

Abstract
Inflammation contributes to the pathogenesis of acute kidney injury (AKI). IL-33 is a proinflammatory cytokine, but its role in AKI is unknown. Here we observed increased protein expression of full-length IL-33 in the kidney following induction of AKI with cisplatin. To determine whether IL-33 promotes injury, we administered soluble ST2 (sST2), a fusion protein that neutralizes IL-33 activity by acting as a decoy receptor. Compared with cisplatin-induced AKI in untreated mice, mice treated with sST2 had fewer CD4 T cells infiltrate the kidney, lower serum creatinine, and reduced acute tubular necrosis (ATN) and apoptosis. In contrast, administration of recombinant IL-33 (rIL-33) exacerbated cisplatin-induced AKI, measured by an increase in CD4 T cell infiltration, serum creatinine, ATN, and apoptosis; this did not occur in CD4-deficient mice, suggesting that CD4 T cells mediate the injurious effect of IL-33. Wildtype mice that received cisplatin and rIL-33 also had higher levels of the proinflammatory chemokine CXCL1, which CD T cells produce, in the kidney compared with CD4-deficient mice. Mice deficient in the CXCL1 receptor also had lower serum creatinine, ATN, and apoptosis than wildtype mice following cisplatin-induced AKI. Taken together, IL-33 promotes AKI through CD4 T cell-mediated production of CXCL1. These data suggest that inhibiting IL-33 or CXCL1 may have therapeutic potential in AKI.
Cisplatin and other platinum derivatives are important chemotherapeutic agents used to treat solid tumors, including ovarian, head and neck, and testicular germ cell tumors.1 A known complication of cisplatin administration is acute kidney injury (AKI).2 The nephrotoxic effect of cisplatin is cumulative and dose-dependent and often necessitates dose reduction or withdrawal.1 Therefore, an understanding of the pathogenesis of cisplatin-induced AKI is important for the development of therapies to prevent this complication.
We have demonstrated that there is proximal tubular apoptosis and necrosis, and a massive increase, in both proinflammatory caspase-1 and proapoptotic caspase-3 in the kidney in cisplatin-induced AKI in mice..3 IL-33 is a proinflammatory cytokine.4 It has been proposed that IL-33 is released from necrotic cells, binds the ST2R on immune cells, and increases secretion of cytokines, with resultant inflammation.5 In apoptosis, caspase-3 cleaves and lessens IL-33 binding to the ST2R.5–8 As IL-33 is modulated by necrosis and apoptosis, and increases in necrosis and apoptosis are features of cisplatin-induced AKI, we determined the effect of cisplatin on IL-33 in mice.
The role of inflammation in AKI is gaining prominence9; specifically, CD4 T-cell inflammation has been described to play an injurious role in cisplatin-induced AKI in mice.10 IL-33 is known to be a chemoattractant for CD4 T cells via the ST2R.11–14 As inflammation with CD4 T cells is a feature of cisplatin-induced AKI, we propose that IL-33 induces CD4 T-cell infiltration in the kidney, with resultant kidney injury. Thus, we developed the hypothesis that injection of sST2, a fusion protein that neutralizes IL-33 activity by acting as a decoy receptor, would be protective against cisplatin-induced AKI by reducing CD4 T cell infiltration in the kidney.
On this background, we sought to determine the role of IL-33 in cisplatin-induced AKI. We hypothesize that IL-33 plays an injurious role in cisplatin-induced AKI. The role of IL-33 has not previously been described in any kidney disease.
RESULTS
Results are on day 3 after cisplatin (25 mg/kg), unless otherwise specified.
Serum IL-33
Serum IL-33, measured by enzyme-linked immunosorbent assay (ELISA), was significantly increased on day 2 compared with vehicle, days 1 and 3 (Figure 1). The increase in serum IL-33 on day 2 preceded the significant increase in serum creatinine that occurs on day 3 after cisplatin (see Methods). IL-33 was undetectable in the urine by ELISA. The lack of IL-33 in the serum on day 3 and the further increase in IL-33 on day 3 in the kidney suggest that the kidney is not a major source of serum IL-33.
Figure 1.
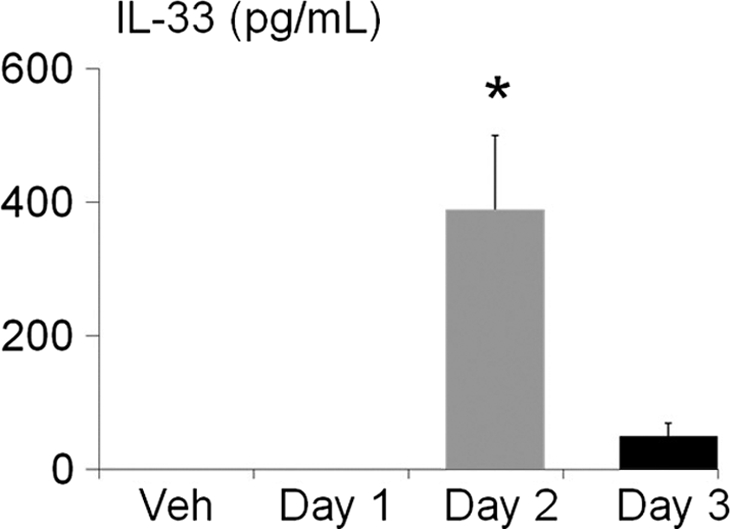
IL-33 in serum. Serum IL-33, measured by Enzyme-linked immunosorbent assay (ELISA), was significantly increased on day 2 of cisplatin-induced AKI compared with vehicle (Veh), days 1 and 3. *P < 0.01 versus vehicle (Veh), days 1 and 3.
