

Abstract
Attenuation of blood-brain barrier (BBB) disruption is one of the therapeutic candidates for treatment of subarachnoid hemorrhage (SAH). In this study, the protective effect of sodium orthovanadate (SOV) on BBB disruption was investigated in SAH using the endovascular perforation model. Fifty-five rats were randomly assigned to sham-operated, SAH treated with saline (as a vehicle) or 10mg/kg SOV groups, and evaluated for neurofunction and Evans blue dye extravasation. The phosphorylation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and mitogen-activated protein kinase (MAPK), and the expression of matrix metalloproteinase-9 (MMP-9), occludin, and collagen-IV were examined by Western blot analyses. Cell death on endothelial cells were revealed by immunofluorescence and terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling (TUNEL) staining. SOV significantly improved neurofunction and reduced Evans blue dye extravasation in brains after SAH. SOV phosphorylated PTEN, decreased phospho-JNK and MMP-9, and preserved occludin expression. SOV also attenuated SAH-induced capillary endothelial cell death. The current study showed that SOV was protective against BBB disruption after SAH possibly via PTEN phosphorylation.
Keywords: subarachnoid hemorrhage, sodium orthovanadate, Phosphatase and tensin homolog deleted on chromosome 10, blood-brain barrier, tight junction
Introduction
Basic and clinical scientific research efforts in subarachnoid hemorrhage (SAH) have investigated early brain injury (EBI) and cerebral vasospasm, with the former recently thought to be important in preventing the latter and the resulting poor outcome (Sehba et al., 2011). Brain edema, which is a main factor for EBI and a determinant for mortality and morbidity after SAH, mainly comes from disruption of the blood brain-barrier (BBB; Suzuki et al., 2010a). Therefore, stabilization of injured BBB is a candidate target for the treatment of SAH.
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) regulates several basic cellular functions, and has been more recently the focus in experimental study of cerebral ischemia (Zhao et al., 2005; Zhou et al., 2008). PTEN is widely expressed in various cell types including endothelial cells. The main substrates of PTEN are inositol phospholipids generated by phosphoinositide 3 kinase (PI3K) activation, with PTEN acting as a major negative regulator of the PI3K/Akt signaling pathway (Zhao et al., 2005). The phosphorylation of PTEN is associated with decreased PTEN function (Zhou et al., 2008), and phosphorylation induced-Akt activation is neuroprotective in cerebral ischemia (Li and Ross, 2007; Zhao et al., 2005). In addition, PTEN is reported to affect c-Jun N-terminal kinase (JNK) pathways (Zhang et al., 2007), which may be involved in the BBB disruption after SAH (Suzuki et al., 2010a). However, we are not aware of an investigation on whether PTEN plays a role in the pathophysiology of early brain injury after SAH.
We recently reported that sodium orthovanadate (SOV), a well-known tyrosine phosphatase inhibitor, ameliorated EBI after SAH through neuronal anti-apoptosis (Hasegawa et al., 2011a). However, it remains unknown whether SOV prevents BBB disruption after SAH. Because SOV can phosphorylate PTEN (Wu et al., 2006), we hypothesized that SOV prevents post-SAH BBB disruption via PTEN phosphorylation. In this study, we demonstrated that SOV modulated PTEN-related signaling pathways, suppressed capillary endothelial cell death, and ameliorated BBB disruption after experimental SAH.
Materials and Methods
Experimental animals
All experiments were approved by the Institutional Animal Care and Use Committee of Loma Linda University. Fifty-five male Sprague-Dawley rats (Harlan, Indianapolis, IN) weighing 300 to 350g were divided randomly into the following groups: sham-operated (sham group: n=15), SAH+saline (vehicle group: n=20), and SAH+10mg/kg of SOV in saline (SOV group: n=20).
Induction of subarachnoid hemorrhage
Anesthesia was induced with 5% isoflurane and maintained with 2.5% isoflurane, 30% oxygen, and 70% medical air via face mask. Blood pressure and blood gas were measured via the left femoral artery. The rectal temperature was monitored and kept at 36.5°C±0.5°C by using a feedback-regulated heating system during surgery.
SAH model was produced as described previously (Hasegawa et al., 2011). Briefly, the left common carotid artery was exposed, 4-0 sharpened nylon sutures were advanced rostrally into the left internal carotid artery until resistance was felt, and perforated at that position. Immediately after surgery, saline or 10mg/kg of SOV was injected intraperitoneally.
