

Abstract
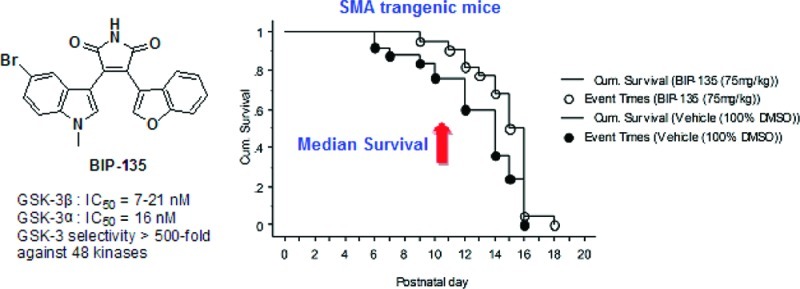
The discovery of upregulated glycogen synthase kinase-3 (GSK-3) in various pathological conditions has led to the development of a host of chemically diverse small molecule GSK-3 inhibitors, such as BIP-135. GSK-3 inhibition emerged as an alternative therapeutic target for treating spinal muscular atrophy (SMA) when a number of GSK-3 inhibitors were shown to elevate survival motor neuron (SMN) levels in vitro and to rescue motor neurons when their intrinsic SMN level was diminished by SMN-specific short hairpin RNA (shRNA). Despite their cellular potency, the in vivo efficacy of GSK-3 inhibitors has yet to be evaluated in an animal model of SMA. Herein, we disclose that a potent and reasonably selective GSK-3 inhibitor, namely BIP-135, was tested in a transgenic Δ7 SMA KO mouse model of SMA and found to prolong the median survival of these animals. In addition, this compound was shown to elevate the SMN protein level in SMA patient-derived fibroblast cells as determined by Western blot, and was neuroprotective in a cell-based, SMA-related model of oxidative stress-induced neurodegeneration.
Keywords: GSK-3 inhibitor, BIP-135, median survival, spinal muscular atrophy, survival motor neuron, Δ7 SMA KO mice
Over the years, glycogen synthase kinase-3 (GSK-3), a member of the serine/threonine kinase family, has been extensively studied as a drug target, because upregulated GSK-3 has been linked to a number of human pathological conditions.1 This enzyme is known to regulate a diverse array of intracellular processes through the phosphorylation of its protein substrates.2 In mammals, GSK-3 is expressed in two highly homologous isoforms, namely GSK-3α and GSK-3β. This multifunctional kinase is constitutively active in resting cells and can be physiologically inhibited by various signaling pathways, including the PI3K/Akt mediated apoptosis cascade in stimulated cells through phosphorylation of residues Ser-21/Ser-9 (GSK-3α/β).3 Inhibition of GSK-3 results in the activation of its downstream constituents, such as β-catenin, c-Jun, and the cyclic AMP response element binding protein (CREB), which consequently upregulate Tcf/Lef gene transcription and CREB-induced gene transcription of neurotrophic factors, such as brain-derived neurotrophic factor (BDNF).4,5 BDNF helps to support the survival of existing neurons and promote neurogenesis.6 Thus, GSK-3 inhibition has shown neuroprotective effects in various models of neurodegenerative disease.7,8 Motor neuron diseases (MNDs), such as amyotrophic lateral sclerosis (ALS), also experienced neuroprotection in vitro.9 Significant delay in symptom onset and an increase in survival time was observed in an animal model of ALS when a GSK-3 inhibitor was administered to the animals.10 As such, neuroprotection, offered by GSK-3 inhibition, is a validated therapeutic strategy in treating many other MNDs, such as spinal muscular atrophy (SMA).