Immunofluorescence for IL-33
Immunofluorescence for IL-33 in the kidney was performed in vehicle-treated mice. On double staining for IL-33 and lectin, IL-33 was clearly seen in glomeruli and also in peritubular areas (Figure 2, A). To determine whether the IL-33 staining in glomeruli and peritubular areas was in endothelium, double IF staining for IL-33 and von Willebrand factor (vWF) was performed. IL-33 co-localized with vWF in glomeruli and in peritubular areas, suggesting staining in peritubular capillaries (Figure 2, B and C). To determine whether IL-33 was present in larger blood vessels, double staining with vascular α-smooth muscle actin and IL-33 was performed. IL-33 staining was seen in the endothelial areas of blood vessels (Figure 2, D).
Figure 2.

Immunofluorescence staining for IL-33 in vehicle-treated mouse kidneys. (A) Staining for lectin (green) and IL-33 (red) demonstrates IL-33 (red) in glomeruli and in the peritubular spaces. Staining for von Willebrand factor (red) and IL-33 (green) demonstrates colocalization of vWF and IL-33 (yellow-orange) in glomeruli and in peritubular capillaries (arrows) in cortex (B) and outer stripe of outer medulla (C). (D) Staining for vascular α-smooth muscle actin (green) and IL-33 (red) demonstrates IL-33 staining on the endothelial surface of blood vessels.
No major differences in IL-33 staining between vehicle and cisplatin-treated mice were seen. However, as the monoclonal Ab (Nessy-1; see Methods) used for IF detects both full-length and cleaved IL-33, and IF is not optimal for quantitation, we used ELISA and immunoblotting rather than IF to quantify full-length and cleaved IL-33.
Immunoblotting and ELISA for IL-33
On ELISA of whole kidney, IL-33 was increased on days 1, 2, and 3 of cisplatin-induced AKI compared with vehicle-treated mice (Figure 3, A). On immunoblot of the whole kidney, there was an increase in both the full-length (34 kD) and cleaved (22 kD) IL-33 on days 2 and 3 of cisplatin-induced AKI compared with vehicle-treated mice (Figure 3, B).
Figure 3.

Cisplatin increases IL-33. (A) On ELISA of whole kidney, IL-33 was increased on days 1, 2, and 3 of cisplatin-induced AKI. *P < 0.05 compared with vehicle-treated (Veh); **P < 0.01 versus Veh. (B) On immunoblot of whole kidney, there was an increase in both the full-length (34 kD) and cleaved (22 kD) IL-33 on days 2 and 3 of cisplatin-induced AKI. (C) On ELISA of nuclear extracts of whole kidney, IL-33 was increased on days 1, 2, and 3 of cisplatin-induced AKI. *P < 0.001 compared with vehicle-treated (Veh). (D) On immunoblot of nuclear extracts of whole kidney, there was an increase in the full-length (34 kD) IL-33 on day 1, 2, and 3 and cleaved (22 kD) IL-33 on day 3 of cisplatin-induced AKI. Representative immunoblots of at least three independent experiments are demonstrated. Equal protein loading was confirmed by staining the same membrane for actin (43 kD) or the nuclear protein Lamin A (69 kD).
IL-33 has nuclear localization in the cell.4 On ELISA of nuclear extracts, IL-33 was more than 100% increased on days 1, 2, and 3 of cisplatin-induced AKI compared with vehicle-treated mice (Figure 3, C). On immunoblot of nuclear extracts, there was an increase in the full-length (34 kD) on days 1, 2, and 3, and cleaved (22 kD) IL-33 on day 3, of cisplatin-induced AKI compared with vehicle-treated mice (Figure 3, D).
Microvascular Endothelial Cells
To determine whether cisplatin causes release of IL-33 in vitro, microvascular endothelial cells in culture were studied. Microvascular endothelial cells were treated with 10 μM cisplatin to induce apoptosis and 50 μM cisplatin to induce necrosis.15 Full-length (34 kD) IL-33 increased in the medium of endothelial cells treated with 50 μM cisplatin compared with endothelial cells treated with the vehicle (Figure 4). No cleaved IL-33 was detected in the medium.
Figure 4.
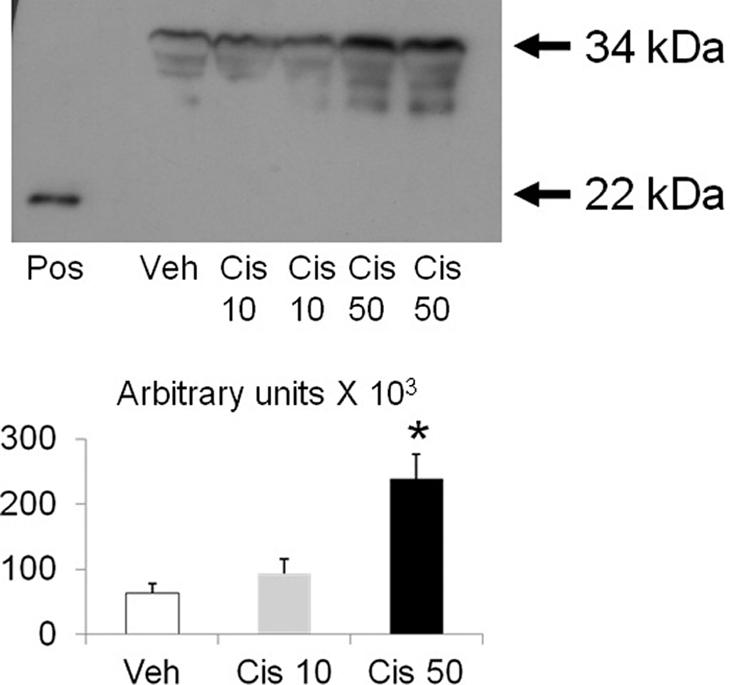
In cisplatin-treated endothelial cells, there is release of the full-length IL-33 into the medium. Microvascular endothelial cells were treated with 10 μM cisplatin that we have described to induce apoptosis and 50 μM cisplatin that produces necrosis.15 On immunoblot of the medium of the cisplatin-treated endothelial cells, there was an increase in the full-length (34 kD) IL-33 with 50 μM cisplatin compared with vehicle-treated. Pos = positive control: recombinant cleaved IL-33 (22 kD). *P < 0.05 versus vehicle (Veh) and Cisplatin 10 μM (Cis 10); n = 3 per group.