SAH severity
The severity of SAH was blindly assessed using the SAH grading scale at the time of euthanasia (Sugawara et al., 2008). Because the severity of brain injures in the endovascular perforation model of SAH was correlated with a SAH grading score and weak SAH animals did not induce brain injury (Hasegawa et al., 2011a), two animals (a total score of ≤7; vehicle: 1, SOV: 1) were excluded from this study.
Neurological scoring and mortality
The neurological function was blindly evaluated one hour before euthanasia using a modified 22-point scoring system of the method described by Garcia et al. (1995). Mortality rate was measured at 24 hours after SAH.
BBB permeability
BBB permeability was evaluated using Evans blue dye extravasation as previously described (Suzuki et al., 2010b). Briefly, Evans blue dye (2%; 5ml/kg) was administered over 2 minutes into the left femoral vein, allowed to circulate for 60 minutes, then the animals were euthanized with perfusion of phosphate-buffered saline intracardially. The brains were divided into four parts: the right and left (ipsilateral to perforation) cerebral hemispheres, cerebellum, and brain stem. Brain specimens were weighed, homogenized in PBS, and centrifuged at 15,000g for 30 minutes. The resultant supernatant was added to an equal volume of trichloroacetic acid. After overnight incubation at 4°C and centrifugation at 15,000g at 4°C for 30 minutes, the supernatant was taken for spectrophotometric quantification of extravasated Evans blue dye at 615 nm.
Western blot analysis
The left cerebral hemisphere was isolated and collected at 24 hours after SAH induction (n=5, respectively). Protein concentration was determined using DC protein assay (Bio-Rad, Hercules, CA). Western blot analysis was evaluated as previously described (Hasegawa et al., 2011a) with the following primary antibodies: anti-phospho-PTEN (1:1000, Cell Signaling Technology, Danvers, MA), anti-phospho- ERK, anti-phospho-p38, anti-phospho-JNK, anti- MMP-9, anti-occludin, and anti-collagen IV (1:200, Santa Cruz Biotechnology, Santa Cruz, CA). The images were scanned and analyzed semi-quantitatively in a blind fashion using ImageJ software. β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) was used as an internal control for all experiments. Changes in phosphorylation of PTEN, ERK, p38, JNK and protein level of MMP-9, occludin, and collagen IV were expressed as a percentage of the sham (at 24 hours after SAH) level/β-actin.
TUNEL staining
Samples from sham-operated, saline- and SOV-treated rats were used (n=4, respectively). The brain specimens were prepared as previously described (Hasegawa et al., 2011a). Ten-micron-thick coronal sections of the brain at the level of bregma-2 mm were cut on cryostat (Leica Microsystems LM3050S) and mounted on poly-L-lysine-coated slides. For checking cell death on endothelial cells, we immunostained the brain sections with anti-von Willebrand factor (vWF) antibody (1:100, Santa Cruz Biotechnology) and then subjected the sections to TUNEL staining with an in situ cell death detection kit (Roche Inc., Mannheim, Germany). Incubation with labeling solution without the enzyme served as a negative labeling control.
Statistical analysis
All values were expressed as the mean±SD. Statistical differences among the various groups were assessed with one-way analysis of variance followed by a Scheffe post-hoc analysis. The differences between the two groups were compared using an unpaired t test. Differences of p<0.05 were considered significant.
Results
Mortality and SAH grade
No statistical differences were observed among the groups with regard to physiological parameters (data not shown). The mortality rate in the vehicle and SOV groups were the same (21.1%; 4 of 19 rats, each) at 24 hours. No sham-operated rats died.
The average of SAH grading scores in SAH rats was similar between groups (11.2±2; vehicle, 11.5±2; SOV; Figure 1A).
Figure 1.
