

Abstract
Homoenolate equivalents are generated by Lewis basic N-heterocyclic carbene catalysts and then protonated to generate efficiently saturated esters from unsaturated aldehydes. This reactivity is extended to the generation of β-acylvinyl anions from alkynyl aldehydes. The asymmetric protonation of a homoenolate equivalent generated from a β,β-disubstituted aldehyde can be accomplished with a chiral N-heterocyclic carbene.
Keywords: carbenes, umpolung, protonations, asymmetric catalysis, esters
Introduction
The discovery and understanding of unconventional reactivity patterns for the construction of organic molecules is an important goal. These advances can greatly facilitate the rapid access to a wide range of molecular targets. Research to promote or catalyze the reversal of a functional group’s polarity, known as umpolung,1–3 has undergone a renaissance, and can provide unusual routes to desirable compounds. Over the last decade, N-heterocyclic carbenes (NHCs) have attracted a growing interest as catalysts or reagents for an expanding set of reactions.4–6 This unique class of Lewis basic catalyst7 has long been known to catalyze the benzoin8,9 and Stetter10,11 reactions by converting an electrophilic carbonyl functional group, such as an aldehyde,12–14 α-keto acid,15,16 or acylsilane,17–19 into nucleophilic acyl anion equivalents.
In a key study reported in 1958,9 Breslow proposed that the intermediate in the thiazolium-catalyzed benzoin reaction was an unusually substituted alkene A (Scheme 1) resulting from the addition of the azolium to the aldehyde. After proton migration, what was the carbonyl carbon of this neutral species is rendered nucleophilic. Our hypothesis in 2004 was that this electron density could be transported to a distal position by virtue of unsaturation (i.e., an alkene or alkyne).20 If successful, this would generate a homoenolate B (Scheme 1) or β-acylvinyl anion reactivity pattern, respectively. Since that time, it has been shown that these unusual nucleophiles can react with a variety of electrophiles.21–30
Scheme 1.

Homoenolate generation and protonation with NHC catalysis
Here we describe the development of our first NHC-catalyzed process, namely the protonation of homoenolate equivalents. This protonation accomplishes an internal redox reaction, wherein the carbon–carbon π-bond of an α,β-unsaturated aldehyde is reduced and the carbonyl is oxidized to yield a saturated ester. To further our initial examination of this interesting reactivity pattern, our efforts to render this protonation enantioselective are also described.
Homoenolate Equivalents Generated by N-Heterocyclic Carbenes (NHCs)
Our investigation of this reactivity began with the protonation of the homoenolate equivalent obtained from cinnamaldehyde31 catalyzed by the carbene derived from benzimidazolium salt 1 (Table 1). Although unnecessary for the generation of the protonated product,32 higher yields were observed with the addition of a Brønsted acid, such as phenol (pKa = 18.0 in DMSO),33 as an additional proton source in conjunction with a nucleophilic alcohol to form the ester and regenerate the carbene catalyst. Although phenol is ostensibly the proton source, it is interesting to note that the homoenolate equivalent is most likely deprotonating the conjugate acid of the base generated through deprotonation of the carbene precursor (in this case DBU, pKa = 13.9 in DMSO).34 The optimal conditions for this reaction demonstrate an important balance of the ability to generate the NHC catalyst through deprotonation of the benzimidazolium salt (pKa = 18.6 in DMSO)35 and the acidity needed to protonate the homoenolate equivalent.
Table 1.
Internal Redox Leading to Saturated Estersa
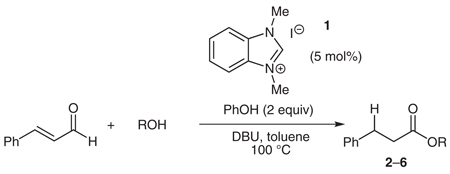 | |||
|---|---|---|---|
| Entry | Product | Yieldb (%) | |
| 1 | 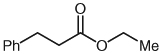 |
2 | 72 |
| 2 | 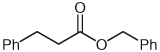 |
3 | 82 |
| 3 | 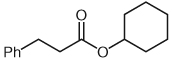 |
4 | 57 |
| 4 | 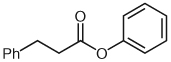 |
5 | 56 |
| 5 | 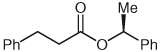 |
6 | 77 |
Reaction conditions: 1 (0.04 mmol), DBU (0.04 mmol), cinnamal-dehyde (0.79 mmol), PhOH (1.6 mmol), ROH (4.0 mmol), toluene (1.6 mmol), 100 °C, 2–6 h.
Isolated yields.
Separate reports by Glorius and co-workers and Bode and co-workers in 2004 described the dimerization of aldehydes in the presence of NHCs through homoenolate addition to form γ-butyrolactones.21,22 Under our homoenolate protonation conditions, none of these undesired products are observed, and formation of the saturated ester is the dominant reaction pathway. Variation of the nucleophilic alcohol allows practical access to a variety of esters (Table 1). Primary, secondary, and aromatic alcohols are all competent nucleophiles in the NHC-catalyzed reaction. Not surprisingly, stereochemical information is preserved in the case when a chiral secondary alcohol is used as the nucleophile to form the ester product 6 (Table 1, entry 5).