Evan's Blue Dye In Vivo Leakage Assay
To determine whether cisplatin causes vascular injury in vivo, an Evan's blue dye in vivo leakage assay was performed, as described in the Methods section. There was increased leakage of dye in the lung on day 2, and in the kidney on days 1 and 2, of cisplatin-induced AKI compared with vehicle-treated controls. Evan's blue dye in the lung (μg/g of lung weight) was 183.7 ± 16.8 in vehicle-treated mice and 287.7 ± 14.1 on day 2 of cisplatin-induced AKI (P < 0.01 versus vehicle-treated mice; n = 5 per group). Evan's blue dye in the kidney (μg/g of kidney weight) was 58.9 ± 3.0 in vehicle-treated mice, 77.3 ± 4 on day 1 of cisplatin-induced AKI (P < 0.05 versus vehicle-treated mice), and 108.1 ± 4 on day 2 of cisplatin-induced AKI (P < 0.01 versus vehicle-treated mice, P < 0.05 versus day 1; n = 5 to 7 per group).
Soluble ST2 (sST2) Injection
To determine whether inhibition of IL-33 protects against cisplatin-induced AKI, mice were injected with soluble ST2 (sST2), a fusion protein that neutralizes IL-33 activity by acting as a decoy receptor. Mice with cisplatin-induced AKI injected with sST2 had a significant decrease in creatinine (Figure 5, A), ATN score (Figure 5, B), and number of apoptotic tubular cells (Figure 5, C).
Figure 5.

sST2 injection protects against cisplatin-induced AKI. Injection of cisplatin plus sST2 (Cis+sST2) resulted in a significant decrease in (A) creatinine, (B) ATN score, and (C) number of apoptotic tubular cells compared with cisplatin plus vehicle for sST2 (Cis+Veh). *P < 0.001 versus Veh; **P < 0.01 versus Cis+Veh; NS versus Veh; n = 5 to 11 per group.
To determine whether inhibition of IL-33 increases survival in cisplatin-induced AKI, mice were injected with sST2 or vehicle on days 1, 2, and 3 after cisplatin injection. On day 4 of cisplatin-induced AKI, 0/10 vehicle-treated mice survived, whereas 3/6 sST2-treated mice survived (P < 0.05). On day 5, 0/6 sST2-treated mice survived. The model of cisplatin-induced AKI is a severe and irreversible model of cisplatin-toxicity. On day 4 after cisplatin injection, mice become severely systemically ill, do not eat or drink, and require sacrifice. The exact cause of death is not known. In addition to AKI, cisplatin also causes liver injury, gastrointestinal ulcerations, myocardial injury, lung injury, and neurotoxicity in animals.16–19 Thus, it is possible that the mice died of other organ toxicities in addition to AKI. While sST2 protected against cisplatin-induced AKI on day 3, injection of sST2 did not provide protection against the severe systemic toxicity of cisplatin.
IL-33 Injection
Injection of rIL-33 into wildtype mice had no effect on serum creatinine or infiltration of CD4 T cells into the kidney on flow cytometry. To determine if rIL-33 aggravated cisplatin-induced AKI, mice were injected with a lower dose of cisplatin (15 mg/kg) that causes a smaller increase in serum creatinine than we have described with 25 mg/kg cisplatin. Injection of rIL-33 plus cisplatin (15 mg/kg) caused an increase in serum creatinine, ATN score, and apoptosis score compared with injection of cisplatin (15 mg/kg) alone (Figure 6, A, B, and C). The increase of serum creatinine, ATN score, and apoptosis score with injection of rIL-33 plus cisplatin (15 mg/kg) was not seen in CD4 T-cell-deficient mice (Figure 6, A, B, and C).
Figure 6.

IL-33 injection aggravates cisplatin-induced AKI. Injection of low-dose cisplatin (15 mg/kg) plus rIL-33 in wildtype mice (WT Cis + IL-33) resulted in a threefold increase in serum creatinine compared with cisplatin alone (WT Cis). The increase in (A) creatinine, (B) ATN score, and (C) apoptotic tubular cells with cisplatin plus rIL-33 in wildtype mice (WT Cis + IL-33) was not seen in cisplatin plus rIL-33 in CD4-T-cell-deficient mice (CD4−/− Cis +IL-33). *P < 0.001 versus WT Cis and CD4 −/− Cis+IL-33; n = 3 to 6 per group.
Flow Cytometry for CD4 T Cells
CD4 T cells were absent in spleens of CD4 −/− mice compared with wildtype mice. In the kidney, the number of CD4 positive cells as a percentage of total live cells is shown in Figure 7, A. In wildtype kidneys, CD4 T cells were increased with cisplatin (25 mg/kg) treatment on days 1, 2, and 3 compared with vehicle treatment alone. Injection of rIL-33 plus cisplatin (15 mg/kg) resulted in a significant increase in CD4 T cells in the kidney compared with cisplatin (15 mg/kg) alone. CD4 T cells in the kidney were absent in CD4 −/− mice treated with cisplatin (15 mg/kg) plus rIL-33, and significantly decreased in wildtype mice treated with cisplatin (25 mg/kg) plus sST2 injection. Representative pictures of flow cytometry are shown in Figures 7, B-G.