SMA is a major cause of lethality among infants in their early stage of life, and it remains as an untreatable disease. Several therapeutic strategies are currently under investigation for correcting some of the symptoms associated with SMA.11,12 A characteristic defect that occurs in SMA is a deficiency in the survival motor neuron (SMN) protein, constituted by the mutation or deletion of the survival motor neuron 1 (SMN1) gene responsible for SMN protein expression. Its absence progresses into the degeneration of the lower motor neurons found in the anterior horn of the spinal cord that is commonly found in SMA patients.11,12 The severity of SMA in patients is inversely dependent on the expression level of SMN2, a paralogue of SMN1.12 All SMA patients contain at least one copy of SMN2, and thus, pharmacologic agents capable of elevating endogenous level of SMN2-derived SMN proteins would be highly desirable for the development of SMA therapeutics. (11,12)
Recently, inhibition of GSK-3 was shown to elevate cellular SMN levels in SMA type I (a severe form of SMA often diagnosed before 6 months of age) patient-derived fibroblasts and also in motor neurons in which the intrinsic SMN levels were diminished by SMN-specific short hairpin RNA (shRNA).13 The connection between GSK-3 inhibition and SMA was first demonstrated in an image-based screen, conducted by Rubin and co-workers, to identify compounds capable of elevating SMN levels in vitro. Independently of these findings, and prior to becoming aware of the then unpublished research of Rubin, we had submitted several of our GSK-3 inhibitors for screening to the SMA Foundation and received reports showing their efficacy in animal models. We now disclose the results of these SMA animal experiments, along with in vitro biology conducted by the Rubin group.
To date, a plethora of GSK-3 inhibitors have been reported, and a few of these are shown in Figure 1A.14−17 Additionally, we have disclosed a variety of selective, maleimide-bearing GSK-3 inhibitors that are active in both cell and animal models of neurological and psychiatric disorders, including Parkinson’s disease and bipolar disorder (Figure 1B).16−18 Based upon the usual cycle of design, synthesis, and kinase testing, we identified a potent ATP-competitive GSK-3 inhibitor, namely 3-(5-bromo-1-methyl-1H-indol-3-yl)-4-(benzofuran-3-yl)pyrrole-2,5-dione, or BIP-135. Ultimately, this compound was tested against a total of 62 kinases (see the Supporting Information).16 BIP-135 was found to be relatively selective for GSK-3β (21 nM). However, due to the high sequence homology between GSK-3α and GSK-3β, it was not surprising to find similar inhibitory activity against both isoforms. Among all the other kinases tested, this compound was found to show some activity toward PKCβ (β1, 980 nM; β2, 219 nM), DYRK1B (590 nM), and PI3Kα (870 nM). Given the structural resemblance of BIP-135 to known PKCβ inhibitors of the staurosporine class and the high homology between PKCβ1 and β2, this off-target activity was not unexpected.19 In the case of the modest inhibition shown by BIP-135 of DYRK1B, this may not be a concern. A number of articles on DYRK1B suggest that it plays a key role in cancer biology.20 In particular, overexpression of the DYRK1B gene appears to be associated with pancreatic cancers as a consequence of its downstream effect on oncogenic K-ras.20 As for PI3Kα, it is one of a number of isozymes in the PI3K family that can activate Akt. The activation of Akt has been shown to downregulate FOXO, a protein that is overexpressed in type I SMA that contributes to muscle atrophy.21 As a weak inhibitor of PI3Kα, it is unlikely that BIP-135 can influence the expression level of the FOXO protein.
Figure 1.

Representative examples of GSK-3 inhibitors. (A) Some examples of GSK-3 inhibitors having diverse chemical scaffolds. (B) Examples of maleimide-bearing GSK-3 inhibitors disclosed previously together with the chemical structure of BIP-135.
As reported previously by Rubin, a set of commercially available GSK-3 inhibitors, including alsterpaullone, CHIR98014, and AR-A014418, were shown to elevate SMN levels in vitro.13 Under the same conditions, Western blot analysis employing BIP-135 led to an elevation in SMN protein levels. Exposure of human SMA patient fibroblasts to a dose of 25 μM of the test compound for 72 h led to a 7-fold increase in SMN levels compared to vehicle treated cells (Figure 2). However, the typical bell-shaped dose–response curve was observed due to some toxicity at higher concentrations.
Figure 2.

SMN levels in fibroblasts from SMA patients after 72 h exposure to BIP-135. (A) Representative Western blot assessing the SMN protein level in cells treated with DMSO, 20, 25, or 30 μM concentrations of BIP-135. (B) Quantification of SMN protein levels from three separate experiments.