Several unsaturated aldehydes are also functional substrates in this reaction, and a variety of benzyl esters can be synthesized by this method (Table 2). Unsaturated aldehydes with alkyl or electron-rich aryl substituents successfully undergo the protonation/acylation sequence. Despite concerns about steric interactions of substituents in the α-position of the aldehyde with the NHC catalyst upon addition to the carbonyl, no significant decrease in yield is observed when an α-substituted aldehyde is employed (Table 2, entry 3). However, elevated temperatures and longer reactions times (2 days) are required. Notably, β,β-disubstituted unsaturated aldehydes (Table 2, entries 5, 6) are also capable of being protonated under these conditions, which provides a natural inspiration for asymmetric variants of this reaction (vide infra).
Table 2.
Reaction of Various Aldehydes with Benzyl Alcohola
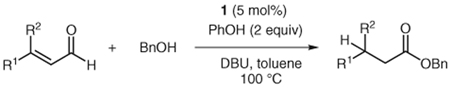 | ||||
|---|---|---|---|---|
| Entry | Aldehyde | Product | Yield (%) | |
| 1 |  |
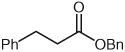 |
3 | 82 |
| 2 | 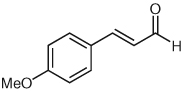 |
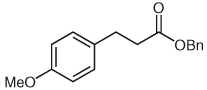 |
7 | 76 |
| 3 | 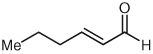 |
 |
8 | 90 |
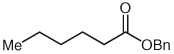
| 4 | 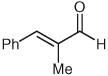 |
 |
9 | 82b |
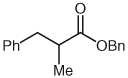
| 5 | 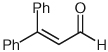 |
 |
10 | 86 |
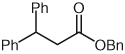
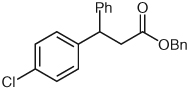
| 6 | 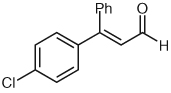 |
 |
11 | 82 |
Reaction conditions: 1 (0.04 mmol), DBU (0.04 mmol), cinnamaldehyde (0.79 mmol), PhOH (1.6 mmol), BnOH (4.0 mmol), toluene (1.6 mmol), 100 °C, 2–6 h.
100 °C, 2 d.
A plausible pathway for this reaction is to proceed first by the addition of the carbene to the carbonyl of the unsaturated aldehyde system (Scheme 2). After intermolecular proton transfer, the homoenolate equivalent III is formed. The β-position, which is now nucleophilic by virtue of being electronically coupled to the benzimidazole ring system, is protonated, most likely by the conjugate acid of the base (DBU). Tautomerization produces an acyl-azolium intermediate IV, which acts as an acylating agent for the nucleophilic alcohol within the reaction. This acylation gives the ester product and regenerates the carbene catalyst. The proton from the acylated alcohol regenerates the protonated base, which is then available to protonate another homoenolate equivalent.
Scheme 2.

Reaction pathway for homoenolate protonation
Evidence for the acyl-azolium intermediate IV as the acylating agent is provided through the kinetic resolution of racemic 1-phenylethanol (Scheme 3). The chiral benzimidazolium salt 12 first described by Diver et al.36 creates a chiral acylating agent similar to intermediate IV. Selective acylation of one enantiomer of the secondary alcohol is accomplished, resulting in an s factor of 4.8 at 40% conversion.31 This selective reactivity induced by the use of a chiral NHC demonstrates the ability of these catalysts to create a chiral environment in their reactions with aldehydes.
Scheme 3.

Kinetic resolution of 1-phenylethanol
Generation of β-Acylvinyl Anions
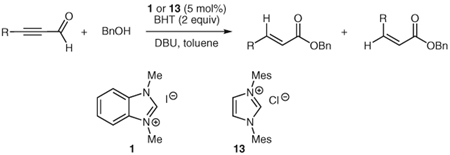
Through a similar pathway, the partial reduction of a triple bond can be conceived. Investigations into this internal redox reaction were carried out by the generation of β-acylvinyl anions through the reaction of NHC catalysts with alk-2-ynals. In this manner, alk-2-ynals can be converted into α,β-unsaturated esters (Table 3), compounds that hold further potential for functionalization through conjugate addition or other synthetic manipulations. Experiments in our laboratories demonstrated this reactivity to be feasible, although limited to aryl substrates or alk-2-ynals with no enolizable protons (Table 3). These results are congruous with those reported in 2005 by Zeitler.37 Control over the double bond stereochemistry is possible through judicious choice of catalyst precursor. In the presence of 2,6-di-tert-butyl-4-methylphenol (BHT), with 1,8-diazabicyclo[5.4.0]undec-7-ene as the base, the benz-imidazolium salt 1 employed in the initial investigations of this reactivity gives a mixture of isomers (Table 3, entries 1, 3, 5) at 100 °C, while the imidazolium salt, 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride (IMes, 13] catalyzes the reaction at lower temperatures (45 °C) than previously observed by Zeitler, and selectively affords the E-isomer (Table 3, entries 2, 4). This marked difference in reactivity and selectivity underscores the profound effect that subtle changes to the balance of acids and bases can have in this catalytic system [the pKa of imidazolium salts similar to IMes (13) have been determined through theoretical calculations to be ~17.0 in DMSO].38
Table 3.