Figure 7.

Flow cytometry for CD4 T cells in kidney. The number of CD4 + cells as a percentage of total live cells in the kidney is demonstrated in panel A. In wildtype kidneys, CD4 T cells were increased with cisplatin (25 mg/kg)-treatment on days 1, 2, and 3 (D1, D2, and D3) compared with vehicle treatment alone (Veh). Injection of rIL-33 plus cisplatin (15 mg/kg; Cis 15 + IL-33) resulted in a significant increase in CD4 T cells in kidney compared with cisplatin (15 mg/kg; Cis 15) alone. CD4 T cells in the kidney were absent in CD4-deficient mice treated with cisplatin (15 mg/kg) plus rIL-33 (Cis 15 + IL-33 CD4 −/−) and significantly decreased in wildtype mice treated with cisplatin (25 mg/kg) plus sST2 injection (Cis 25+sST2). *P < 0.05 versus vehicle-treated (Veh); **P < 0.05 versus cisplatin 15 mg/kg (Cis 15) or cisplatin 25 mg/kg (Cis 25); n = 3 to 6 per group. Representative pictures of flow cytometry are demonstrated in panels B to I. In Figures B to I, CD4 T cells are represented by Quadrant 2 (Q2), which includes cells strongly positive for CD3 and CD4. B, vehicle-treated; C, cisplatin 25 mg/kg day 1; D, cisplatin 25 mg/kg day 2; E, cisplatin 25 mg/kg day 3; F, cisplatin 15 mg/kg; G, cisplatin 15 mg/kg plus rIL-33 injection; H, cisplatin 15 mg/kg plus rIL-33 injection in CD4-deficient mice; I = cisplatin 25 mg/kg plus sST2 injection.
ST2 is known to be expressed on Th2 but not Th1 cells.11 Flow cytometry for CD4 and ST2 was performed in the kidney. In vehicle-treated mice, the percentage of CD4 T cells that were also ST2 positive was 48% ± 10 (n = 3). On day 3 after cisplatin, the percentage of CD4 T cells that were also ST2 positive was 97% ± 1 (n = 3). Thus, most of the CD4 T cells detected on flow cytometry in the kidney in cisplatin-induced AKI were also ST2 positive, indicating that these cells were Th2 CD4 T cells.
The number of neutrophils in the kidney is increased by rIL-33 added to low-dose cisplatin. The number of neutrophils per high power field (hpf) on day 3 was 0 in vehicle-treated mice, 0.02 in mice treated with cisplatin 15 mg/kg, and 13.4 in mice treated with cisplatin 15 mg/kg plus rIL-33 (P < 0.001 versus vehicle and cisplatin 15 mg/kg, n = 5 to 13 per group). The number of neutrophils in the kidney in cisplatin-induced AKI is decreased by injection of sST2. The number of neutrophils per hpf on day 3 was 0 in vehicle-treated mice, 2.4 in mice treated with cisplatin 25 mg/kg (P < 0.01 versus vehicle-treated), and 0.02 in mice treated with cisplatin 25 mg/kg plus sST2 (P < 0.01 versus cisplatin 25 mg/kg, n = 5 to 13 per group). In our model of cisplatin-induced AKI, neutrophils are undetectable in the kidney in vehicle-treated mice and on days 1 and 2 after cisplatin.3 In addition, renal myeloperoxidase (MPO) activity in the kidney was found to be very low (less than 0.05 OD/min/mg protein) and was not significantly different between vehicle-treated mice and on days 1 and 2 after cisplatin.3 We have previously demonstrated that neutrophil depletion in the kidney using the neutrophil-depleting antibody RB6–8C5 is not protective against cisplatin-induced AKI.20
Depletion of CD4 T cells
To confirm the role of CD4 T cells in cisplatin-induced AKI, mice were depleted of CD4 T cells. Mice depleted of CD4 T cells (CD4 −/− mice or GK1.4 antibody injection) had less cisplatin-induced AKI, demonstrating the role of CD4 T cells in cisplatin-induced AKI. CD4 −/− mice treated with cisplatin had significantly lower creatinine (Figure 8, A) and apoptosis scores (Figure 8, C) than wildtype mice treated with cisplatin. The ATN score in CD4 −/− mice treated with cisplatin (Figure 8, B) was lower than wildtype mice treated with cisplatin but did not reach statistical significance. Mice treated with GK1.5 antibody plus cisplatin had a significantly lower creatinine than mice treated with cisplatin plus vehicle (Figure 8, D).
Figure 8.

Depletion of CD4 T cells protects against cisplatin-induced AKI. CD4 −/− mice treated with cisplatin (CD4 −/− Cis) had a significantly lower (A) creatinine and (C) apoptosis score than wildtype mice treated with cisplatin (WT Cis). (B) The ATN score in CD4 −/− mice treated with cisplatin (CD4 −/− Cis) was lower than wildtype mice treated with cisplatin (WT Cis) but did not reach statistical significance. (D) Mice treated with GK1.5 antibody plus cisplatin (GK1.5 Cis) had a significantly lower creatinine than mice treated with cisplatin plus vehicle (Veh Cis). *P < 0.001 versus Veh; **P < 0.05 versus WTCis or VehCis; n = 4 to 8 per group.
