

Abstract
Background:
Rosai–Dorfman disease (RDD) was first described in 1969 as an idiopathic histiocytic proliferative disorder. It commonly presents as a massive and painless adenopathy. Until 1990, extranodal involvement of the central nervous system (CNS) was rare and reported in less than 5% of the total number of patients with extranodal RDD. Complete removal of CNS RDD has been achieved in many cases.
Case Description:
We report a case of an isolated intracranial RDD in a 53-year-old man. The patient had an episode of generalized seizures. Imaging studies of the brain were compatible with a meningioma en plaque. The mass was exposed by a right frontotemporal craniotomy. The tumor was adhered tightly to the adjacent cerebral cortex and was permeated by pial arteries of the brain surface. The sacrificing of these arteries was inevitable in order to achieve the total removal of the tumor. The patient had incomplete left hemiparesis after the surgery. Brain computed tomography (CT) imaging revealed a postoperative hemorrhage and a low-density lesion in the right frontal lobe. The patient was postoperatively diagnosed with isolated central nervous system RDD.
Conclusion:
Although the complete removal of dural-based lesions without any neurological deficits has been performed in many cases, the treatment of cases with high risks, such as the present case, indicates conservative excisions and adjuvant radiotherapy with or without chemotherapy.
Keywords: En plaque meningioma, intracranial, meningioma, Rosai–Dorfman disease
INTRODUCTION
Rosai–Dorfman disease (RDD) was first described in 1969 as an idiopathic histiocytic proliferative disorder.[13] It commonly presents as a massive and painless adenopathy. RDD commonly involves the cervical nodes, and extranodal involvement is seen in 43% of the cases.[7] Rarely, sites other than the lymph nodes are involved, including the skin, the orbits, and the breast. Until 1990, extranodal involvement of the central nervous system (CNS) was rare and reported in less than 5% of the total number of patients with extranodal RDD. Isolated CNS RDD without other involvement is more exceptional.[2]
However, there has been a significant increase in the number of isolated CNS RDD cases that have been reported in the last decade. Over 100 cases of CNS RDD have now been reported.[1] Complete removal has been achieved in many cases. However, the mass is sometimes adhered tightly to the cerebral cortex. We report on a case of isolated CNS RDD that mimicked meningioma en plaque and that had a high risk for the total removal of the mass.
CASE REPORT
A 53-year-old homeless man was admitted to the hospital due to a right femoral neck fracture. His medical history included noninsulin-dependent diabetes mellitus. A physical examination performed at admission was unremarkable. There was no fever, lymphadenopathy, or other neurological defects. Laboratory tests revealed the following results: white blood cells, 4330 cells/mL; hemoglobin, 9.2 g/dL; and platelet count, 29,900 platelets/mL. C-reactive protein was negative, and only normochromic-normocytic anemia was present. He successfully underwent surgery for the femoral neck fracture, which was performed by an orthopedic surgeon. However, he had an episode of generalized seizures during the postoperative course, and phenytoin was administered. After this episode of generalized seizures, he was referred to our department for further examination and treatment.
A computed tomography (CT) examination of the head that was conducted without contrast showed a high-density and extraaxial mass in the right parietal convexity, and peritumoral brain edema was clearly observed [Figure 1]. Enhanced magnetic resonance imaging (MRI) revealed an extraaxial, dural-based, and homogeneously enhanced mass with clear borders that was compatible with a meningioma en plaque [Figure 2]. The mass was iso- to hypointense on fluid-attenuated inversion recovery and T2 imaging sequences and iso- to hyperintense on T1 imaging. It was strongly suspected that the patient had a meningioma en plaque.The mass was exposed with a right frontotemporal craniotomy. The tumor appeared to be a meningioma en plaque, and it was extraaxial, xanthochromic, firm, nonaspiratable, and it seemed to have a high vascularity and was dural based. The tumor was tightly adhered to the adjacent cerebral cortex and was permeated by many pial arteries and veins of the brain surface. It is very difficult to preserve these pial vessels during the total removal of a tumor.
Figure 1.
