·min) (21). In all these cases, the suppression of ACTH levels occurred after a time delay of 20–40 min, which did not shorten with increasing steroid dose. More recently, we showed that injections of either methylprednisolone (8, 9) or aldosterone (9) have rapid suppressive actions on basal HPA activity in conscious rats. Using a single dose of methylprednisolone (250 μg), we showed that corticosterone levels significantly declined below preinjection baseline 33 min after steroid administration and remained low for 4–5 h (9). Using a higher dose of methylprednisolone (500 μg), we showed that plasma corticosterone levels significantly differed from saline-injected control animals 45–230 min after treatment (8). This is a similar duration to that measured in the current study (significant effect 80–275 min after injection).
In contrast to the limited animal data, several reports have described the effect of glucocorticoid administration on basal HPA activity in man. Infusions of cortisol have been shown to either have no effect (infusions of 6 and 12 mg/h cortisol for 2 h) (43) or an inhibitory effect (29–33) on plasma ACTH/β-lipotropin. For example, infusions of 2–6 mg/h of cortisol performed during the early part of the day (i.e. near the diurnal peak similar to the present study) caused a decline in ACTH levels after a delay of 30–45 min (29). Apparent recovery of ACTH secretion during constant infusion led to the suggestion that this was a rate-sensitive process, although persistent suppression at higher doses indicated it was also level dependent. This has been largely supported by subsequent studies. For example, in a group of eight normal subjects, Goodwin et al. (31) showed that a bolus infusion of 7 mg/kg cortisol caused suppression of ACTH levels with onset 15–60 min after the infusion. A similar result was found using a much lower dose of steroid (33); a fall in ACTH levels was detected 30–60 min after the start of a 2-h iv infusion of 15 mg cortisol. We recently showed that bolus injection of 10 mg methylprednisolone caused a significant decline in ACTH after 60 min and cortisol after 70 min when tested in the morning (i.e. during the diurnal acrophase) (10).
Measures of onset delay
It is important to note that to examine glucocorticoid-mediated suppression of basal HPA activity, the present studies were mostly performed during the late light phase (1630–1800 h) when basal levels of corticosterone rise in anticipation of the dark phase (in contrast to most other studies performed during the nadir of HPA activity). Under these conditions, measures from saline-treated animals showed corticosterone levels progressively changed over the sampling period, possibly due to the combined effects of the diurnal pattern and experimental procedures. Thus, to accurately resolve the temporal dynamics of glucocorticoid-induced suppression, each drug-induced response was normalized to the control (saline-treated) data and onset measured as a significant departure from 1. On this basis, the shortest delay between iv injection of a glucocorticoid agonist and a significant change in plasma corticosterone was 40 min (measured after injection of 50 μg methylprednisolone). This delay will be the combined time required for the inhibitory process to occur (effect) and the time for circulating corticosterone to be cleared (detection). The clearance half-life of corticosterone in the rat is very fast [measured here at <5 min compared with previous estimates by ourselves of 8.6 min (37) and others of 9.3 min (44)]; therefore, we suggest that much of this delay is due to the time for the inhibitory mechanism to occur. However, an unexpected finding was a dose-related rapid and transient increase in corticosterone that was most evident at the higher agonist doses (Figs. 2 and 3). This suggests that any lower-dose-associated HPA suppression occurring earlier than 40 min may have been masked by an opposing stimulation. This interaction between stimulatory and inhibitory effects of steroid injection might account for the finite delay in HPA suppression observed both in the present and earlier studies in animals (21, 26–28) and man (29, 33). Thus, although rapid negative corticosteroid feedback occurring within the 5 to 300 min time range has been incorporated into models of the dynamic basal activity of the HPA (18), it is clear from the present data that suppression of basal HPA activity by the selective GR agonists methylprednisolone and dexamethasone does not occur in this time frame. Although it is possible that ligand affinity for the p-glucoprotein (multidrug resistance protein 1) at the blood-brain barrier may have contributed to the slower dynamics of this response, the failure to shorten the onset delay even at high doses and the fact that the transport efficiency of p-glycoprotein for dexamethasone is the same as cortisol (45) suggests that this is not a factor masking rapid inhibition.