CXCL1 (Also Known as IL-8 or KC)
CD4 T cells produce CXCL1. CXCL1 was increased in the kidney by day 2 after cisplatin (Figure 9, A). CXCL1 was increased in wildtype mice but not in CD4 T cell −/− mice treated with low-dose cisplatin (15 mg/kg) plus rIL-33 (Figure 9, B). The increase in CXCL1 in cisplatin-induced AKI was decreased in CD4 T cell −/− mice (Figure 9, C). IL-33 does not increase CXCL1 in normal mice. IL-33 (pg/mg) was 18.7 ± 7.1 in vehicle-treated wildtype mice and 19.0 ± 2.7 in mice treated with rIL-33 (n = 3, P > 0.05). To demonstrate that CXCL1 is a mediator of cisplatin-induced AKI, CXCR2 −/− mice that lack the receptor for CXCL1 were studied. CXCR2 is not specific for CXCL1 and binds other Glu-leu-Arg ELR +CXC chemokines like CXCL2 (MIP-2).21,22 However, CXCL2 has a lower affinity for CXCR2 than CXCL1 and is less able to initiate signal transduction and a biologic effect than CXCL121,22 CXCR2 −/− mice with cisplatin-induced AKI had a significantly lower creatinine (Figure 9, D), ATN score (Figure 9, E), and apoptosis score (Figure 9, F) in tubular cells compared with wildtype mice with cisplatin-induced AKI.
Figure 9.

CXCR2 −/− mice are protected against cisplatin-induced AKI. (A) CXCL1 was increased in the kidney by day 2 after cisplatin (Cis day 2). *P < 0.01 versus vehicle (Veh) and day 1 after cisplatin (Cis day 1). (B) rIL-33 injection in wildtype mice (WT + IL-33) did not increase CXCL1 compared with vehicle injection in wildtype mice (WT Veh). CXCL1 was significantly higher in wildtype mice treated with low-dose cisplatin (15 mg/kg; WT Cis) compared with wildtype mice treated with the vehicle for cisplatin (WT Veh). CXCL1 was increased in wildtype mice treated with low-dose cisplatin (15 mg/kg) plus rIL-33 (WT Cis IL33) compared with wildtype mice treated with low-dose cisplatin (15 mg/kg) alone (WT Cis). The increase in CXCL1 seen in wildtype mice treated with low-dose cisplatin (WT Cis) and wildtype mice treated with low-dose cisplatin plus rIL-33 (WT Cis IL33) was not seen in CD4 T cell −/− mice treated with low-dose cisplatin (CD4 −/− Cis) or low-dose cisplatin plus rIL-33 (CD4 −/− Cis IL-33). **P < 0.001 versus all other groups; *P < 0.01 versus Veh. (C) The increase in CXCL1 in cisplatin-treated (25 mg/kg) wildtype mice AKI (WT Cis) was decreased in cisplatin-treated CD4 T cell −/− mice (CD4 −/− Cis). *P < 0.001 versus vehicle-treated (Veh) and CD4−/− Cis. (D–F) CXCR2 −/− mice (CR2 −/−) that lack the receptor for CXCL1 have significantly lower creatinine, ATN score, and apoptosis in tubular cells compared with cisplatin-treated wildtype mice (WT Cis).*P < 0.001 versus Veh; **P < 0.01 versus WT Cis; n = 4 per group.
To further establish a link between IL-33, CD4 T cells, and CXCL1, the effect of rIL-33 on CD4 T cells in vitro to produce CXCL1 was performed as described in the Methods. In vitro, rIL-33 stimulated CD4 T cells to produce CXCL1. CXCL1 was higher in cisplatin-treated CD4 T cells compared with vehicle-treated CD4 T cells. rIL-33 did not result in a further increase in CXCL1 in cisplatin-treated CD4 T cells. CXCL1 (pg/mg) was 4.0 in vehicle-treated CD4 T cells, 12.3 in rIL-33-treated CD4 T cells (P < 0.01 versus vehicle-treated), 11.0 in cisplatin-treated CD4 T cells (P < 0.01 versus vehicle-treated), and 12.0 in cisplatin plus rIL-33-treated CD4 T cells (P < 0.01 versus vehicle-treated; (n = 3 per group). We have previously demonstrated the presence of CXCL1 in mouse kidneys in proximal tubules and macrophages in the outer stripe of the outer medulla.23 The in vitro experiment demonstrates CXCL1 in normal CD4 T cells and an increase in CXCL1 in CD4 T cells treated with rIL-33.
Time Sequence of Events in Cisplatin-Induced AKI
Figure 10 demonstrates the time sequence of events in cisplatin-induced AKI.
Figure 10.
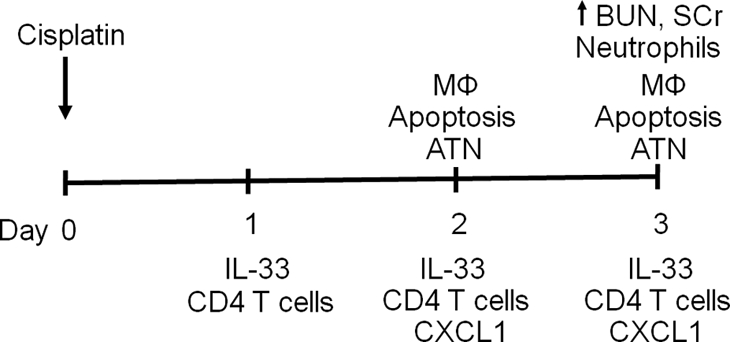
Time sequence of events in cisplatin-induced AKI. There is ATN and tubular apoptosis on day 2 and ATN and renal failure on day 3 of cisplatin-induced AKI.3 There is increased neutrophil infiltration on day 3 but not on days 1 and 2 of cisplatin-induced AKI.3 There is increased macrophage infiltration on day 2.29 In the present study, we demonstrate increased IL-33 in the kidney on days 1, 2, and 3; increased CD4 T cell infiltration in the kidney on days 1, 2, and 3; and increased CXCL1 in the kidney on days 2 and 3. The increase in IL-33 on day 1 precedes the ATN, tubular apoptosis, inflammation, and renal failure. Mφ, macrophage; BUN, blood urea nitrogen; SCr, serum creatinine.