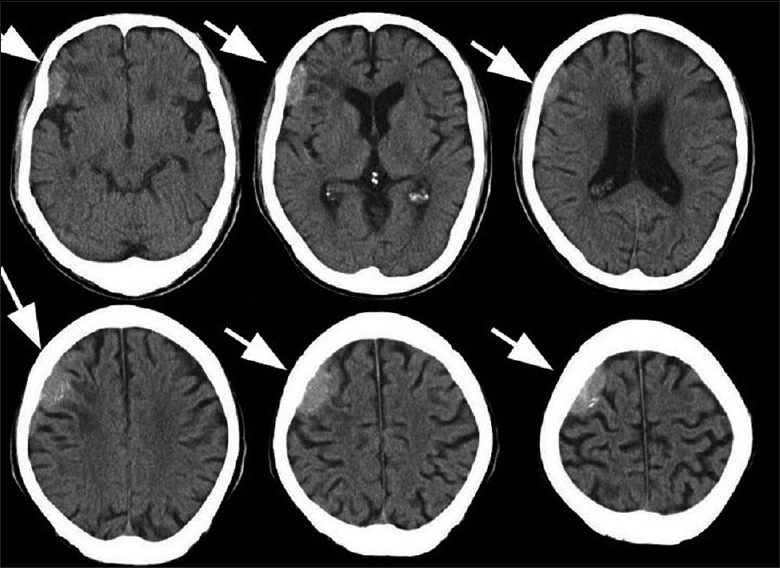
Preoperative brain computed tomography scan conducted without contrast showing a high-density and extraaxial mass in the right parietal convexity and peritumoral brain edema
Figure 2.
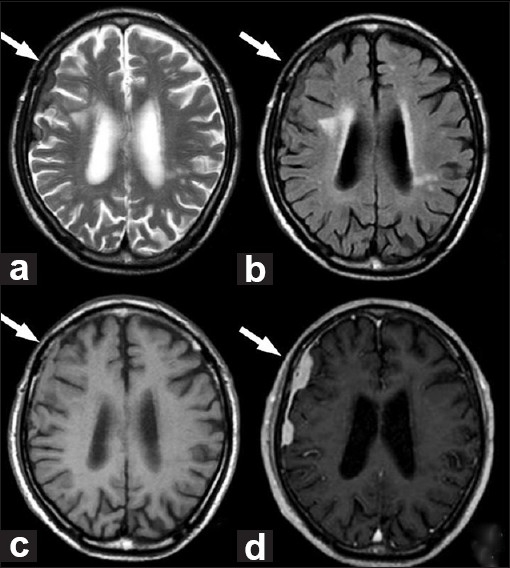
(a) T2-weighted magnetic resonance images. (b) Fluid-attenuated inversion recovery. (c) T1-weighted images. (d) T1-weighted magnetic resonance image with gadolinium
A frozen section of the lesion showed inflammatory cell infiltration, which mainly consisted of lymphocytes and plasma cells, and the presence of these cells was initially interpreted as some kind of hematologic disorder or inflammatory pseudotumor [Figure 3a]. Paraffin-embedded sections, however, showed a hypercellular pattern with features of polymorphous and mixed inflammatory infiltrates that were composed mainly of histiocytes in a background of collagen fibers [Figure 3b]. The cytoplasm in some histiocytes was foamy and eosinophilic. Some histiocytes were seen to engulf viable lymphocytes, which was thought to reflect emperipolesis (lymphophagocytosis) [Figure 3c].
Figure 3.
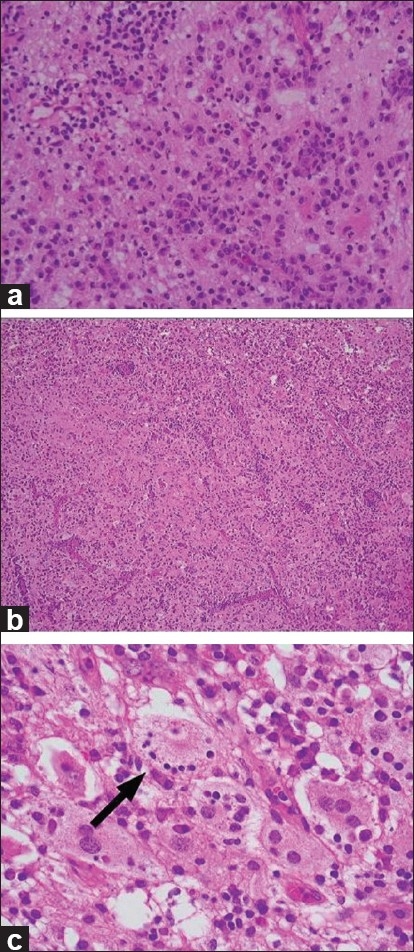
(a) Photomicrographs of the frozen sections showing inflammatory cell infiltration, which consisted of lymphocytes and plasma cells, which was initially interpreted as a hematologic disorder or an inflammatory pseudotumor. (b) Paraffin-embedded sections show a hypercellular pattern with features of polymorphous and mixed inflammatory infiltrate that was composed of mainly histiocytes in a background of collagen fibers. (c) The cytoplasm in some histiocytes was foamy and eosinophilic. Some histiocytes were seen to engulf viable lymphocytes, and this was thought to be indicative of emperipolesis (arrow)
These histiocytes were immunopositive for S-100 protein and CD68, but negative for CD1α [Figure 4]. All of these findings were consistent with extranodal RDD.