Effects of BIP-135 in a Model of Oxidative Stress
As oxidative stress also appears to play a role in the dysfunction of motor neurons in SMA, we accordingly sought to explore the possibility that BIP-135 might also work in preventing cell loss under such conditions.22−24 Specifically, it has been suggested that free radicals generated under conditions of oxidative stress lead to the production of reactive lipid aldehydes, resulting in increased levels of cell damage and cellular death in cells that have lower SMN levels.25 Thus, to examine the therapeutic efficacy of BIP-135 in a neuronal model of oxidative stress, primary immature cortical neurons were exposed to the glutamate analogue homocysteic acid (HCA; 5 mM), which was used to deplete the cortical neurons of the antioxidant glutathione. Since glutathione is a major cellular antioxidant, its depletion allows the gradual accumulation of endogenously produced oxidants, thereby inducing neuronal degeneration over a period of approximately 24 h.23 Using this model of oxidative stress, the survival of the HCA-treated cortical neurons was reduced to approximately 25% (Figure 3). On the other hand, the BIP-135 treated cells were protected to the extent of about 80% at a concentration of 20 μM. No significant toxicity was observed when the neurons were exposed to BIP-135 alone, in the absence of HCA (Figure 3A). Other commercially available GSK-3 inhibitors, such as AR-A014418, also provided some protection in this model; however, BIP-135 was able to protect at a lower concentration, demonstrating that BIP-135 is a superior neuroprotective agent in this model of oxidative stress (Figure 3B and C). Micrographs taken of the cortical neurons after 24 h exposure to the GSK-3 inhibitors, BIP-135, SB-216763, or AR-A014418, either alone or in combination with HCA are shown in Figure 3D. These images visually reflect the important neuroprotective effect of BIP-135.
Figure 3.

Acute treatment of primary cortical neurons with BIP-135 protects from oxidative stress. Graphs showing the survival of cortical neurons upon exposure to the GSK-3 inhibitors: (A) BIP-135, (B) AR-A014418, or (C) SB-216763, in the absence (blue bar) or presence (red bar) of HCA (5 mM) after 48 h, using the MTT assay. Data are presented as percent of control ± SEM. (D) Representative micrographs showing live/dead staining of primary cortical neurons after 48 h incubation with BIP-135 (c, d), AR-A014418 (e, f), or SB-216763 (g, h) in the absence (a, c, e, g) or presence (b, d, f, h) of HCA (5 mM). Live and dead neurons were stained with calcein AM (green fluorescence) and ethidium homodimer (red fluorescence), respectively. The control contains no GSK-3 inhibitors.
The dependence of this oxidative stress model on impaired cysteine/cystine transport and glutathione depletion is well-documented.26 However, one could argue that the observed neuroprotective effects of BIP-135 may be due to the inhibition of one or both of these two processes. To rule out these possibilities, the total intracellular glutathione levels (reduced glutathione and oxidized glutathione) were measured after the neurons were treated with HCA, with or without the GSK-3 inhibitors. The data obtained revealed that the GSK-3 inhibitors did not alter the overall glutathione levels (Figure 4), demonstrating that the observed neuroprotection afforded by BIP-135 is independent of antioxidant production. A possible explanation to the observed neuroprotection is the activation of BDNF by GSK-3 inhibition, which has offered neuroprotection in many other models of neurodegenerative diseases.6 This compound may also increase a neuron’s resistance to oxidative damage by disrupting the regulatory role of GSK-3 in destabilizing the antiapoptotic protein Bcl-2 in the intrinsic apoptotic pathway,6 and thus enhance the overall Bcl-2 level that leads to neuronal survival.27,28 These findings further support the possible therapeutic use of BIP-135 in SMA, at least in an animal model, where Bcl-2 deficiency was found on the spinal cord of SMA mice.28 In fact, increasing evidence has suggested that neuronal oxidative stress plays a significant role in cell death and dysfunction associated with neurological diseases, including SMA.23,24 Thus, neuroprotection in this oxidative model, demonstrated by BIP-135, may also be a useful predictor in qualifying other GSK-3 inhibitors for use in treating SMA.
Figure 4.