It is important to note that this apparent initial stimulation was not a nonspecific response to the injections, because it was both dose dependent and did not occur in animals treated with saline or 5 μg methylprednisolone. Furthermore, the low cross-reactivity of methylprednisolone and dexamethasone in our assay could not account for the large elevations in corticosterone-like immunoreactivity, particularly because the effect was most marked with dexamethasone, which displays no cross-reactivity. Interestingly, a study of normal human volunteers showed a trend for a similar immediate and transient increase in ACTH levels after injection of a dose of cortisol similar to our highest dose of methylprednisolone (7 mg/kg equivalent to 2 mg/rat) (31), an effect that is not seen with a much lower dose (25 mg bolus equivalent to 100 μg/rat) (34). One possible mechanism for this stimulation may be an interaction with CRH. In cultured equine pituitary cells, cortisol has been shown to inhibit ACTH release in conditions without CRH and have no effect with low levels of CRH (2 pm) but to potentiate ACTH release at higher (20 pm) CRH levels (46). Because endogenous CRH levels would be elevated at the time of injection as a drive to the diurnal rhythm of HPA activity, such an interaction might also occur in vivo.
GR involvement
Data obtained using various receptor ligands indicate that the rapid suppression of HPA activity is mediated through a GR-like mechanism. We have shown (9) that although the mineralocorticoid receptor (MR)-selective agonist aldosterone (250 μg) has a transient suppressive effect on corticosterone levels, injection of methylprednisolone (which has one tenth of the MR activity of aldosterone but >20-fold greater GR activity) (47) had a much greater and long-lasting effect, indicating that GR activation may play the predominant role in fast feedback mechanisms. This is borne out by the current data showing the effect of dexamethasone that, although having similar MR activity to methylprednisolone, has a 6- to 12-fold greater GR potency (47) and was approximately 10-fold more effective at suppressing HPA activity. Furthermore, the GR antagonist RU38486 attenuated the effects of methylprednisolone and at high dose displayed evidence of a stimulation, suggesting that GR activation by endogenous corticosteroids may have a role in regulating HPA activity during the diurnal acrophase. That rapid suppression is a GR-mediated phenomenon is consistent with our studies in man (10), which showed that the MR antagonist spironolactone did not affect the suppression of ACTH by methylprednisolone (10 mg/subject), whereas RU38486 attenuated this effect. There are few other data on the fast actions of GR-selective ligands on basal HPA activity with which to compare. However, iv injection of 15 μg/kg dexamethasone in conscious cats has been shown to cause marked suppression of cortisol 2 h later (the first time point measured) (48), and sc injection of 25 or 50 μg/kg dexamethasone to rats significantly reduces corticosterone 90 min later (11). Interestingly, despite being more potent than methylprednisolone, dexamethasone showed a similar finite minimal delay to inhibition (∼40 min) and a similar rapid and transient stimulation, again supporting the existence of a rapid GR-mediated activation. It is, however, important to be aware of the caveat that both methylprednisolone and dexamethasone have markedly different effects on GR-DNA binding dynamics to native hormones cortisol and corticosterone (49), and this is very likely to contribute to the biological effects of these ligands. Furthermore, the possible importance of cooperativity between MR and GR, and even the potential importance of MR-GR heterodimerization remains unexplored.
Site of fast inhibitory actions
Rat studies have shown attenuation of the HPA response when corticosteroids are peripherally administered shortly before exposure to a variety stressors, e.g. restraint (14, 50), air puff startle (24), insulin-induced hypoglycemia (21), and photic and acoustic stressors (51), although not to others, e.g. hemorrhage (24). The present protocol (injection of 500 μg methylprednisolone 15 min before a 30-min noise stress) is similar to that which we used previously to examine attenuation of stress-induced HPA activity (2000 μg administered 40 min before a 10-min noise stress) (23) and confirms that inhibitory mechanisms are activated within a rapid time frame. Likewise, Ginsberg et al. (14) showed that treatment with corticosterone (5 mg/kg) or a GR agonist RU28362 (150 μg/kg) 1 h before restraint attenuates the neuroendocrine response. The present data show that although acute glucocorticoid administration suppressed corticosterone secretion, injection of exogenous CRH was able to reactivate cortisol release, even at the peak of corticosteroid-induced suppression. The fact that exogenous CRH was ineffective before steroid administration (presumably due to the already high endogenous CRH drive during the diurnal acrophase) but was effective once the system was inhibited is consistent with a central site of action. Furthermore, methylprednisolone blocked stress-induced corticosterone release and PVN activation (measured by levels of c-fos mRNA) but not the response to CRH. These data clearly indicate a suprapituitary site of action and suggest either that the limited brain penetration of prednisolone demonstrated in the mouse (52) does not operate in the rat or that the amount of glucocorticoid that enters is sufficient to have a functional effect.