Protonation of β-Acylvinyl Anionsa
 | ||||
|---|---|---|---|---|
| Entry | Aldehyde | Catalyst | Yieldb (%) | RatiocE/Z |
| 1 | 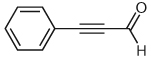 |
1 | 65 | 3:1 |
| 2 | 13 | 67 | >20:1 | |
| 3 | 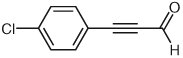 |
1 | 56 | 3:1 |
| 4 | 13 | 59 | >20:1 | |
| 5 | 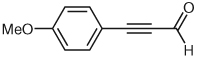 |
1 | 10 | 4:1 |
| 6 | 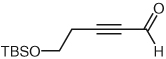 |
1 | 0 | – |
Reaction conditions: step 1: 1 or 13 (0.04 mmol), DBU (0.04 mmol), toluene, 23 °C, 10 min, step 2: addition of aldehyde (0.43 mmol), BHT (0.85 mmol), BnOH (2.2 mmol), 100 °C (for 1) or 45 °C (for 13), 12–36 h.
Isolated yields.
Determined by 500 MHz 1H NMR spectroscopy.
Stereoselective Protonation of Homoenolate Equivalents
One of the significant advantages of the use of NHC catalysts over traditional Lewis base catalysts, such as cyanide,39–41 is the availability of enantiopure chiral NHC precursors for stereoselective reactions.42–45 Chiral NHCs have been successfully applied to the asymmetric protonation of the α-position of α,α-dichloroaldehydes,46 but to date we are unaware of any asymmetric protonation of homoenolate equivalents generated by NHCs. The protonation of these reactive intermediates appeared to be a challenging manifold for asymmetric synthesis due to (a) the small nature of the electrophile (i.e., proton) and (b) the distance between the catalyst’s reactive site (the carbonyl) and the newly formed stereogenic center at the β-position.
Due to this anticipated difficulty in projecting asymmetric induction to the β-position, several chiral bases [cinchona alkaloid derivatives, (−)-sparteine, or alkylated Simpkins’ base derivatives47] were employed with the goal that their conjugate acids would enantioselectively protonate the homoenolate equivalent. These and other chiral proton sources (e.g., BINOL48 or TADDOL49) were not selective when combined with an achiral catalyst such as IMes (13) or 1. Thus, our attempts to render this protonation of NHC-generated homoenolates stereoselective turned to chiral NHC catalysts (Figure 1). We began with a survey of chiral triazolium precatalysts. A range of selectivities are observed (Table 4) in the protonation of β-methylcin-namaldehyde (14), with the best catalysts 18 and 19 being derived from geminal diphenyl-substituted derivatives of the amino acids phenylalanine and tryptophan (Table 4, entries 4, 5).
Figure 1.

Chiral NHC precursors
Table 4.
Asymmetric Protonation of Homoenolate Equivalentsa
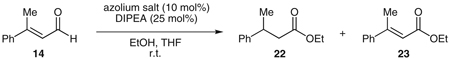 | |||
|---|---|---|---|
| Entry | Azolium salt | Yieldb (%) of 22 + 23 | ee (%) of 22 |
| 1 | 15 | 51 | 33 |
| 2 | 16 | –c | 9d |
| 3 | 17 | –c | 23d |
| 4 | 18 | 58 | 55 |
| 5 | 19 | 45 | 59 |
| 6 | 20 | 0 | – |
| 7 | 21 | 0 | – |
Reaction conditions: 15–21 (0.02 mmol), DIPEA (0.05 mmol), 14 (0.20 mmol), EtOH (0.41 mmol), THF (1.0 mL), r.t., 12–24 h.
Isolated yields.
Not isolated.
Assessed by HPLC of crude reaction mixture.
The relative success of these catalysts demonstrates the remote nature of the newly formed stereogenic center. As the size of the sidearm group of the NHC is increased, greater selectivity is observed (Table 4, entries 1, 4, 5). Geminal disubstitution with larger groups (entries 2–4) restricts the rotation of the blocking group and also projects it into the vicinity of the protonation site, increasing the selectivity.
The reaction yields two inseparable products, the desired saturated ester 22 and the unsaturated ester 23 resulting from the oxidation of the aldehyde. We were initially surprised to observe this oxidation product 23, which results from the Cannizzaro-like generation of a hydride equivalent from the aldehyde50–54 (Scheme 4, V to VII). The generation of a reducing equivalent oxidizes the NHC-aldehyde adduct to an acyl-azolium intermediate VII, which then acylates ethanol to generate the undesired side product. While we have utilized this type of reactivity in a beneficial manner for the reductions of activated ketones,51 the minimization of this pathway in these enantio-selective protonations has been problematic.
Scheme 4.

Generation of oxidized side product 23
The most detrimental aspect of this reduction pathway is that the NHC-catalyzed oxidation reduces another equivalent of the starting aldehyde. For this undesired reaction, two full equivalents of starting material aldehyde are consumed for every equivalent of the unsaturated ester generated. The inability thus far to eliminate this side reaction is the primary cause of the observed low yields. The partitioning of tetrahedral intermediates such as V between homoenolate equivalents (such as VI) and acyl azoliums (VII) is a fascinating aspect of carbene catalysis. The modulation of these outcomes by tuning the electronic and steric properties of potential catalysts should open new avenues of discovery using N-heterocyclic carbenes for both new bond-forming reactions and redox processes.