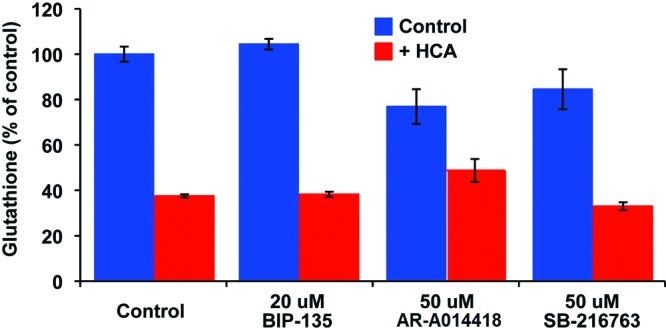
Total intracellular glutathione levels of the primary cortical neuron cultures after 8 h incubation with BIP-135, AR-A014418, or SB-216763 in the absence (blue bar) or presence (red bar) of HCA (5 mM). The control contains no GSK-3 inhibitor.
SMA Animal Studies
While the in vitro results are promising, the possibility to move a compound such as BIP-135 forward to the clinic requires a demonstration that it works in animal models of the disease. To date, animal models of SMA have been limited in large part because SMA is a disease exclusive to humans. In general, murine models of SMA often require homozygous mutation or deletion of the Smn gene, followed by insertion of the SMN2 gene with or without SMN2 cDNA lacking exon 7 (SMN2Δ7+/+).29 This will ensure that the correct phenotypes of the SMA disease are present in the mice. In the present study, the latter murine species (with SMNΔ7+/+) was used to analyze the in vivo effect of BIP-135. In addition, this particular strain of SMA transgenic mice mimics the phenotypes that resembles the human SMA Type I disease.30
In the Δ7 SMA neonatal mouse model of SMA,30,31 the impact of BIP-135 on several phenotypes observed within the human SMA disease, such as (1) loss of motor function, (2) body weight deficiency, and (3) survival, were investigated using three different doses (25, 75, and 125 mg/kg). Among these doses, the in vivo potency of BIP-135 is most promising at 75 mg/kg (intraperitoneal injection, 100% DMSO as vehicle). At this dose, this GSK-3 inhibitor did not appear to be toxic and was well-tolerated by the animals (no decrease in body weight) (Figure 5B). More importantly, BIP-135 caused a modest extension in the median survival of SMA KO animals by two days, suggesting a valid in vivo protective effect for this compound when the median lifespan of these animals was approximately 14 days (Figure 5A). The motor function of the transgenic animals was evaluated based on their performances in the geotaxis and tube test (Figure 5C and D).31 In brief, geotaxis test examined the ability of the animal to orient itself from a downward- to an upward-facing position when placed on an inclined platform. Tube test analyzed the hind-limb strength of the animals when rising from a laying to a standing position. Based on the results gathered from the geotaxis and tube test, BIP-135 did not improve the overall motor function, but it did increase the number of SMA KO animals that completed the geotaxis test (Figure 5C, b). Due to the short life span of these animals (≤14 days), it may be too difficult to detect motor function improvement from the BIP-135 treatment, especially when the end point of the model is the death of the animals. A mouse model with milder SMA phenotypes (type III or IV) and longer lifespan may be more amenable to evaluate the therapeutic effect of BIP-135 in SMA as well as its effect on SMN protein level in vivo.32
Figure 5.

Effect of BIP-135 on several phenotypes of SMA disease that are observed in transgenic mice. (A) Kaplan–Meier survival curve of SMA KO mice treated once a day with BIP-135 at 75 mg/kg or vehicle (100% DMSO) from postnatal day 0 to 21. Post hoc Logrank and Wilcoxon tests showed significant differences between the two treatment groups (p = 0.0294 and 0.0266, respectively). The mean survival for KOs treated with BIP-135 was 14.7 ± 0.4 and 12.8 ± 0.6 days for vehicle treated control [F(1,45) = 5.314, p = 0.0258)]. (B) Effects of GSK-3 inhibitor BIP-135 at 75 mg/kg or its vehicle once a day on the SMA KO body weight. KO animals treated with BIP-135 showed an improvement in body weight in comparison to vehicle treated control (B). ANOVA indicated a significant main effect for treatment [F(1,43) = 4.33, p = 0.0435)] and no significant treatment/age [F(15,540) = 1.43, p = 0.1289)] or treatment/gender/age interactions [F(30,540) = 1.12, p = 0.3016]. Improvement in body weight measures was observed after around P6. (C) Geotaxis test results for KO mice receiving BIP-135 at 75 mg/kg or its vehicle once a day. (a) Latency to complete the negative geotaxis test. There were no significant treatment effects [F(1,43) = 3.50, p = 0.0683] or treatment/age interaction [F(3,111) = 1.81, p = 0.1487] in the latency to reorient upward in the geotaxis test between the two groups. (b) Percent of KO mice that completed the geotaxis test. Chi square test (4.625) indicated significant treatment effect at P12 (p = 0.0315), where more KO animals treated with BIP-135 completed the geotaxis test in comparison to the vehicle treated controls. (D) Tube test results for KO mice receiving BIP-135 at 75 mg/kg or its vehicle once a day. The tube test was performed at postnatal day 6, 8, 10, and 12. (a) Time spent hanging at the edge of the tube; (b) number of pulls; (c) hind-limb strength score; (d) tube test score. There was no significant treatment effect in the tube test parameters.