DISCUSSION
The IL-33/ST2 pathway has been described as a novel biomarker in vascular injury as well as a therapeutic target.24,25 Serum levels of ST2 predict mortality and clinical outcome after acute myocardial infarction26 and are a biomarker of worsening heart failure.27 Serum IL-33 levels are increased in patients with active rheumatoid arthritis.28 Serum levels of IL-33 were increased in mice with cisplatin-induced AKI, suggesting diffuse endothelial injury due to cisplatin. The increase in serum IL-33 preceded the increase in ATN and serum creatinine. In this regard, we have previously demonstrated that serum von Willebrand factor, also a marker of endothelial injury, is also increased on day 2 in mice with cisplatin-induced AKI.29 Also, vascular injury (e.g., thrombotic microangiopathy) has been reported in patients receiving cisplatin.30, 31 In the present study, we demonstrate leakage of Evan's blue dye from the blood vessels into the kidney on days 1 and 2 after cisplatin. Thus, serum ST2 or IL-33 as a biomarker of vascular injury in cisplatin-induced AKI merits further investigation.
Next we determined the localization of IL-33 in the kidney. IL-33 was predominantly expressed in glomeruli, blood vessels, and peritubular capillaries in the kidney. Next we determined the intracellular localization of IL-33 in the kidney. IL-33 is identical to nuclear factor expressed in high endothelial venules (NF-HEV).32 IL-33 in the nucleus may activate the transcription factor AP-1 as well as nuclear factor–kappa B (NF-κB).24 Thus, IL-33 serves as an intracellular nuclear factor with transcriptional regulatory properties as well as a proinflammatory cytokine. The present study demonstrates that IL-33 in the kidney is predominantly expressed in nuclear extracts. The exact function of IL-33 in the nucleus has not been established.5
When cells undergo caspase-3-mediated apoptosis, caspase-3 cleaves IL-33 into a less active cleaved form (22 kD on immunoblot).5–8 We think that the predominant process in cisplatin-induced AKI is an increase in synthesis of the full-length form of IL-33 rather than caspase-3 cleavage of IL-33 into a less active form. Evidence for the former is as follows: (1) an increase of the full-length form of IL-33 on day 1 rather than the cleaved form of IL-33 in the kidney (Figure 3, D), (2) release of full-length IL-33 rather than cleaved IL-33 into the medium of endothelial cells (Figure 4), and (3) if IL-33 were being predominantly cleaved and inactivated by caspase-3, then IL-33 would not be injurious. Specific IL-33 inhibition is protective against cisplatin-induced AKI, demonstrating the injurious role of IL-33 (Figure 5). A recent study demonstrates that calpain can also cleave IL-33 into a less active form.33 However, we have demonstrated an increase in calpain activity in cisplatin-treated endothelial cells.15 We also demonstrated that calpain causes endothelial necrosis, as calpain inhibition is protective against cisplatin-induced necrosis,15 suggesting that calpain-mediated inactivation of IL-33 is not the dominant process in cisplatin-treated endothelial cells. The present study is unique, as it demonstrates that there is an increase in IL-33 protein in cisplatin-induced AKI in vivo. IL-33 has also been described as a cytokine mainly involved in allergic inflammation and asthma.34 The present study is also unique in that it demonstrates an effect of a nonimmune stimulus, cisplatin, on IL-33.
To demonstrate the injurious role of IL-33, mice injected with rIL33 had worse cisplatin-induced AKI, and mice injected with soluble sST2, a fusion protein that neutralizes IL-33 activity by acting as a decoy receptor, had a significant reduction in cisplatin-induced AKI. Next we determined the effect of IL-33 on inflammation in the kidney. The role of inflammation in AKI is gaining prominence.9 Our published data in cisplatin-induced AKI demonstrate that although there is an increase in neutrophils and macrophages in the kidney, neither neutrophil nor macrophage depletion is protective.20,29 It has been demonstrated that CD4 T cell −/− mice are functionally and histologically protected against cisplatin-induced AKI.10,35 The major proinflammatory effect of IL-33 is to induce CD4 T-cell Th2 cell responses via the ST2R.11 In the present study, we demonstrate that administration of rIL-33 worsens cisplatin-induced AKI in wildtype but not CD4 −/− mice, suggesting that IL-33-mediated injury is dependent on CD4 T cells. To confirm the injurious role of IL-33 in cisplatin-induced AKI, mice were injected with soluble ST2 (sST2), a fusion protein that neutralizes IL-33 activity by acting as a decoy receptor. Injection of sST2 significantly reduced the number of CD4 T cells in the kidney in cisplatin-induced AKI and protected against cisplatin-induced AKI. To confirm the injurious effect of CD4 T cells, CD4 −/− mice or injection of the GK1.5 antibody was protective against cisplatin-induced AKI.
Renal TNF-α, IL-1β, and CXCL1 are increased in cisplatin-induced AKI.10 IL-1β inhibition is not protective against cisplatin-induced AKI.20 TNF-α inhibition has been shown to be a mediator of cisplatin-induced AKI.36,37 TNF-α promotes caspase-1 activation via the inflammasome.38 The major effect of CXCL1 is to cause neutrophil recruitment. The exact role of neutrophils in AKI is controversial.9,20,39 However, in addition to neutrophil recruitment, CXCL1 can directly induce apoptosis of cells in vitro via release of intracellular Ca, FasL expression.40 Thus, the injurious effect of CXCL1 in cisplatin-induced AKI may be related to neutrophil recruitment or direct cellular injury. Our finding that CXCL1 is a mediator of cisplatin-induced AKI may result in potential new therapies for cisplatin-induced AKI. For example, the novel CXCL1 inhibitor, reparixin, and the CXCR2 inhibitor, repertaxin, have completed phase 2 studies (see http://clinicaltrials.gov).