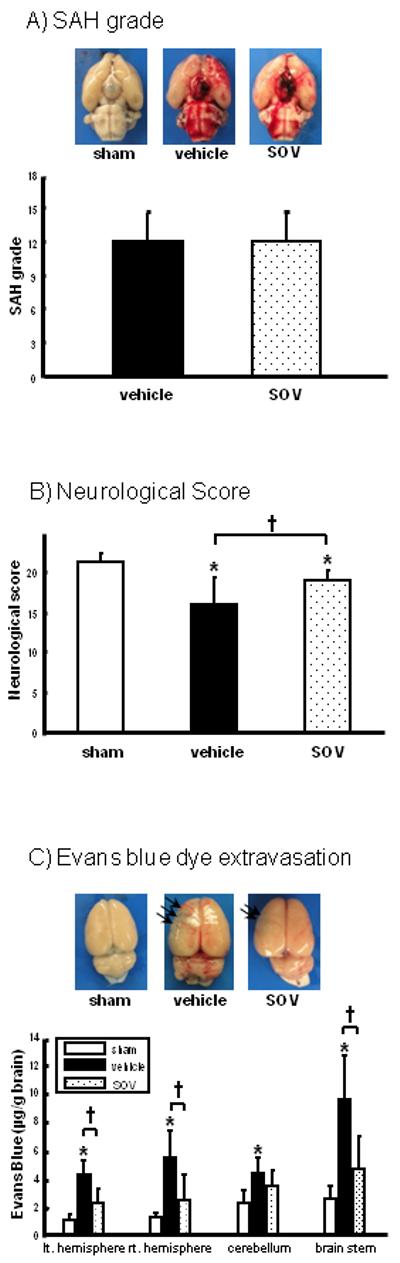
SAH grade (A; n=15, respectively), neurological score (B; n=15, respectively), and Evans blue dye extravasation (C: n=6, respectively) in groups treated with saline (vehicle), 10mg/kg of sodium orthovanadate (SOV), and/or sham-operation at 24 hours after SAH. Upper pictures in (A) and (C) are representative brain specimens of each experiment and arrows in (C) indicate the areas of Evans blue dye extravasation in ipsilateral SAH brain. Values are the mean±SD; *P<0.05 vs. sham, †P<0.05 vs. vehicle, ANOVA.
*; p<0.05 versus sham, †; p<0.05 versus vehicle
Neurological score and Evans blue dye extravasation
Although neurological score deteriorated in SAH-animals compared with sham animals at 24 hours, a significant improvement was observed in the SOV group compared with the vehicle group at 24 hours after SAH (Figure 1B).
Evans blue dye extravasation in the vehicle group significantly increased in all brain regions, and was significantly attenuated by SOV (Figure 1C).
Phosphorylation of PTEN and mitogen-activated protein kinases (MAPKs)
To address whether SOV modulated PTEN phosphorylation, we evaluated phosphorylation levels of PTEN in the ipsilateral hemisphere after SAH. PTEN was significantly phosphorylated by SOV compared with the vehicle group (Figure 2B). Next, the phosphorylation levels of all MAPKs including extracellular signal-regulated kinase (ERK), p38, and JNK were measured, because SOV activated not only Akt but also MAPKs, especially ERK (Hasegawa et al., 2003). In this study, phosphorylation levels of JNK were upregulated by SAH, whereas decreased by SOV significantly, and the phosphorylation levels of ERK and p38 were not significantly changed by SAH and SOV (Figure 2C-E).
Figure 2.
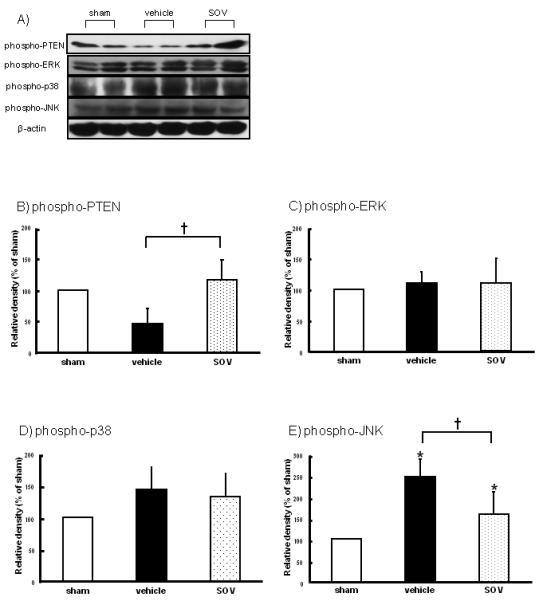
Phosphorylation levels of phosphatase and tensin homolog deleted on chromosome 10 (phospho-PTEN), extracellular signal-regulated kinase (phospho-ERK), p38 (phospho-p38), and c-Jun N-terminal kinase (phospho-JNK) in the left hemisphere at 24 hours after SAH (n=5, respectively). Representative Western blots (A), and quantitative analysis of phospho-PTEN (B), phospho-ERK (C), phospho-p38 (D), and phospho-JNK (E) in groups treated with saline (vehicle), 10mg/kg of SOV, and sham-operation. The band density values are calculated as a ratio of that of β-actin, and the values from the sham-operation are used as 100%. Values are the mean±SD; *P<0.05 vs. sham, †P<0.05 vs. vehicle, ANOVA.