In summary, we have demonstrated that BIP-135 is a potent and reasonably selective GSK-3 inhibitor that is neuroprotective in a cortical neuron model of oxidative stress. More importantly, BIP-135 was able to elevate SMN protein levels in vitro and was found to extend the median survival period of transgenic mice bearing a severe SMA phenotype. This is the first report of a GSK-3 inhibitor to exert protective effects in an animal model of SMA, and this finding will hopefully support other efforts to test validated GSK-3 inhibitors in SMA. In addition, the evaluation of BIP-135 in a less severe mouse model of SMA is being pursued.
Methods
Drug
To examine the in vitro and in vivo effects of BIP-135 in SMA, this compound was resynthesized using our previously reported methods (see the Supporting Information).15 The staurosporine-related maleimide BIP-135 is a potent GSK-3 inhibitor (IC50 = 7– 21 nM, tested in the presence of 10 μM ATP). The overall purity of BIP-135 used in both the in vitro and in vivo experiments exceeds 98% according to high-performance liquid chromatography.
Kinase Selectivity
All kinase inhibition assays were conducted at Reaction Biology Corporation, Inc. (http://www.reactionbiology.com).
Animals
Male and female SMN2+/+;SMN2Δ7+/+;Smn+/– /(heterozygote knockout for Smn gene, HET) mice were purchased from Jackson Laboratories, Bay Harbor, ME (stock number 5025) and were bred to generate a self-sustaining colony of SMN2+/+;SMN2Δ7+/+;Smn+/– breeder mice. The breeder mice then generated the SMN2+/+; SMN2Δ7+/+; Smn–/– (SMA model mice, homozygote knockout for Smn, KO) as well as the SMN2+/+; SMN2Δ7+/+; Smn+/+ (wild type for Smn gene, WT) and SMN2+/+; SMN2Δ7+/+; Smn+/– (HET) control mice for behavioral phenotyping. All mice included in the present study were homozygous for human SMN2+/+ and SMN2Δ7+/+. One male was housed with 1–3 female mice until vaginal plugs were observed. The male was then removed. Pregnant females were housed individually in Plexiglas cages and were provided with nesting materials and enriched environments containing a plastic igloo, a flexible gnaw bone, and “Envirodri” bedding. Food and water were available ad libitum. All mice were maintained at a temperature of 21 °C on a 12 h light/dark cycle. All studies were approved by an Institutional Animal Care and Use Committee (IACUC) established at PsychoGenics Inc. according to the rules set out by the Public Health Service Office of Laboratory Animal Welfare.
At birth (defined as postnatal day 0, or P0), litters were randomly culled to 10 with equal numbers of males and females removed. Pups were tattooed using nontoxic ink applied under the skin and a tail snip sample was taken for genotyping. Genotyping was performed by Transnetyx Inc. Genotype data were normally available within 48 h after birth. Once the genotype results are known, the litters were further culled to a maximum of 8 pups per litter by removing the HET animals at P3. Litters with less than 6 pups at P3 were voided (thus litter size used ranged from 6 to 8 pups). Both body weight and survival were monitored daily for these mice. Mice were dosed once a day between 08:30 and 09:30 a.m. via intraperitoneal injection (IP) starting at P3 and continued until the KO pups died. Body weights for unused (not dosed) littermate WT and HET were taken at P10, P12, and P14 only to monitor the litter and dam overall health. Motor function assessments (negative geotaxis followed by the tube test) were performed in the afternoon at least 4 h post morning drug injection. The study end point for each KO mouse was death of the animal.