Although glucocorticoid feedback may operate at several sites, including hippocampus, a PVN site of action is supported by recent evidence of rapid suppression of restraint-induced HPA activity after direct intra-PVN injection of dexamethasone (53), although in this study, stress-induced Fos expression was unaffected. Other studies have also shown that restraint-induced c-fos mRNA or Fos protein induction in the PVN are unaffected by acute GR agonist treatment but noted that CRH heteronuclear RNA induction was blocked (14, 50). Compared with the present data, these differences in the effect of acute corticosteroids on Fos may be due to the use of 30 min restraint compared with the relatively mild stress of 30 min noise used here. Alternatively, it may be that under certain conditions, the PVN may still be activated, and the target for steroid feedback is the pituitary. In this regard, Hinz and Hirschelmann (12) showed that CRH-induced ACTH secretion could be attenuated by iv treatment of rats with a range of glucocorticoid agonists (cortisol, corticosterone, RU28362, and dexamethasone) administered at the same time as the CRH injection. Interestingly, this suppressive action was unaffected by previous treatment with 250-fold excess of RU38486 given 45 min before the steroid (12), suggesting that this pituitary effect differs from the GR-like effect we report. Although it is possible that endogenous corticosterone has additional effects to those of the selective GR agonists used here, studies of rapid effects of glucocorticoids have indicated that dexamethasone has similar effects to cortisol (12) and is able to rapidly block stress-induced HPA activity when injected centrally (53). The site of rapid glucocorticoid feedback may also be species specific. In man, we recently showed that a dose of 10 mg methylprednisolone (equivalent to about 100 μg/kg) not only suppressed basal ACTH and cortisol but also completely blocked the response to 1 μg/kg CRH (10), indicating that one site of action for rapid negative feedback was the pituitary. In contrast, the present data show that a considerably higher dose of methylprednisolone (500 μg, equivalent to about 1500 μg/kg) despite completely suppressing corticosterone secretion failed to block the response to exogenous CRH. It will be interesting to explore the mechanisms underlying this difference in pituitary sensitivity.
Acknowledgments
This work was supported by Wellcome Trust Program Grant 051846 to S.L.L. and C.D.I. and Wellcome Trust Prize Studentship 055348 to M.H.A.
Disclosure Summary: M.H.A., S.A.W., R.J.W., S.L.L., and C.D.I. have nothing to declare.
Footnotes
- GR
- Glucocorticoid receptor
- HPA
- hypothalamo-pituitary-adrenal
- MR
- mineralocorticoid receptor
- PVN
- paraventricular nucleus.