*; p<0.05 versus sham, †; p<0.05 versus vehicle
Expression of matrix metalloproteinase-9 (MMP-9), occludin, and collagen IV
To examine whether SOV affected the stabilization of BBB after SAH, we measured BBB-related proteins such as MMP-9, occludin, and collagen IV. MMP-9 was increased, and occludin and collagen IV were decreased in the vehicle group. SOV attenuated an increase in MMP-9 and significantly inhibited the degradation of occludin, but not collagen IV (Figure 3).
Figure 3.
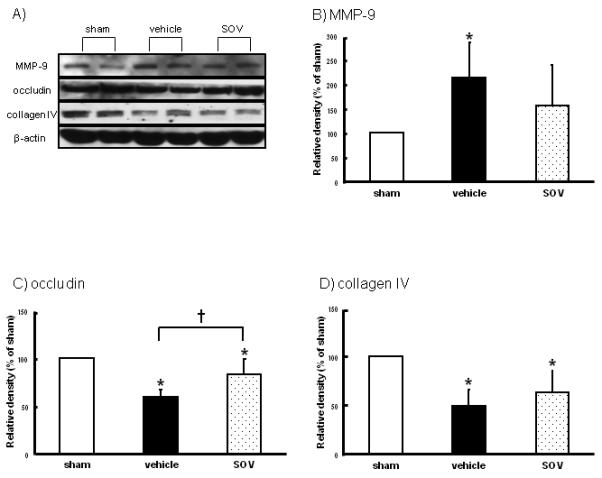
Expression levels of matrix metalloproteinase-9 (MMP-9), occludin, and collagen IV in the left hemisphere at 24 hours after SAH (n=5, respectively). Representative Western blots (A), and quantitative analysis of MMP-9 (B), occludin (C), and collagen IV (D) expressions in groups treated with saline (vehicle), 10mg/kg of SOV, and sham-operation. The band density values are calculated as a ratio of that of β-actin, and the values from the sham-operation are used as 100%. Values are the mean±SD; *P<0.05 vs. sham, †P<0.05 vs. vehicle, ANOVA.
*; p<0.05 versus sham, †; p<0.05 versus vehicle
Immunohistochemical evaluations of endothelial cell death
Terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end-labeling (TUNEL)-positive endothelial cells were increased after SAH, immunofluorescence study showed that SOV reduced endothelial cell death compared with the vehicle group (Figure 4).
Figure 4.
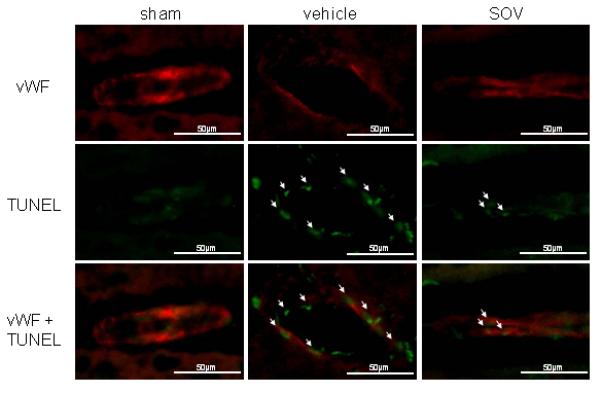
Expression Of Endothelial Cell Death After Sah In The Area In Coronal Sections At Bregma-2Mm In Groups Treated With Saline (Vehicle), 10Mg/Kg Of Sov, And Sham-Operation (N=4). Arrows Indicate Tunel-Positive Endothelial Cells. Vwf: Von Willebrand Factor, Tunel: Terminal Deoxynucleotidyl Transferase-Mediated Uridine 5′-Triphosphate-Biotin Nick End-Labeling, Scale Bar: 50μM
Discussion
SOV is known to be a non-selective protein tyrosine phosphatase inhibitor, and we reported that the drug prevented neuronal apoptosis after cerebral ischemia and SAH (Hasegawa et al., 2003; Hasegawa et al., 2011a). In the current study, we showed for the first time that SOV attenuated BBB disruption and improved neurofunction after SAH. The effects were associated with the inhibition of post-SAH PTEN dephosphorylation, JNK activation, MMP-9 induction, and the preservation of occludin which is a tight junction protein. In addition, SOV was demonstrated to reduce TUNEL-positive endothelial cells. We speculated that all observed modulations of signaling pathway might come from PTEN phosphorylation by SOV.