The Δ7 SMA mouse model closely mimics the human SMA genotype and results in mouse phenotype that resembles the human SMA Type I disease. A battery of tests was used to assess the effect of GSK-3 inhibition on the body weight, life span, and motor function (negative geotaxis and hind limb suspension tests, aka tube test) in the KO animals. Ten KO females and 13 males were treated with the GSK-3 inhibitor BIP-135 at 75 mg/kg, and 12 females and 13 males with vehicle (100% DMSO). As much as possible, littermate KO animals received different treatments. BIP-135 was dissolved in DMSO and injected once per day with a dosing volume of 2.5 mL/kg, IP. One female KO treated with BIP-135 was found dead at P4 (i.e., a day after receiving the first dose). This animal showed significant delay in body weight growth prior to the commencement of the treatment and thus was excluded from the analysis.
Motor Function Tests
Motor function tests (negative geotaxis and tube test) were performed as previously described.31
Statistical Analysis
Survival evaluation in the SMA study was performed using Kaplan–Meier analysis with Logrank (Mantel–Cox) and Breslow–Gehan–Wilcoxon as the post hoc tests. Body weight, negative geotaxis, and tube test parameters were analyzed using the Mixed Effects Model (also known as Mixed ANOVA Model) which is more robust to missing values caused by fatalities over time, and is based on likelihood estimation rather than moment estimation as in the typical repeated-measures ANOVA analysis. The treatment and gender were analyzed as independent and trials and age as dependent factors. Mixed model ANOVA was followed by simple effect and Tukey’s post hoc tests when indicated. ANOVAs were performed using the PROC MIXED procedure in SAS 9.1.3 (SAS Institute, Cary, NC). Values are presented as mean ± SEM. A p-value of <0.05 was considered statistically significant.
Primary Neurons, Cell Cultures, and Neuronal Viability Assays (Oxidative Stress-Induced Neurodegeneration)
Primary neurons, cell cultures, and neuronal viability assays were prepared and performed as previously described.26,33
Intracellular Total Glutathione Measurements
Intracellular total glutathione [glutathione (GSH) + oxidized glutathione (GSSG)] measurements of primary neuron cultures were determined using the GSH-Glo Glutathione Assay kit (Promega) according to the manufacturer’s protocol.26
SMA Patient Fibroblast, Cell Cultures, Immunostaining, and Immunoblot Analysis
SMA patient fibroblast, cell cultures, immununostaining, and immunoblot analysis were prepared and performed as previously described.13
Acknowledgments
We would like to give our thanks to Dr. Brett Langley for his input on the writing of the manuscript, as well as his work on the oxidative-stress induced model of neurodegeneration and glutathione measurement.
Glossary
Abbreviations
- SMA
spinal muscular atrophy
- GSK-3
glycogen synthase kinase-3
- MND
motor neuron disease
- ALS
amyotrophic lateral sclerosis
- SMN1
survival motor neuron 1
- SMN2Δ7
survival motor neuron 2 cDNA lacking exon 7
- HCA
homocysteic acid
- GSH
glutathione
- GSSG
oxidized glutathione
- IP
intraperitoneal
- CREB
cyclic AMP response element binding protein
- BDNF
brain-derived neurotrophic factor
- shRNA
short hairpin RNA
- SMN
survival motor neuron
Supporting Information Available
Synthetic scheme and kinase selectivity table of BIP-135. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Synthesis, purification, and kinase selectivity of BIP-135 were completed by P.C.C. and I.N.G. Preparation and phenotypic analysis of the SMA transgenic mice were performed by B.F.E. and S.R. Immunostaining and immunoblotting of SMA patient-derived fibroblast were accomplished by N.R.M and L.L.R. Experiment design, data analysis, writing, and editing were completed by P.C.C., I.N.G., B.F.E, S.R., N.R.M., L.L.R., and A.P.K.