The protonation reactions using chiral azolium precata-lysts are carried out using ethanol without an additional proton source. The use of phenol or other exogenous proton source yields a lower ratio of homoenolate reactivity to oxidation without any improvement in enantioselectivity. N,N-Diisopropylethylamine (Hünig’s base) is used to generate the active carbene catalyst. Chiral imidazolium catalysts 2055 and 2156 were also surveyed but gave no homoenolate reactivity with this substrate.
Since our previous studies indicated that reactivity varied significantly with minor manipulation of the acid/base balance in the reaction, the role of the base was investigated in an effort to suppress the oxidation side product. A range of bases was surveyed with 10 mol% of NHC precursor 18 (Table 5). Using N,N-diisopropylethylamine at 25 mol% yields the best results (Table 5, entry 4), but still contains a significant amount of the undesired oxidation product. Although not the only factor determining the divergent reactivity, the pKa of the base in this reaction is clearly very important. Stronger bases yield more oxidation product (Table 5, entries 5, 6), while bases with a pKa in the range of 9 to 11 yield the best ratios of homoenolate to oxidation reactivity.
Table 5.
Variation of Base in Asymmetric Protonation of Homoenolatesa
| Entry | Base | pKa | Equiv or mol% | Ratiob 22/23 | ee (%) of 22 |
|---|---|---|---|---|---|
| 1 | 1-methyl-1H–imidazole | 7.0 | 1 | 23 only | – |
| 2 | N,N,α-trimethylbenzylamine | 8.9 | 40 mol% | 1:1 | 50 |
| 3 | (–)-sparteine | 10.1 | 40 mol% | 1.5:1 | 52 |
| 4 | DIPEA | 11.0 | 25 mol% | 3.4:1 | 55 |
| 5 | DBU | 12.0 | 40 mol% | 1:3 | 40 |
| 6 | t-BuOK | 17.0 | 40 mol% | 23 only | – |
Reaction conditions: 18 (0.02 mmol), base, 14 (0.20 mmol), EtOH (0.4 mmol), THF (1.0 mL), r.t., 12–24 h.
Ratios determined by 1H NMR of purified mixtures.
It is interesting to note that minimal variation in enantioselectivity is observed, even with the use of chiral bases such as (−)-sparteine. The weakest bases, such as 1-methyl-1H–imidazole (Table 5, entry 1), may be unable to generate the catalytic carbene in meaningful quantities, as these reactions were extremely sluggish and low yielding. These results reinforce the delicate nature of the balance of acids and bases involved in the generation of these homoenolate equivalents.
Efforts to eliminate the oxidation side product 23 by manipulating the electronic nature of intermediate I through the use of different substrates and NHC catalysts have not yet achieved an optimal process. However, we report the first asymmetric protonation of NHC-generated homoenolate equivalents in up to 59% ee with the use of chiral triazolium precatalyst 19 and β-methylcinnamaldehyde (14).
Conclusion
The internal redox reaction resulting from the protonation of homoenolate equivalents catalyzed by N-heterocyclic carbenes is a useful transformation with significant potential. In addition to the synthesis of saturated esters without using activating reagents or acid chlorides, this umpolung reactivity can be extended to the generation of β-acylvinyl anions in select cases. The enantioselective protonation of these intermediates is also possible, but under current conditions, its development is hampered by the generation of an oxidation side product through a Cannizzaro-like pathway. This promising reaction is also limited in selectivity (up to 59% ee) by the remote nature of the newly formed stereocenter. Current efforts are directed toward accessing new NHC precursors that will attenuate the generation of the undesired oxidized product, as well as the development of NHC-catalyzed oxidations and reductions inspired by this unexpected side reaction. From its biochemical origins and early benzoin and Stetter applications, the field of carbene catalysis is rapidly expanding and in doing so provides new avenues for reaction development.
All reactions were carried out under an N2 atmosphere in flame-dried glassware with magnetic stirring. THF, Et2O, CH2Cl2, DMF, and toluene were purified by passage through a bed of activated alumina.57 Reagents were purified prior to use unless otherwise stated following the guidelines of Perrin and Armarego.58 Purification of reaction products was carried out by flash chromatography using EM Reagent silica gel 60 (230–400 mesh). Analytical TLC was performed on EM Reagent 0.25 mm silica gel 60-F plates. Visualization was accomplished with UV light and anisaldehyde, CAN, KMnO4, or phosphomolybdic acid stain followed by heating. IR spectra were recorded on a Perkin Elmer 1600 series FT-IR spectro-photometer. 1H NMR spectra were recorded on a Varian Inova 500 (500 MHz) or Mercury 400 (400 MHz) spectrometer and are reported using solvent as an internal standard (CDCl3 at δ = 7.26). Proton-decoupled 13C NMR spectra were recorded on a Varian Inova 500 (125 MHz) or Mercury 400 (100 MHz) spectrometer and are reported using solvent as an internal standard (CDCl3 at δ = 77.0). MS data were obtained on a Varian 1200 Quadrupole Mass Spectrometer.