Figure 4.
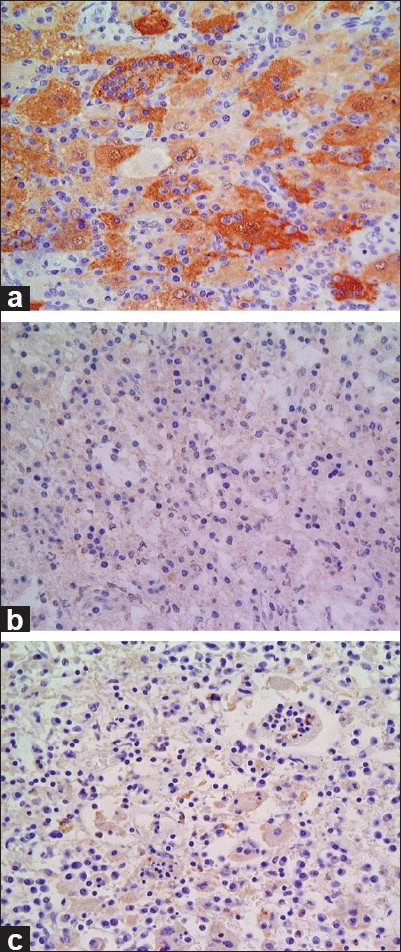
Photomicrograph of immunostaining. These histiocytes were immunopositive for (a) S-100 protein and (b) CD68, but negative for (c) CD1α
The patient had incomplete left hemiparesis after surgery. A brain CT examination conducted without contrast revealed postoperative hemorrhage and a low-density lesion in the right frontal lobe, which seemed to be due to the sacrificed pial arteries from the brain-tumor interface. His seizures completely ceased.
In order to find evidence of extra-CNS RDD, we performed a whole-body CT examination postoperatively, and findings of lymphadenopathy or other extranodal involvements were not found. The patient has been free of seizures since the surgery, and he recovered from his hemiparesis 6 months after the surgery.
DISCUSSION
It is known that intracranial RDD may mimic a meningioma in clinical and radiographic findings as well as in surgical findings. Despite the significant increase in the number of reported CNS RDD cases, isolated intracranial RDD remains an underdiagnosed entity. This is, at least in part, due to the fact that neurosurgeons typically encounter meningiomas rather than RDD. Approximately 75% of all CNS RDD cases are intracranial, and 90% cases involve the leptomeninges.[4]
In the present case, the typical clinical and laboratory findings of systemic RDD, such as lymphadenopathy and an elevation of the erythrocyte sedimentation rate, were not present. Only normocytic anemia was indicative of systemic RDD. Whole-body CT showed that among patients with intracaranial RDD, lymphadenopathy and associated systemic disease were absent in 70% and 52% of the cases, respectively.[10,14]
Intracranial RDD mimics meningioma in many aspects of the radiographic appearances and it is still challenging to differentiate between these two diseases. RDD, which displays as an isodense and dura mater-seated mass, is often accompanied by marked peritumoral brain edema on the contiguous cerebral surface on CT and resembles meningioma. The presence of calcification has not been reported in RDD.
RDD lesions are seen as iso- to hyperintense with clear borders on MRI T1 sequences and iso- to hypointense on MRI T2 sequences, and they display obvious enhancement after contrast administration.[19] The existence of low-signal intense T2 foci within the lesion has been reported as a characteristic feature of RDD. However, the low-signal foci may represent the existence of free radicals that are produced by macrophages during phagocytosis.[17,18] This feature was not found in our case.The correct diagnosis of RDD primarily relies on histological and immunohistochemical findings. Microscopic examination reveals a polymorphous infiltration of histiocytes, lymphocytes, and plasma cells in fibrous stroma. The RDD histiocytes contained single, and sometimes multiple, nuclei that were larger and more hyperchromatic than those of usual histiocytes.[5] Many RDD histiocytes had voluminous and eosinophilic cytoplasm. Lymphophagocytosis by RDD histiocytes, which is a characteristic finding of RDD, is called emperipolesis. It is a nonspecific but diagnostic feature of RDD. However, these characteristic findings of microscopic examinations are sometimes unclear in frozen sections that are examined for intraoperative diagnosis. Actually, it was difficult to diagnose RDD with frozen sections during the operation in our case.Chen reported that crush cytology is useful when performed alone or in conjunction with frozen sections in the intraoperative diagnosis of intracranial RDD.[5] He also reported that the emperipolesis and nuclear details in the RDD histiocytes were more visible in the crush smears than in the frozen sections. Another author reported that cases of RDD in bone lesions were diagnosed by microscopic findings of emperipolesis in fine needle aspiration smears.[16] Crush cytology is especially useful in cases that are difficult to be diagnosed with frozen sections. The intraoperative diagnosis of RDD is essential for preventing postoperative neurological deficits in cases with high risks for the total removal, as in our case.Although therapy remains controversial, most intracranial RDD has been treated surgically with satisfactory results. Recurrence after surgery has been reported in only 14% of the cases.[1] In our review, most cases of intracranial RDD mimicking meningioma along the convexity have been resected successfully without any postoperative neurological deficits.