The work was supported by NIH grants 1R01-MH07294001 (to A.P.K.) and 1-P01NS066888 (to L.L.R.), the Harvard Stem Cell Institute, the NINDS, and the Spinal Muscular Atrophy Foundation.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cohen P.; Goedert M. (2004) GSK3 inhibitors: Development and therapeutic potential. Nat. Rev. Drug Discovery 3, 479–487. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Frame S. (2002) The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2, 769–776. [DOI] [PubMed] [Google Scholar]
- Woodgett J. R. (1990) Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould T. D.; Mangi H. K. (2005) Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology 30, 1223–1237. [DOI] [PubMed] [Google Scholar]
- Tanis K. Q.; Duman R. S. (2007) Intracellular signaling pathways pave roads to recovery from mood disorders. Ann. Med. 39, 531–544. [DOI] [PubMed] [Google Scholar]
- Chuang D.-M.; Wang Z.; Chiu C.-T. (2011) GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front. Mol. Neurosci. 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden K. R.; Trojanowski J. Q.; Lee V. M.-Y. (2009) Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discovery 8, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chico L. K.; Van Eldik L. J.; Watterson D. M. (2009) Targeting protein kinase in central nervous system disorders. Nat Rev Drug Discovery 8, 892–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S. H.; Lee Y. B.; Kim K. S.; Kim H. J.; Kim M.; Lee Y. J.; Kim J.; Lee K. W.; Kim S. H. (2005) Role of GSK-3β activity in motor neuronal cell death induced by G93A or A4V mutant hSOD1 gene. Eur. J. Neurosci. 22, 301–309. [DOI] [PubMed] [Google Scholar]
- Koh S. H.; Kim Y.; Hwang S.; Lee C. H.; Kim S. H. (2007) Inhibition of glycogen synthase kinase-3 suppresses the onset of symptoms and disease progression of G93A-SOD1 mouse model of ALS. Exp. Neurol. 205, 336–346. [DOI] [PubMed] [Google Scholar]
- Sumner C. J. (2006) Therapeutics development for spinal muscular atrophy. NeuroRx 3, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskoui M.; Kaufmann P. (2008) Spinal muscular atrophy. Neurotherapeutics 5, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhortova N. R.; Hayhurst M.; Cerqueira A.; Sinor-Anderson A. D.; Zhao W. N.; Heiser P. W.; Arvanites A. C.; Davidow L. S.; Waldon Z. O.; Steen J. A.; Lam K.; Ngo H. D.; Rubin L. L. (2011) A screen for regulator of survival of motor neuron protein levels. Nat. Chem. Biol. 7, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L.; Flajolet M.; Greengard P. (2004) Pharmacological inhibitors of glycogen synthase kinase 3. Trends Phamacol. Sci. 25, 471–480. [DOI] [PubMed] [Google Scholar]
- Gaisina I. N.; Gallier F.; Ougolkov A. V.; Kim K. H.; Kurome T.; Guo S.; Holzle D.; Luchini D. N.; Blond S. Y.; Billadeau D. D.; Kozikowski A. P. (2009) From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase-3β inhibitors that suppress proliferation and survival of pancreatic cancer cell. J. Med. Chem. 52, 1853–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A. P.; Gaisina I. N.; Hongbin Y.; Petukhov P. A.; Blond S. Y.; Fedolak A.; Caldarone B.; McGonigle P. (2007) Structure-based design leads to the identification of lithium mimetics that block mania-like effects in rodents. Possible new GSK-3β therapies for bipolar disorders. J. Am. Chem. Soc. 129, 8328–8332. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P.; Gaisina I. N.; Petukhov P. A.; Sridhar J.; King L. T.; Blond S. Y.; Duka T.; Rusnak M.; Sidhu A. (2006) Highly potent and specific GSK-3β inhibitors that block tau phosphorylation and decrease α-synuclein protein expression in a cellular model of Parkinson’s disease. ChemMedChem 1, 256–266. [DOI] [PubMed] [Google Scholar]