PTEN is a lipid phosphatase, and the phosphorylation of PTEN leads to a decrease both in function and stability of PTEN (Lu et al., 2003; Koul et al., 2002). PTEN negatively regulates the PI3K/Akt pathway, while it was reported that PTEN played a critical role in the functional coupling between JNK and Akt pathways (Zhang et al., 2007). Bpv (pic), a vanadate compound, was shown to phosphorylate PTEN, deactivate JNK and enhance Akt phosphorylation, resulting in significant rescue of hippocampal CA1 neurons in cerebral ischemia (Zhang et al., 2007). Also, SOV was shown to downregulate PTEN and activate PI3K-Akt signaling in ischemic brain injury (Wu et al., 2006). We previously demonstrated that SOV reduced brain injury after SAH was associated with Akt activation (Hasegawa et al., 2011a). In this study, we showed that SOV phosphorylated PTEN, in addition to Akt activation and JNK inactivation, in brain tissues after SAH. Thus, we speculated that BBB protection by SOV might come from PTEN phosphorylation, followed by Akt activation and JNK inactivation.
Kusaka et al. (2004) reported that all MAPKs such as JNK, ERK, and p38 were upregulated and contributed to the altered permeability of the BBB and the resultant brain edema formation after SAH, associated with vascular endothelial growth factor (VEGF) upregulation. However, other studies reported that only JNK was consistently activated and associated with the BBB disruption (Suzuki et al., 2010a) and that brain VEGF levels were unchanged after SAH (Hasegawa et al., 2011a). JNK is activated by various stimulants, including oxidative stress and inflammatory response, and upregulates the apoptotic cascade (Fassbender et al., 2001; Hasegawa et al., 2011b; Hirashima et al., 1997). In the current study, we also demonstrated that only JNK was activated among the MAPKs in brains after SAH, associated with BBB permeability. This may mean that activation of JNK is the most important factor in BBB disruption after SAH.
It was reported that brain edema formation was an independent risk factor for mortality and poor outcome after SAH (Claassen et al., 2002). The formation of the vasogenic brain edema in SAH occurs at the microvascular level via the opening of endothelial tight junctions, disruption of the basal lamina, and injury of the endothelium, all of which constitute the BBB. Occludin is the only integral membrane protein exclusively localized at the tight junction (Furuse et al., 1993) and the presence of the protein at the tight junction is correlated with increased barrier function and decreased paracellular permeability. MMP-9 is upregulated in brain injury and is closely associated with BBB disruption (Rosenberg et al., 1998),22 because activated MMP-9 directly damages neurovascular substrates including tight junction proteins such as occludin (Yang et al., 2007). Also, MMP-9 activation has been reported to be involved in JNK-related pathways after SAH (Suzuki et al., 2010a; Yatsushige et al., 2007), as shown in this study. Although we did not assess a direct pathway between JNK and MMP-9, we speculated that SOV preserved occludin through deactivation of JNK and MMP-9. On the other hand, collagen IV, which is known as a basal lamina protein, was not preserved by SOV in this study. This may explain why SOV did not fully prevent post-SAH BBB disruption and neuronal dysfunction in this study. Moreover, apoptosis in endothelial cell is known to result in BBB disruption with an increase in endothelial MMP-9 in experimental SAH (Park et al., 2004). We demonstrated that SOV reduced capillary endothelial cell death after SAH in this study. So, all these findings support that SOV is protective against SAH-induced BBB disruption.
This study is somewhat limited. First, it was not designed to demonstrate that SOV certainly affects endothelial PTEN, although SOV treatment clearly protected capillary endothelial cell death. Second, only immediate treatment after SAH with SOV was tested in this study, so, to be more translational, a late treatment as well as long-term outcomes should be examined in future preclinical evaluation. Third, SOV is a non-selective protein tyrosine phosphatase inhibitor, and has been reported to be neuroprotective (Hasegawa et al., 2003; Hasegawa et al., 2011a). Therefore, this study cannot completely exclude the possibility that observed findings in this study might be secondary to other neuroprotective effects of SOV, and that other molecular mechanisms might also contribute to the attenuation of BBB disruption.
In conclusion, the present investigation demonstrated that SOV attenuated SAH-induced BBB disruption through PTEN phosphorylation. Further studies are needed to provide collecting evidences to use the drug as a treatment of human SAH.
Acknowledgments
Grant information: This study was partially supported by grants (NS053407) from the National Institutes of Health to J.H.Z.