Homoenolate Equivalents Generated by NHCs
3-(4-Chlorophenyl)cinnamaldehyde was prepared according to Kirsch and co-workers.59,60 4-Chlorocinnamaldehyde was prepared according to a procedure analogous to Moloney and co-workers.61 4-Methoxycinnamaldehyde was purchased from Acros and the remaining aldehydes and IMes (13) were commercially available from Sigma-Aldrich and purified before use.
Benzyl 3-Phenylpropanoate (3); Typical Procedure
A screw-capped tube was charged with the benzimidazolium salt 1 (11 mg, 0.04 mmol) in a N2-filled dry box. The tube was removed from the box and placed under a positive pressure of N2. The tube was charged with toluene (1.0 mL), DBU (6 µL, 0.04 mmol), and lastly, distilled cinnamaldehyde (104 mg, 0.79 mmol). The mixture was allowed to stir for 5 min after which PhOH (150 mg, 1.6 mmol) in toluene (0.6 mL) was added via syringe followed by the addition of BnOH (429 mg, 4 mmol). The mixture was heated at 100 °C for 2–6 h until cinnamaldehyde was consumed (GC). The mixture was cooled to r.t., diluted with CH2Cl2 (20 mL) and washed with H2O (20 mL). The aqueous layer was washed with CH2Cl2 (3 × 30 mL) and the combined organic extracts were dried (anhyd Na2SO4), filtered, and concentrated in vacuo. The resulting residue was purified by flash column chromatography (silica gel).
Spectral data for compounds 2,62 3,63 4,64 5,65 and 866 matched those found in the literature.
(S)-1-Phenylethyl 3-Phenylpropanoate (6)
Yellow oil; yield: 155 mg (77%); Rf = 0.70 (Et2O–hexanes, 1:4).
IR (film): 3446, 3030, 2981, 2868, 1732, 1248 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.36–7.17 (m, 10 H), 5.89 (q, J = 6.7 Hz, 1 H), 2.96 (t, J = 7.3 Hz, 2 H), 2.66 (t, J = 5.7 Hz, 2 H), 1.51 (d, J = 6.7 Hz, 3 H).
13C NMR (125 MHz, CDCl3): δ = 172.4, 141.8, 140.7, 129.6, 128.8, 128.7, 128.5, 128.0, 126.4, 126.3, 36.3, 31.1, 22.4.
LR-MS (ACPI): m/z [M + H]+ calcd for C17H19O2: 255.3; found: 255.4.
Benzyl 3-(4-Methoxyphenyl)propanoate (7)
Light yellow oil; yield: 127 mg (76%); Rf = 0.6 (Et2O–hexanes, 1:4).
IR (film): 3448, 3033, 2951, 2835, 1734, 1247 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.40–7.30 (m, 5 H), 7.10 (d, J = 7.94 Hz, 2 H), 6.82 (d, J = 7.63 Hz, 2 H), 5.11 (s, 2 H), 3.79 (s, 3 H), 2.92 (t, J = 7.63 Hz, 2 H), 2.65 (t, J = 7.63 Hz, 2 H).
13C NMR (125 MHz, CDCl3): δ = 173.3, 158.5, 136.4, 132.9, 129.7, 129.0, 128.7, 128.6, 114.3, 66.7, 55.7, 36.7, 30.6.
LR-MS (APCI): m/z [M + H]+ calcd for C17H19O3: 271.3; found: 271.4.
Benzyl 2-Benzylpropanoate (9)
Light tan oil; yield: 150 mg (82%); Rf = 0.71 (EtOAc–hexanes, 1:4).
IR (film): 3448, 3064, 3030, 3974, 2936, 1735, 1245 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.35–7.14 (m, 10 H), 5.07 (s, 2 H), 3.04 (dd, J = 13.3, 7.0 Hz, 1 H), 2.80 (q, J = 7.3 Hz, 1 H), 2.69 (dd, J = 13.4, 7.3 Hz, 1 H), 1.18 (d, J = 6.7 Hz, 3 H).
13C NMR (125 MHz, CDCl3): δ = 176.4, 139.7, 136.5, 129.5, 129.0, 128.8, 128.6, 128.5, 126.8, 66.6. 42.0, 40.2, 17.3.
LRMS (APCI): m/z [M + H]+ calcd for C17H19O2: 255.3; found: 255.3.
Benzyl 3,3-Diphenylpropanoate (10)
Light yellow oil; yield: 113 mg (82%); Rf = 0.70 (Et2O–hexanes, 1:4).
IR (film): 3448, 3061, 3030, 2953, 2921, 1735, 1258 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.38–7.14 (m, 15 H), 5.02 (s, 2 H), 4.56 (t, J = 8.2 Hz, 1 H), 3.12 (d, J = 7.9 Hz, 2 H).
13C NMR (125 MHz, CDCl3): δ = 172.1, 143.8, 136.2, 129.6, 129.0, 128.9, 128.8, 128.7, 128.6, 128.5, 128.4, 128.1, 127.0, 66.8, 47.5, 41.3.
Benzyl 3-(4-Chlorophenyl)-3-phenylpropanoate (11)
Light yellow oil; yield: 94 mg (86%); Rf = 0.85 (CH2Cl2–hexanes, 5:1).
IR (film): 3449, 3030, 2955, 1735, 1254 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.31–7.14 (m, 14 H), 5.02 (s, 2 H), 4.54 (t, J = 7.9 Hz, 1 H), 3.08 (d, J = 7.7 Hz, 2 H).