References
- 1. Jingami H, Matsukura S, Numa S, Imura H. 1985. Effects of adrenalectomy and dexamethasone administration on the level of prepro-corticotropin-releasing factor messenger ribonucleic acid (mRNA) in the hypothalamus and adrenocorticotropin/β-lipotropin precursor mRNA in the pituitary in rats. Endocrinology 117:1314–1320 [DOI] [PubMed] [Google Scholar]
- 2. Kovács KJ, Mezey E. 1987. Dexamethasone inhibits corticotropin-releasing factor gene expression in the rat paraventricular nucleus. Neuroendocrinology 46:365–368 [DOI] [PubMed] [Google Scholar]
- 3. Drouin J, Sun YL, Nemer M. 1989. Glucocorticoid repression of pro-opiomelanocortin gene transcription. J Steroid Biochem 34:63–69 [DOI] [PubMed] [Google Scholar]
- 4. Lightman SL, Young WS., 3rd 1989. Influence of steroids on the hypothalamic corticotropin-releasing factor and preproenkephalin mRNA responses to stress. Proc Natl Acad Sci USA 86:4306–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakai Y, Usui T, Tsukada T, Takahashi H, Fukata J, Fukushima M, Senoo K, Imura H. 1991. Molecular mechanisms of glucocorticoid inhibition of human proopiomelanocortin gene transcription. J Steroid Biochem Mol Biol 40:301–306 [DOI] [PubMed] [Google Scholar]
- 6. Ma XM, Aguilera G. 1999. Differential regulation of corticotropin-releasing hormone and vasopressin transcription by glucocorticoids. Endocrinology 140:5642–5650 [DOI] [PubMed] [Google Scholar]
- 7. Dallman MF. 2005. Fast glucocorticoid actions on brain: back to the future. Front Neuroendocrinol 26:103–108 [DOI] [PubMed] [Google Scholar]
- 8. Spiga F, Harrison LR, Wood SA, MacSweeney CP, Thomson FJ, Craighead M, Grassie M, Lightman SL. 2008. Effect of the glucocorticoid receptor antagonist Org 34850 on fast and delayed feedback of corticosterone release. J Endocrinol 196:323–330 [DOI] [PubMed] [Google Scholar]
- 9. Atkinson HC, Wood SA, Castrique ES, Kershaw YM, Wiles CCR, Lightman SL. 2008. Corticosteroids mediate fast feedback of the rat hypothalamic-pituitary-adrenal axis via the mineralocorticoid receptor. Am J Physiol 294:E1011–E1022 [DOI] [PubMed] [Google Scholar]
- 10. Russell GM, Henley DE, Leendertz J, Douthwaite JA, Wood SA, Stevens A, Woltersdorf WW, Peeters BWMM, Ruigt GSF, White A, Veldhuis JD, Lightman SL. 2010. Rapid glucocorticoid receptor-mediated inhibition of hypothalamic-pituitary-adrenal ultradian activity in healthy males. J Neurosci 30:6106–6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cole MA, Kim PJ, Kalman BA, Spencer RL. 2000. Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology 25:151–167 [DOI] [PubMed] [Google Scholar]
- 12. Hinz B, Hirschelmann R. 2000. Rapid non-genomic feedback effects of glucocorticoids on CRF-induced ACTH secretion in rats. Pharm Res 17:1273–1277 [DOI] [PubMed] [Google Scholar]
- 13. Sapolsky RM, Krey LC, McEwen BS. 1984. Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc Natl Acad Sci USA 81:6174–6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ginsberg AB, Campeau S, Day HE, Spencer RL. 2003. Acute glucocorticoid pretreatment suppresses stress-induced hypothalamic-pituitary-adrenal axis hormone secretion and expression of corticotropin-releasing hormone hnRNA but does not affect c-fos mRNA or Fos protein expression in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol 15:1075–1083 [DOI] [PubMed] [Google Scholar]
- 15. Jacobson L, Sapolsky R. 1991. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev 12:118–134 [DOI] [PubMed] [Google Scholar]
- 16. Feldman S, Weidenfeld J. 1995. Neural mechanisms involved in the corticosteroid feedback effects on the hypothalamo-pituitary-adrenocortical axis. Prog Neurobiol 45:129–141 [DOI] [PubMed] [Google Scholar]
- 17. Herman JP, Ostrander MM, Mueller NK, Figueiredo H. 2005. Limbic system mechanisms of stress regulation: Hypothalamo-pituitary-adrenocortical axis. Prog Neuro-Psychopharmacol Biol Psychiatry 29:1201–1213 [DOI] [PubMed] [Google Scholar]