In summary, cisplatin induces an increase in IL-33 in endothelial cells in culture and an increase in IL-33 in the kidney that precedes the increase in serum creatinine in cisplatin-induced AKI. Inhibition of IL-33 with sST2 provided functional and histologic protection against cisplatin-induced AKI, demonstrating that IL-33 is a mediator of cisplatin-induced AKI. Administration of rIL-33 worsened cisplatin-induced AKI in wildtype but not CD4 −/− mice, suggesting that IL-33-mediated injury is dependent on CD4 T cells. To determine a mechanism of CD4 T-cell-induced injury, CXCL1 was studied. CXCL1 was increased in wildtype mice but not in CD4 T cell −/− mice treated with cisplatin plus rIL-33. Inhibition of the action of CXCL1 in CXCR2 −/− mice was protective against cisplatin-induced AKI, demonstrating the injurious role of CXCL1 in cisplatin-induced AKI. Thus, IL-33-mediated cisplatin-induced AKI is dependent on CD4 T cell-mediated production of CXCL1. Inhibition of IL-33 or CXCL1 offers therapeutic potential in cisplatin-induced AKI in humans.
CONCISE METHODS
Cisplatin Administration
For all of the mouse studies, 8- to 10-wk-old male C57BL/6 mice weighing 20 to 25 g were used. All experiments were conducted with adherence to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal protocol was approved by the Animal Care and Use Committee of the University of Colorado Health Sciences Center. Mice were maintained on a standard diet, and water was freely available. Mice were housed five per cage under a 12-h light-and-dark schedule for at least 1 wk before cisplatin administration. Six hours before cisplatin administration, food and water were withheld. Cisplatin (cis-Diamminedichloro-platinum [II]; Aldrich, Milwaukee, WI) was freshly prepared on the day of administration in sterile normal saline at a concentration of 1 mg/ml. Mice were given either 25 mg/kg or 15 mg/kg body wt of cisplatin or vehicle (saline) intraperitoneally, after which the mice again had free access to food and water.
We described this model of cisplatin-induced ARF in detail elsewhere.3 Briefly, after 25 mg/kg cisplatin injection, blood urea nitrogen (BUN) and serum creatinine are normal on day 1 and slightly increased on day 2. On day 3 after cisplatin injection, renal dysfunction, renal neutrophil infiltration, and acute tubular necrosis scores are severe. Serum creatinine, ATN score, and apoptosis score on day 3 are increased after cisplatin (15 mg/kg; Figure 6, A, B, and C) but are significantly lower compared with 25 mg/kg (Figure 5, A, B, and C).
Histologic Examination
Paraformaldehyde (4%)-fixed and paraffin-embedded kidneys were sectioned at 4 μm and stained with periodic acid-Schiff (PAS) by standard methods. All histologic examinations were performed by the renal pathologist in a blinded fashion. Histologic changes due to acute tubular necrosis (ATN score) were evaluated in the outer stripe of the outer medulla on PAS-stained tissue and were quantified by counting the percent of tubules that displayed cell necrosis, loss of brush border, cast formation and tubule dilation, as follows: 0 = none, 1 = <10%, 2 = 10 to 25%, 3 = 26 to 45%, 4 = 46 to 75%, and 5 = >75%. At least 10 fields (×250) were reviewed for each slide.
Neutrophil infiltration was quantitatively assessed on PAS-stained tissue by the renal pathologist by counting the number of neutrophils per high power field (×400), as we have previously described.3 At least 10 fields were counted in the outer stripe of the outer medulla for each slide.
Morphologic criteria were used to count apoptotic cells on PAS-stained tissue by the pathologist experienced in the evaluation of renal apoptosis. Morphologic characteristics included cellular rounding and shrinkage, nuclear chromatin compaction, and formation of apoptotic bodies.41 Apoptotic tubular cells were quantitatively assessed per 10 high-power fields (×400) in the outer stripe of the outer medulla by the renal pathologist in a blinded fashion.
Endothelial Cells in Culture
MS1 (MILE SVEN 1) mouse endothelial pancreatic islet cells were purchased from American Type Culture Collection (ATCC®, Manassas, VA; catalog no. CRL-2279). The line retains the properties of endothelial cells, including uptake of acetylated LDL and expression of both factor-VIII-related antigen and VEGFR. Cells were grown in Dulbecco's Modified Eagle's Medium (DMEM). Experiments were performed when the plates became at least 80% confluent.
We have described the model of cisplatin-induced injury to microvascular endothelial cells in detail elsewhere.15 Briefly, cells treated with 10 μM of cisplatin demonstrate apoptosis, and cells treated with 50 μM cisplatin demonstrate necrosis. After cisplatin treatment, the medium was removed from the cells. Proteins in the medium of the cells were precipitated using trichloroacetic acid (TCA).
IL-33 and CXCL1 ELISA
Both the mouse IL-33 ELISA (catalog no. M3300) and CXCL1/KC ELISA (catalog no. MKC 00B) kits were obtained from R&D Systems (Minneapolis, MN). ELISA was performed according to the manufacturer's instructions.
Nuclear Extracts
Nuclear extracts of whole kidney were prepared from fresh kidney using a NE-PER Nuclear and cytoplasmic extraction kit (Thermo Scientific). Nuclear extracts were immunoblotted with the nuclear protein Lamin A (Santa Cruz Biotechnology, Santa Cruz, CA) (catalog no. sc-20681) as a loading control.