13C NMR (125 MHz, CDCl3): δ = 171.9, 143.3, 142.2, 136.1, 132.8, 129.5, 129.2, 129.0, 128.7. 128.6, 128.0, 127.2, 114.6, 66.9, 46.9, 41.2.
Generation of β-Acylvinyl Anions
Phenylpropynal and 5-(tert-butyldimethylsiloxy)pent-2-ynal were prepared according to a procedure analogous to that of Kuroda et al.67 (4-Chlorophenyl)propynal and (4-methoxyphenyl)propynal were synthesized by a procedure analogous to that of Roy and Banerjee.68
Benzyl Prop-2-enoates (Table 3); General Procedure
A screw-capped tube was charged with IMes (13, 15 mg, 0.04 mmol) or benzimidazolium salt 1 (12 mg, 0.04 mmol) in an N2-filled dry box. The tube was removed from the box and placed under a positive pressure of N2. The tube was charged with toluene (1.5 mL) and DBU (6 µL, 0.04 mmol) and the mixture was allowed to stir at 23 °C for 10 min after which the aldehyde (0.43 mmol) and BHT (187 mg, 0.85 mmol, 2 equiv) were added, followed by the addition of BnOH (227 µL, 2.2 mmol, 5 equiv). The mixture was heated at 100 °C or 45 °C for 12–36 h until the aldehyde was consumed (TLC). The mixture was cooled to r.t., diluted with CH2Cl2 (20 mL) and washed with H2O (20 mL). The aqueous layer was washed with CH2Cl2 (3 × 20 mL) and the collected organic layers were dried (anhyd Na2SO4), filtered and concentrated in vacuo. The resulting residue was purified by flash column chromatography (silica gel).
Spectral data for benzyl cinnamate69 and benzyl 4-methoxy-cinnamate70 matched those found in the literature.
Benzyl 4-Chlorocinnamate
Orange solid; yield: 68 mg (59%); Rf = 0.64 (EtOAc–hexanes, 1:4).
IR (film): 3056, 3041, 2953, 1715, 1165, 822, 697 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.67 (d, J = 16.1 Hz, 1 H), 7.34– 7.46 (m, 9 H), 6.46 (d, J = 16.1 Hz, 1 H), 5.26 (s, 2 H).
13C NMR (125 MHz, CDCl3): δ = 166.7, 143.9, 136.4, 136.1, 133.0, 129.4, 129.3, 128.8, 128.5, 118.6, 66.7.
LRMS (APCI): m/z [M + H]+ calcd for C16H14ClO2: 272.1; found: 272.3.
Stereoselective Protonation of Homoenolate Equivalents
β-Methylcinnamaldehyde (14) was prepared according to Oh and co-workers.71 Triazolium catalysts were prepared by a route analogous to that of Rovis and co-workers.72 Imidazolium salts 2055 and 2156 were prepared according to procedures of Glorius and Hermann and co-workers.
Ethyl 3-Methylbutanoate (22) and Ethyl 3-Methylbut-2-enoate (23) (Table 4); General Procedure
A triazolium salt (0.02 mmol) was dissolved in THF (1.0 mL) followed by sequential addition of DIPEA (9 µL, 0.05 mmol), β-methylcinnamaldehyde (14, 30 mg, 0.20 mmol), and abs EtOH (24 µL, 0.41 mmol). The mixture was stirred at r.t. for 12–24 h and monitored (TLC, 30% Et2O–hexanes). Upon consumption of starting aldehyde, the mixture was diluted with CH2Cl2 and washed with H2O and brine. The organic layer was dried (Na2SO4) and evaporated. The residue was purified by flash chromatography (silica gel, 5% Et2O–hexanes) to give 22 as well as unsaturated ester 23 as an inseparable mixture. Enantioselectivity was assessed by chiral HPLC (OD-H column). Spectral data for both 22 and 23 were identical to those previously reported.
Acknowledgment
Financial support for this work was provided by NIGMS (RO1 GM73072), Abbott Laboratories, Amgen, AstraZeneca, Glaxo-SmithKline, the Sloan Foundation, and Boehringer-Ingelheim. B.E.M. was supported by a GAANN Fellowship and A.C. thanks Dow Chemical Company for a graduate fellowship. FMCLithium and Sigma-Aldrich provided generous reagent support. Funding for the NU Integrated Molecular Structure Education and Research Center (IMSERC) has been furnished in part by the NSF (CHE-9871268). We thank Dr. Steve Wittenberger, Professor Regan Thomson, and Eric M. Phillips for helpful discussions.
Biographies

Brooks Maki received a Bachelor of Arts degree in chemistry and mathematics from Gustavus Adolphus College in 2004. His graduate research, at Northwestern University under the direction of K. A. Scheidt, is focused on NHC-catalyzed redox reactions. His graduate studies have been funded in part by a GAANN fellowship from the U.S. Department of Education.

Audrey Chan obtained her degree in chemistry from Cornell University in 2002. After working at Merck, Rahway under Dr. Sheryl Devenham, she joined the Scheidt laboratory in 2003. Her graduate research is directed toward the discovery of new NHC-catalyzed reactions, including homoenolate protonation, homoenolate annulations with azomethine imines, homoenolate aminations, and carbene-catalyzed hydro-acylations. She is currently the Dow Chemical Company fellow at Northwestern.