- 18. Keenan DM, Licinio J, Veldhuis JD. 2001. A feedback-controlled ensemble model of the stress-responsive hypothalamo-pituitary-adrenal axis. Proc Natl Acad Sci USA 98:4028–4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Walker JJ, Terry JR, Lightman SL. 2010. Origin of ultradian pulsatility in the hypothalamic-pituitary-adrenal axis. Proc R Soc B 277:1627–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abe K, Critchlow V. 1977. Effects of corticosterone, dexamethasone and surgical isolation of the medial basal hypothalamus on rapid feedback control of stress-induced corticotropin secretion in female rats. Endocrinology 101:498–505 [DOI] [PubMed] [Google Scholar]
- 21. Keller-Wood M. 1990. Fast feedback control of canine corticotropin by cortisol. Endocrinology 126:1959–1966 [DOI] [PubMed] [Google Scholar]
- 22. Young EA, Akana S, Dallman MF. 1990. Decreased sensitivity to glucocorticoid fast feedback in chronically stressed rats. Neuroendocrinology 51:536–542 [DOI] [PubMed] [Google Scholar]
- 23. Nolan LA, Windle RJ, Wood SA, Kershaw YM, Lunness HR, Lightman SL, Ingram CD, Levy A. 2000. Chronic iodine deprivation attenuates stress-induced and diurnal variation in corticosterone secretion in female Wistar rats. J Neuroendocrinol 12:1149–1159 [DOI] [PubMed] [Google Scholar]
- 24. Thrivikraman KV, Nemeroff CB, Plotsky PM. 2000. Sensitivity of glucocorticoid-mediated fast-feedback regulation of the hypothalamic-pituitary-adrenal axis is dependent upon stressor specific neurocircuitry. Brain Res 870:87–101 [DOI] [PubMed] [Google Scholar]
- 25. Spiga F, Harrison LR, Wood SA, Atkinson HC, MacSweeney CP, Thomson F, Craighead M, Grassie M, Lightman SL. 2007. Effect of the glucocorticoid receptor antagonist Org 34850 on basal and stress-induced corticosterone secretion. J Neuroendocrinol 19:891–900 [DOI] [PubMed] [Google Scholar]
- 26. Rotsztejn W, Lalonde J, Normand M, Fortier C. 1975. Feedback inhibition of adrenocorticotropin release by corticosterone infusions in the adrenalectomised rat. Can J Physiol Pharmacol 53:475–478 [DOI] [PubMed] [Google Scholar]
- 27. Cowan JS, Windle WJ. 1978. Progressive suppression of adrenocorticotropin secretion in resting adrenalectomized dogs by low stepwise infusions of cortisol. Endocrinology 103:1173–1182 [DOI] [PubMed] [Google Scholar]
- 28. Cowan JS, Layberry RA. 1983. Feedback suppression of ACTH secretion by cortisol in dogs: Lags after large signals equal those following very small signals. Can J Physiol Pharmacol 61:1281–1288 [DOI] [PubMed] [Google Scholar]
- 29. Reader SC, Alaghband-Zadeh J, Daly JR, Robertson WR. 1982. Negative, rate-sensitive feedback effects on adrenocorticotrophin secretion by cortisol in normal subjects. J Endocrinol 92:443–448 [DOI] [PubMed] [Google Scholar]
- 30. Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. 1991. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry 48:693–699 [DOI] [PubMed] [Google Scholar]
- 31. Goodwin GM, Muir WJ, Seckl JR, Bennie J, Carroll S, Dick H, Fink G. 1992. The effects of cortisol infusion upon hormone secretion from the anterior pituitary and subjective mood in depressive illness and in controls. J Affect Disord 26:73–83 [DOI] [PubMed] [Google Scholar]
- 32. Kellner M, Holsboer F, Heuser I. 1995. Intermediate glucocorticoid feedback of corticotropin secretion in patients with major depression. Psychiatry Res 59:157–160 [DOI] [PubMed] [Google Scholar]
- 33. Posener JA, Schildkraut JJ, Williams GH, Schatzberg AF. 1997. Cortisol feedback effects on plasma corticotropin levels in healthy subjects. Psychoneuroendocrinology 22:169–176 [DOI] [PubMed] [Google Scholar]
- 34. Boscaro M, Paoletta A, Scarpa E, Barzon L, Fusaro P, Fallo F, Sonino N. 1998. Age-related changes in glucocorticoid fast feedback inhibition of adrenocorticotropin in man. J Clin Endocrinol Metab 83:1380–1383 [DOI] [PubMed] [Google Scholar]
- 35. Boudinot FD, Jusko WJ. 1986. Dose-dependent pharmacokinetics of prednisolone in normal and adrenalectomized rats. J Pharmacokinet Biopharm 14:453–467 [DOI] [PubMed] [Google Scholar]