However, a mass lesion of the intracranial RDD often tightly adheres to the adjacent cerebral cortex and its complete removal is sometimes difficult. Our case report is unique in that the mass was located along the convexity and was directly permeated by many pial arteries of the brain surface. It was impossible to preserve these penetrating arteries in the total removal of the tumor. Our patient had postoperative left hemiparesis that was thought to be due to secondary cortical ischemia caused by the sacrificed penetrating pial arteries of the cortex. In a study of meningioma, Inamura et al. and Michael et al. reported that the development of the vascular supply from intrinsic cerebral arteries on angiography significantly correlated with severe peritumoral brain edema.[9,11] The pial blood supply to the tumor has been considered a causative factor in peritumoral edema in meningioma. The pial supply of meningiomas also reflects the adherence of the tumor surface to the adjacent cerebral cortex.
We observed both peritumoral brain edema on the CT examination and the penetrating vascular supply from intrinsic cerebral arteries in our intracranial RDD case. This relationship, which is reported for meningiomas, is consistent with our case. In addition, we consider that the findings of peritumoral brain edema on the CT examination in intracranial RDD is an effective predictor of penetrating blood vessels and the severe adherence between the mass surface and the adjacent cerebral cortex, which is just as likely as with meningioma. The finding of peritumoral brain edema in intracranial RDD is considered a risk factor for total removal. Considering the good prognosis after surgery of partially resected intracranial RDD, complete removal should be avoided in cases with high risk, as our case.
Postoperative intraparenchymal hemorrhage was observed in our case. We consider that venous outflow impairment due to sacrificed pial vein is one of the most considerable causes of the intraparenchymal hemorrhage. Parenchymal damage due to drag force and compression during peeling procedure of the adherent tumor, and ischemia due to sacrificed pial arteries are also considered as exacerbating factors. The adjuvant treatment modalities for recurrent intracranial lesions or residual lesions, i.e. radiosurgery and systemic corticosteroids, have been reported with success.[3,8,12,15] Petzold et al. reported on the resolution of a relapsing intracranial RDD that was treated by fractionated irradiation.[12] El Majdoub et al. reported on a case of intracranial RDD that was completely treated by stereotactic interstitial radiosurgery alone.[6] Hinduja et al. reported on a case of intraorbital RDD who underwent surgical debulking and was successfully treated with postoperative radiation therapy for the residual lesion.[16] However, data on the long-term outcomes of patients with CNS involvement are still limited. Adeleye et al. reported that follow-up examinations were 1 year or less in 40% of the cases with radical removals, and only 17% reported outcomes of 3 years or longer after treatment.[1] Due to the high radiosensitivity and good prognosis of intracranial RDD, cases with high risks for the total removal, as our case, are thought to have good indications for the avoidance of complete removal and postoperative adjuvant radiotherapy. Intraoperative diagnosis and the knowledge of RDD as a differential diagnosis of meningioma are necessary to perform an operation for the suspicion of meningioma.
Footnotes
Available FREE in open access from: http://www.surgicalneurologyint.com/text.asp?2012/3/1/1/92161
Contributor Information
Ryosuke Tomio, Email: tomy0807@hotmail.com.
Makoto Katayama, Email: makoto.katayama@gmail.com.
Nobuo Takenaka, Email: takenaka-n@city.kawasaki.jp.
Tomoyuki Imanishi, Email: ns-imanishi@kmh.gr.jp.