Professor Karl Scheidt obtained his Ph.D. with William Roush from Indiana University in 1999. After an NIH postdoctoral fellowship under the direction of David A. Evans (Harvard University), he joined the faculty of Northwestern University (Evanston, IL, USA) in 2002. His research focuses on the development of new organic methodology and the synthesis of bioactive molecules. He is a recent Sloan Foundation Fellow and Novartis Lecturer as well as the recipient of awards from Abbott Laboratories, Amgen, AstraZeneca, Boehringer-Ingelheim, GlaxoSmith-Kline, and 3M.
References
- 1.Seebach D. Angew. Chem., Int. Ed. Engl. 1979;18:239. [Google Scholar]
- 2.Eisch JJ. J. Organomet. Chem. 1995;500:101. [Google Scholar]
- 3.Pohl M, Lingen B, Muller M. Chem. Eur. J. 2002;8:5289. doi: 10.1002/1521-3765(20021202)8:23<5288::AID-CHEM5288>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 4.Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]
- 5.Marion N, Diez-Gonzalez S, Nolan IP. Angew. Chem. Int. Ed. 2007;46:2988. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]
- 6.Zeitler K. Angew. Chem. Int. Ed. 2005;44:7506. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]
- 7.Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 8.Ugai T, Tanaka R, Dokawa T. J. Pharm. Soc. Jpn. 1943;63:296. [Google Scholar]
- 9.Breslow R. J. Am. Chem. Soc. 1958;80:3719. [Google Scholar]
- 10.Stetter H, Kuhlmann H. Chem. Ber. 1976;109:2890. [Google Scholar]
- 11.Enders D, Breuer K, Runsink J, Teles JH. Helv. Chim. Acta. 1996;79:1899. [Google Scholar]
- 12.Sheehan JC, Hunneman DH. J. Am. Chem. Soc. 1966;88:3666. [Google Scholar]
- 13.Enders D, Niemeier O, Balensiefer T. Angew. Chem. Int. Ed. 2006;45:1463. doi: 10.1002/anie.200503885. [DOI] [PubMed] [Google Scholar]
- 14.Knight RL, Leeper FJ. J. Chem. Soc., Perkin Trans. 1. 1998:1891. [Google Scholar]
- 15.Stetter H, Lorenz G. Chem. Ber. 1985;118:1115. [Google Scholar]
- 16.Myers MC, Bharadwaj AR, Milgram BC, Scheidt KA. J. Am. Chem. Soc. 2005;127:14675. doi: 10.1021/ja0520161. [DOI] [PubMed] [Google Scholar]
- 17.Mattson AE, Scheidt KA. Org. Lett. 2004;6:4363. doi: 10.1021/ol0481129. [DOI] [PubMed] [Google Scholar]
- 18.Mattson AE, Bharadwaj AR, Scheidt KA. J. Am. Chem. Soc. 2004;126:2314. doi: 10.1021/ja0318380. [DOI] [PubMed] [Google Scholar]
- 19.Mattson AE, Bharadwaj AR, Zuhl AM, Scheidt KA. J. Org. Chem. 2006;71:5715. doi: 10.1021/jo060699c. [DOI] [PubMed] [Google Scholar]
- 20.Our preliminary studies of generating homoenolates using N-heterocyclic carbenes and the protonation of these unusual intermediates was first described in a lecture by K.A.S. at the 2004 Natural Products Gordon Research Conference (Tilton, NH) on Thursday, July 29, 2004.
- 21.Burstein C, Glorius F. Angew. Chem. Int. Ed. 2004;43:6205. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]
- 22.Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- 23.He M, Bode JW. Org. Lett. 2005;7:3131. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]
- 24.Burstein C, Tschan S, Xie XL, Glorius F. Synthesis. 2006:2418. [Google Scholar]
- 25.Nair V, Poonoth M, Vellalath S, Suresh E, Thirumalai R. J. Org. Chem. 2006;71:8964. doi: 10.1021/jo0615706. [DOI] [PubMed] [Google Scholar]
- 26.Nair V, Vellalath S, Poonoth M, Suresh E. J. Am. Chem. Soc. 2006;128:8736. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 27.Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2008;130:2416. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seayad J, Patra PK, Zhang Y, Ying JY. Org. Lett. 2008;10:953. doi: 10.1021/ol800003n. [DOI] [PubMed] [Google Scholar]
- 30.Chan A, Scheidt KA. J. Am. Chem. Soc. 2008;130:2740. doi: 10.1021/ja711130p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan A, Scheidt KA. Org. Lett. 2005;7:905. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]
- 32.Sohn SS, Bode JW. Org. Lett. 2005;7:3873. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 33.Bordwell FG, Mccallum RJ, Olmstead WN. J. Org. Chem. 1984;49:1424. [Google Scholar]