Footnotes
The authors report no conflicts of interest.
References
- Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33:1225–1232. doi: 10.1161/01.str.0000015624.29071.1f. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schütt S, Fritzinger M, Horn P, Vajkoczy P, Kreisel S, Brunner J, Schmiedek P, Hennerici M. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry. 2001;70:534–537. doi: 10.1136/jnnp.70.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JH, Liu KF, Ho KL. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke. 1995;26:636–642. doi: 10.1161/01.str.26.4.636. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Hamada J, Morioka M, Yano S, Kawano T, Kai Y, Fukunaga K, Ushio Y. Neuroprotective effect of postischemic administration of sodium orthovanadate in rats with transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2003;23:1040–1051. doi: 10.1097/01.WCB.0000085160.71791.3F. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Suzuki H, Altay O, Zhang JH. Preservation of tropomyosin-related kinase B (TrkB) signaling by sodium orthovanadate attenuates early brain injury after subarachnoid hemorrhage in rats. Stroke. 2011a;42:477–483. doi: 10.1161/STROKEAHA.110.597344. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Suzuki H, Sozen T, Altay O, Zhang JH. Apoptotic mechanisms for neuronal cells in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl. 2011b;110:43–48. doi: 10.1007/978-3-7091-0353-1_8. [DOI] [PubMed] [Google Scholar]
- Hirashima Y, Nakamura S, Endo S, Kuwayama N, Naruse Y, Takaku A. Elevation of platelet activating factor, inflammatory cytokines, and coagulation factors in the internal jugular vein of patients with subarachnoid hemorrhage. Neurochem Res. 1997;22:1249–1255. doi: 10.1023/a:1021985030331. [DOI] [PubMed] [Google Scholar]
- Li L, Ross AH. Why is PTEN an important tumor suppressor? J Cell Biochem. 2007;102:1368–1374. doi: 10.1002/jcb.21593. [DOI] [PubMed] [Google Scholar]
- Koul D, Jasser SA, Lu Y, Davies MA, Shen R, Shi Y, Mills GB, Yung WK. Motif analysis of the tumor suppressor gene MMAC/PTEN identifies tyrosines critical for tumor suppression and lipid phosphatase activity. Oncogene. 2002;21:2357–2364. doi: 10.1038/sj.onc.1205296. [DOI] [PubMed] [Google Scholar]
- Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24:916–925. doi: 10.1097/01.WCB.0000125886.48838.7E. [DOI] [PubMed] [Google Scholar]
- Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, McMurray JS, Fang X, Yung WK, Siminovitch KA, Mills GB. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003;278:40057–40066. doi: 10.1074/jbc.M303621200. [DOI] [PubMed] [Google Scholar]
- Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Sehba FA, Pluta RM, Zhang JH. Metamorphosis of subarachnoid hemorrhage research: from delayed vasospasm to early brain injury. Mol Neurobiol. 2011;43:27–40. doi: 10.1007/s12035-010-8155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke. 2010a;41:1783–1790. doi: 10.1161/STROKEAHA.110.586537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Ayer R, Sugawara T, Chen W, Sozen T, Hasegawa Y, Kanamaru K, Zhang JH. Protective effects of recombinant osteopontin on early brain injury after subarachnoid hemorrhage in rats. Crit Care Med. 2010b;38:612–618. doi: 10.1097/CCM.0b013e3181c027ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DN, Pei DS, Wang Q, Zhang GY. Down-regulation of PTEN by sodium orthovanadate inhibits ASK1 activation via PI3-K/Akt during cerebral ischemia in rat hippocampus. Neurosci Lett. 2006;404:98–102. doi: 10.1016/j.neulet.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yatsushige H, Ostrowski RP, Tsubokawa T, Colohan A, Zhang JH. Role of c-Jun N-terminal kinase in early brain injury after subarachnoid hemorrhage. J Neurosci Res. 2007;85:1436–1448. doi: 10.1002/jnr.21281. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wu DN, Han D, Zhang GY. Critical role of PTEN in the coupling between PI3K/Akt and JNK1/2 signaling in ischemic brain injury. FEBS Lett. 2007;581:495–505. doi: 10.1016/j.febslet.2006.12.055. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Qian L, Chou T, Iadecola C. Neuroprotection by PGE2 receptor EP1 inhibition involves the PTEN/AKT pathway. Neurobiol Dis. 2008;29:543–551. doi: 10.1016/j.nbd.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]