REFERENCES
- 1.Adeleye AO, Amir G, Fraifeld S, Shoshan Y, Umansky F, Spektor S. Diagnosis and management of Rosai–Dorfman disease involving the central nervous system. Neurol Res. 2010;32:572–8. doi: 10.1179/016164109X12608733393836. [DOI] [PubMed] [Google Scholar]
- 2.Andriko JA, Morrison A, Colegial CH. Rosai-Dorfman disease isolated to the central nervous system: A report of 11 cases. Mod Pathol. 2001;14:172–8. doi: 10.1038/modpathol.3880278. [DOI] [PubMed] [Google Scholar]
- 3.Antonius JI, Farid SM, Baez-Giangreco A. Steroid-responsive Rosai-Dorfman disease. Pediatr Hematol Oncol. 1996;13:563–70. doi: 10.3109/08880019609030873. [DOI] [PubMed] [Google Scholar]
- 4.Bhandari A, Patel PR, Patel MP. Extranodal Rosai-Dorfman disease with multiple spinal lesions: A rare presentation. Surg Neurol. 2006;65:308–11. doi: 10.1016/j.surneu.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 5.Chen KT. Crush cytology of Rosai-Dorfman disease of the central nervous system: A report of 2 cases. Acta Cytol. 2003;47:1111–5. doi: 10.1159/000326659. [DOI] [PubMed] [Google Scholar]
- 6.El Majdoub F, Brunn A, Berthold F, Sturm V, Maarouf M. Stereotactic interstitial radiosurgery for intracranial Rosai-Dorfman disease: A novel therapeutic approach. Strahlenther Onkol. 2009;185:109–12. doi: 10.1007/s00066-009-1911-1. [DOI] [PubMed] [Google Scholar]
- 7.Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the entity. Semin Diagn Pathol. 1990;7:19–73. [PubMed] [Google Scholar]
- 8.Geara AR, Ayoubi MA, Achram MC, Chamseddine NM. Rosai-Dorfman disease mimicking neurofibromatosis: Case presentation and review of the literature. Clin Radiol. 2004;59:625–30. doi: 10.1016/j.crad.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Inamura T, Nishio S, Takeshita I, Fujiwara S, Fukui M. Peritumoral brain edema in meningiomas-influence of vascular supply on its development. Neurosurgery. 1992;31:179–85. doi: 10.1227/00006123-199208000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Kattner KA, Stroink AR, Roth TC, Lee JM. Rosai-Dorfman disease mimicking parasagittal meningioma: Case presentation and review of literature. Surg Neurol. 2000;53:452–7. doi: 10.1016/s0090-3019(00)00197-x. [DOI] [PubMed] [Google Scholar]
- 11.Michael B, Lars W, Andreas RL, Ajay KW, Dirk P, Holger O, et al. The importance of pial blood supply to the development of peritumoral brain edema in meningiomas. J Neurosurg. 1997;87:368–73. doi: 10.3171/jns.1997.87.3.0368. [DOI] [PubMed] [Google Scholar]
- 12.Petzold A, Thom M, Powell M, Plant GT. Relapsing intracranial Rosai-Dorfman disease. J Neurol Neurosurg Psychiatry. 2001;71:538–41. doi: 10.1136/jnnp.71.4.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy.A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63–70. [PubMed] [Google Scholar]
- 14.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy: A pseudolymphomatous benign disorder.Analysis of 34 cases. Cancer. 1972;30:1174–80. doi: 10.1002/1097-0142(197211)30:5<1174::aid-cncr2820300507>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 15.Shaver EG, Rebsamen SL, Yachnis AT, Sutton LN. Isolated extranodal intracranial sinus histiocytosis in a 5-year-old boy: A case report. J Neurosurg. 1993;79:769–73. doi: 10.3171/jns.1993.79.5.0769. [DOI] [PubMed] [Google Scholar]
- 16.Shaoying L, Zhijie Y, Nirag J, Darshana J. Fine needle aspiration diagnosis of Rosai-Dorfman disease in an osteolytic lesion of bone. Cytojournal. 2010;7:12. doi: 10.4103/1742-6413.65058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simos M, Dimitrios P, Philip T. A new clinical entity mimicking meningioma diagnosed pathologically as Rosai-Dorfman disease. Skull Base Surg. 1998;8:87–92. doi: 10.1055/s-2008-1058581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Udono H, Fukuyama K, Okamoto H, Tabuchi K. Rosai-Dorfman disease presenting multiple intracranial lesions with unique findings on magnetic resonance imaging: A case report. J Neurosurg. 1999;91:335–9. doi: 10.3171/jns.1999.91.2.0335. [DOI] [PubMed] [Google Scholar]
- 19.Yin W, Xiang G, Weijun T, Chengchuan J. Rosai-Dorfman disease isolated to the central nervous system: A report of six cases. Neuropathology. 2010;30:154–8. doi: 10.1111/j.1440-1789.2009.01045.x. [DOI] [PubMed] [Google Scholar]