- 34.Lepore SD, Khoram A, Bromfield DC, Cohn P, Jairaj V, Silvestri MA. J. Org. Chem. 2005;70:7443. doi: 10.1021/jo051040u. [DOI] [PubMed] [Google Scholar]
- 35.Bordwell FG. Acc. Chem. Res. 1988;21:456. [Google Scholar]
- 36.Diver ST, Rivas FM, Giessert AJ. J. Org. Chem. 2002;67:1708. doi: 10.1021/jo016251n. [DOI] [PubMed] [Google Scholar]
- 37.Zeitler K. Org. Lett. 2006;8:637. doi: 10.1021/ol052826h. [DOI] [PubMed] [Google Scholar]
- 38.Magill AM, Cavell KJ, Yates BF. J. Am. Chem. Soc. 2004;126:8717. doi: 10.1021/ja038973x. [DOI] [PubMed] [Google Scholar]
- 39.Wöhler F, Liebig J. Ann. Pharm. (Lemgo, Ger.) 1832;3:294. [Google Scholar]
- 40.Adams R, Marvel CS. Org. Synth. Coll. Vol. I. London: John Wiley & Sons; 1932. p. 94. [Google Scholar]
- 41.Ide WS, Buck JS. Org. React. 1949;4:269. [Google Scholar]
- 42.Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew. Chem. Int. Ed. 2007;46:3107. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2007;129:10098. doi: 10.1021/ja073987e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He M, Struble JR, Bode JW. J. Am. Chem. Soc. 2006;128:8418. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]
- 45.He M, Uc GJ, Bode JW. J. Am. Chem. Soc. 2006;128:15088. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]
- 46.Reynolds NT, Rovis T. J. Am. Chem. Soc. 2005;127:16406. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]
- 47.Cain CM, Cousins RPC, Coumbarides G, Simpkins NS. Tetrahedron. 1990;46:523. [Google Scholar]
- 48.Brunel JM. Chem. Rev. 2005;105:857. doi: 10.1021/cr040079g. [DOI] [PubMed] [Google Scholar]
- 49.Seebach D, Beck AK, Heckel A. Angew. Chem. Int. Ed. 2001;40:92. [PubMed] [Google Scholar]
- 50.Maki BE, Chan A, Phillips EM, Scheidt KA. Org. Lett. 2007;9:371. doi: 10.1021/ol062940f. [DOI] [PubMed] [Google Scholar]
- 51.Chan A, Scheidt KA. J. Am. Chem. Soc. 2006;128:4558. doi: 10.1021/ja060833a. [DOI] [PubMed] [Google Scholar]
- 52.Castells J, Llitjos H, Morenomanas M. Tetrahedron Lett. 1977;18:205. [Google Scholar]
- 53.Inoue H, Higashiura K. J. Chem. Soc., Chem. Commun. 1980:549. [Google Scholar]
- 54.Miyashita A, Suzuki Y, Nagasaki I, Ishiguro C, Iwamoto K, Higashino T. Chem. Pharm. Bull. 1997;45:1254. [Google Scholar]
- 55.Glorius F, Altenhoff G, Goddard R, Lehmann C. Chem. Commun. 2002:2704. doi: 10.1039/b208045a. [DOI] [PubMed] [Google Scholar]
- 56.Baskakov D, Herrmann WA, Herdtweck E, Hoffmann SD. Organometallics. 2007;26:626. [Google Scholar]
- 57.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 58.Perrin DD, Armarego WL. Purification of Laboratory Chemicals. 3rd ed. Oxford: Pergamon Press; 1988. [Google Scholar]
- 59.Hesse S, Kirsch G. Synthesis. 2001:755. [Google Scholar]
- 60.Kirsch G, Prim D, Leising F, Mignani G. J. Heterocycl. Chem. 1994;31:1005. [Google Scholar]
- 61.Baldwin JE, Mackenzie Turner SC, Moloney MG. Tetrahedron. 1994;50:9411. [Google Scholar]
- 62.Kurono N, Sugita K, Takasugi S, Tokuda M. Tetrahedron. 1999;55:6097. [Google Scholar]
- 63.Kopecky DJ, Rychnovsky SD. J. Org. Chem. 2000;65:191. doi: 10.1021/jo9914521. [DOI] [PubMed] [Google Scholar]
- 64.Kita Y, Akai S, Yamamoto M, Taniguchi M, Tamura Y. Synthesis. 1989:334. [Google Scholar]
- 65.Barton P, Laws AP, Page MI. J. Chem. Soc., Perkin Trans. 2. 1994:2021. [Google Scholar]
- 66.Peterson PE, Stepanian M. J. Org. Chem. 1988;53:1903. [Google Scholar]
- 67.Kuroda H, Hanaki E, Izawa H, Kano M, Itahashi H. Tetrahedron. 2004;60:1913. [Google Scholar]
- 68.Banerjee M, Roy S. Org. Lett. 2004;6:2137. doi: 10.1021/ol0493352. [DOI] [PubMed] [Google Scholar]
- 69.Waddell TG, Rambalakos T, Christie KR. J. Org. Chem. 1990;55:4765. [Google Scholar]
- 70.Imashiro R, Seki M. J. Org. Chem. 2004;69:4216. doi: 10.1021/jo049893u. [DOI] [PubMed] [Google Scholar]
- 71.Kim N, Kim KS, Gupta AK, Oh CH. Chem. Commun. 2004:618. doi: 10.1039/b316446b. [DOI] [PubMed] [Google Scholar]
- 72.Kerr MS, de Alaniz JR, Rovis T. J. Org. Chem. 2005;70:5725. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]