- 36. Seale JV, Wood SA, Atkinson HC, Bate E, Lightman SL, Ingram CD, Jessop DS, Harbuz MS. 2004. Gonadectomy reverses the sexually diergic patterns of circadian and stress-induced hypothalamic-pituitary-adrenal axis activity in male and female rats. J Neuroendocrinol 16:516–524 [DOI] [PubMed] [Google Scholar]
- 37. Windle RJ, Wood SA, Shanks N, Lightman SL, Ingram CD. 1998. Ultradian rhythm of basal corticosterone release in the female rat: dynamic interaction with the response to acute stress. Endocrinology 139:443–450 [DOI] [PubMed] [Google Scholar]
- 38. Lightman SL, Windle RJ, Julian MD, Harbuz MS, Shanks N, Wood SA, Kershaw YM, Ingram CD. 2000. Significance of pulsatility in the HPA axis. Novartis Foundation Symp 227:244–257 [DOI] [PubMed] [Google Scholar]
- 39. Windle RJ, Wood SA, Kershaw YM, Lightman SL, Ingram CD. 2010. Reduced stress responsiveness in pregnancy: Relationship with pattern of forebrain c-fos mRNA expression. Brain Res 1358:102–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones MT, Brush FR, Neame RL. 1972. Characteristics of fast feedback control of corticotrophin release by corticosteroids. J Endocrinol 55:489–497 [DOI] [PubMed] [Google Scholar]
- 41. Jones MT, Hillhouse EW, Burden JL. 1977. Dynamics and mechanisms of corticosteroid feedback at the hypothalamus and anterior pituitary gland. J Endocrinol 73:405–417 [DOI] [PubMed] [Google Scholar]
- 42. Kaneko M, Hiroshige T. 1978. Fast, rate-sensitive corticosteroid negative feedback during stress. Am J Physiol 234:R39–R45 [DOI] [PubMed] [Google Scholar]
- 43. Krishnan KR, Ritchie JC, Manepalli AN, Saunders W, Li SW, Venkataraman S, Nemeroff CB, Carroll BJ. 1991. Fast feedback regulation of ACTH by cortisol. Prog Neuropsychopharmacol Biol Psychiatry 15:523–529 [DOI] [PubMed] [Google Scholar]
- 44. Hess GD, Riegle GD. 1972. Effects of chronic ACTH stimulation on adrenocortical function in young and aged rats. Am J Physiol 222:1458–1461 [DOI] [PubMed] [Google Scholar]
- 45. Yates CR, Chang C, Kearbey JD, Yasuda K, Schuetz EG, Miller DD, Dalton JT, Swaan PW. 2003. Structural determinants of P-glycoprotein-mediated transport of glucocorticoids. Pharm Res 20:1794–1803 [DOI] [PubMed] [Google Scholar]
- 46. Livesey JH, Evans MJ, Mulligan R, Donald RA. 2000. Interactions of CRH, AVP and cortisol in the secretion of ACTH from perifused equine anterior pituitary cells: “permissive” roles for cortisol and CRH. Endocr Res 26:445–463 [DOI] [PubMed] [Google Scholar]
- 47. Grossmann C, Scholz T, Rochel M, Bumke-Vogt C, Oelkers W, Pfeiffer AF, Diederich S, Bahr V. 2004. Transactivation via the human glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV-1 cells: a comparison of their glucocorticoid and mineralocorticoid properties. Eur J Endocrinol 151:397–406 [DOI] [PubMed] [Google Scholar]
- 48. Peterson ME, Graves TK. 1988. Effects of low dosages of intravenous dexamethasone on serum cortisol concentrations in the normal cat. Res Vet Sci 44:38–40 [PubMed] [Google Scholar]
- 49. Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. 2009. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol 11:1093–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Osterlund C, Spencer RL. 2011. Corticosterone pretreatment suppresses stress-induced hypothalamic-pituitary-adrenal axis activity via multiple actions that vary with time, site of action and de novo protein synthesis. J Endocrinol 208:311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weidenfeld J, Feldman S. 1993. Glucocorticoid feedback regulation of adrenocortical responses to neural stimuli: role of CRF-41 and corticosteroid type I and type II receptors. Neuroendocrinology 58:49–56 [DOI] [PubMed] [Google Scholar]
- 52. Karssen AM, Meijer OC, van der Sandt IC, De Boer AG, De Lange EC, De Kloet ER. 2002. The role of the efflux transporter P-glycoprotein in brain penetrance of prednisolone. J Endocrinol 175:251–260 [DOI] [PubMed] [Google Scholar]
- 53. Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. 2010. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signalling. Endocrinology 151:4811–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]