- LaPointe N. E.; Morfini G.; Pigino G.; Gaisina I. N.; Kozikowski A. P.; Binder L. I.; Brady S. T. (2009) The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J. Neurosci. Res. 87, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Sagawa S.; Hoshi J.; Shimoma F.; Yasue K.; Ubukata M.; Ikemoto T.; Hase Y.; Takahashi M.; Sasase T.; Ueda N.; Matsushita M.; Inaba T. (2006) Synthesis, SAR studies, and pharmacological evaluation of 3-anilino-4-(3-indolyl)maleimides with conformationally restricted structure as orally bioavailable PKCβ-selective inhibitors. Bioorg. Med. Chem. 14, 5781–5794. [DOI] [PubMed] [Google Scholar]
- Mercer S. E.; Friedman E. (2006) Mirk/Dyrk1B: A multifunctional dual-specificity kinase involved in growth arrest, differentiation, and cell survival. Cell Biochem. Biophys. 45, 303–315. [DOI] [PubMed] [Google Scholar]
- Millino C.; Fanin M.; Vettori A.; Laveder P.; Mostacciuolo M. L.; Angelini C.; Lanfranchi G. (2009) Different atrophy-hypertrophy transcription pathways in muscles affected by severe and mild spinal muscular atrophy. BMC Med. 7, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M.; Araki S.; Arai N.; Kumada S.; Itoh M.; Tamagawa K.; Oda M.; Morimatsu Y. (2002) Oxidative stress and disturbed glutamate transport in spinal muscular atrophy. Brain Dev. 24, 770–775. [DOI] [PubMed] [Google Scholar]
- Ratan R. R.; Murphy T. H.; Baraban J. M. (1994) Oxidative stress induces apoptosis in embryonic cortical-neurons. J. Neurochem. 62, 376–379. [DOI] [PubMed] [Google Scholar]
- Barnham K. J.; Masters C. L.; Bush A. I. (2004) Neurodegenerative disease and oxidative stress. Nat. Rev. Drug Discovery 3, 205–214. [DOI] [PubMed] [Google Scholar]
- Acsadi G.; Lee I.; Khaidakov M.; Pecinova A.; Parker G. C.; Hüttemann M. (2009) Mitochondrial dysfunction in a neural cell model of spinal muscular atrophy. J. Neurosci. Res. 87, 2748–2756. [DOI] [PubMed] [Google Scholar]
- Langley B.; D’Annibale M. A.; Suh K.; Ayoub I.; Tolhurst A.; Bastan B.; Yang L.; Ko B.; Fisher M.; Cho S.; Beal M. F.; Ratan R. R. (2008) Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J. Neurosci. 28, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natasha G.; Brandom K. G.; Young E. C.; Young P. J. (2008) Valproate and spinal muscular atrophy (review). Mol. Med. Rep. 1, 161–165. [PubMed] [Google Scholar]
- Tsai L. K.; Tsai M. S.; Ting C. H.; Li H. (2008) Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J. Mol. Med. 86, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Sproule D. M.; Kaufmann P. (2010) Therapeutic development in spinal muscular atrophy. Ther. Adv. Neurol. Disord. 3, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T. T.; Pham L. T.; Butchbach M. E.; Zhang H. L.; Monani U. R.; Coovert D. D.; Gavrilina T. O.; Xing L.; Bassell G. J.; Burghes A. H. (2005) SMNDelta7, the major product of the centrometric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845–857. [DOI] [PubMed] [Google Scholar]
- El-Khodor B. F.; Edgar N.; Chen A.; Winberg M. L.; Joyce C.; Brunner D.; Suárez-Fariñas M.; Heyes M. P. (2008) Identification of a battery of tests for drug candidate evaluation in the SMN Delta7 neonate model of spinal muscular atrophy. Exp. Neurol. 212, 29–43. [DOI] [PubMed] [Google Scholar]
- Gladman J. T.; Bebee T. W.; Edwards C.; Wang X.; Sahenk Z.; Rich M. M.; Chandler D. S. (2010) A humanized Smn gene containing the SMN2 nucleotide alteration in exon 7 mimics SMN2 splicing and the SMA disease phenotype. Hum. Mol. Genet. 19, 4239–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivieccio M. A.; Brochier C.; Willis D. E.; Walker B. A.; D’Annibale M. A.; McLaughlin K.; Siddiq A.; Kozikowski A. P.; Jaffrey S. R.; Twiss J. L.; Ratan R. R.; Langley B. C. (2009) HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U.S.A. 106, 19599–19604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.