Immunoblot Analysis
Whole kidney was homogenized in radioimmunoprecipitation assay (RIPA) buffer plus proteinase inhibitors and immunoblotted, as described previously.15 A monoclonal Ab (Nessy-1) that recognizes mouse, human, and rat full-length IL-33 (34 kD) and the cleaved IL-33 (22 kD) was used (catalog no. ALX-804-840, Alexis Biochemicals, San Diego, CA). Mouse rIL-33, amino acids Ser109-Ile266, was used as a positive control (BioLegend, San Diego, CA). A rabbit polyclonal antibody raised against amino acids 76 to 375 of actin of human origin (43 kD) was used as a loading control (catalog no. sc-10731, Santa Cruz Biotechnology, Santa Cruz, CA).
Immunofluorescence Studies
Kidney tissues were embedded in OCT, snap-frozen in liquid nitrogen, and stored at −80 °C until sectioning. Five-μm cryostat sections were fixed in 70% acetone/30% methanol and prepared for immunofluorescence studies, as described previously.15 A monoclonal Ab (Nessy-1) that recognizes mouse, human, and rat full-length IL-33 (34 kD) and the cleaved IL-33 (22 kD) was used (catalog no. ALX-804-840, Alexis Biochemicals, San Diego, CA). A rabbit polyclonal antibody to vWF, which specifically reacts with the cytoplasm of endothelial cells, was used (Abcam, Cambridge, MA; catalog no. ab6994). An Alexa Fluor® 488 conjugate of wheat germ agglutinin, a widely used lectin, was obtained from Invitrogen (Carlsbad, CA; catalog no. W11261). A fluorescein isothiocyanate (FITC) conjugated monoclonal anti-α-smooth muscle actin antibody was obtained from Sigma (St. Louis, MO; product no. F3777).
IL-33 Administration
One μg of mouse rIL-33, amino acids Ser109-Ile266, (BioLegend, San Diego, CA) or vehicle (PBS) was injected intraperitoneally, twice a day, on days 1, 2, and 3 after cisplatin administration. The same dosage and route of administration of rIL-33 has been demonstrated to reduce the development of atherosclerosis in mice.25
Mouse T-cell CD4 Isolation
Spleens from C57BL/6 mice were crushed in culture dishes to obtain a single-cell suspension. The splenocytes were resuspended in cold RPMI, and red blood cells (RBCs) were lysed using ACK lysis buffer (Quality Biologic, Inc.). The resulting suspension was filtered through a sterile cell strainer (70μm) to remove cellular debris, washed twice with phosphate-buffered saline (PBS) and counted. T cells were enriched by passing splenocyte suspensions over a T-cell CD4 Subset column kit (R&D Systems, catalog no. MCD4C-1000). The purity of the population was confirmed by flow cytometry analysis and routinely reached >90%.
CD4 T cells were resuspended in appropriate volume of RPMI with 0.5% fetal calf serum and 100 U/ml penicillin, and 100 U/ml streptomycin. CD4 T cells were incubated at 37 °C and 5% CO2 during 24 h, with 2000pg/ml of recombinant murine IL-33 or vehicle control (0.1% BSA in 1x PBS). To avoid cytokine release to the media, CD4 T cells were treated with Brefeldin A (eBiosciences) for the last 8 h of the incubation. CXCL1 production by CD4+ cells was performed by a CXCL1/KC ELISA kit (catalog no. MKC 00B) obtained from R&D Systems (Minneapolis, MN) as per the manufacturer's instruction and corrected for protein. The detection limit of CXCL1 is 2.0 pg/ml.
sST2 Administration
The sST2 fusion protein (muST2-huFc) was obtained from Dirk Smith (Amgen, Seattle, WA). Administration and dose of sST2 was based on previous studies.25 One hundred μg of sST2 in PBS or vehicle (hu IgG Fc control) was administered on days 1, 2, and 3 of cisplatin administration.
GK1.5 Antibody Administration
GK1.5 monclonal antibody was obtained from Bioxcell (West Lebanon, NH). Mice were injected with 10 mg/kg of the rat IgG monoclonal antibody GK1.5 intraperitoneally on days −7 and −2 before cisplatin administration.42 Administration of the GK1.5 antibody is known to result in CD4 T cell depletion.43 We have previously demonstrated the effect of GK1.5 to cause CD4 T cell depletion in lymph nodes and kidneys.42 Control animals received 10mg/kg intraperitoneally of the rat IgG (Sigma) at the same time points.
Flow Cytometry
To determine the effect of cisplatin on CD4 T cell population, flow cytometric detection of CD45, CD3, and CD4 was performed on fresh samples of spleen and kidney, as described in detail.44 The samples were incubated with following antibodies: (1) per-CP conjugated anti-CD45 (eBioscience, catalog no. 45-0451-80); (2) APC conjugated anti-CD4 (catalog no. 11-0033-81); (3) PE conjugated anti-CD3 (catalog no. 11-0033-81). Samples were first gated for live cells. Of the live cells, high FITC signals were gated for CD45+ events. CD45+ events were set as gating limit at 10,000 events, and 15,000 events for kidney and spleen, respectively. High PE signals were gated for CD3+ events, and, of that, population high APC signal events were analyzed, which represent CD4+ cells.
CD4 T cell −/− and CXCR2 −/− mice
Mice homozygous for the CD4 tm1Mak targeted mutation in the C57BL/6 strain were obtained from the Jackson Laboratory. Cell surface expression of CD4 protein is not detected on thymocytes and lymph nodes from homozygous mice. Age-, weight-, and sex-matched C57BL/6 mice were used as controls. Mice homozygous for the CXCR2 mutation in the C57BL/6 strain were obtained by breeding and genotyping heterozygous mice obtained from the Jackson Laboratory, as described.45,46 Age-, sex-, and weight-matched littermates were used as controls.
In Vivo Leakage Assay
Mice were injected with Evan's blue dye, combined with albumin, via the tail vein. After 60 min, in separate experiments, the lung was perfused with normal saline via the right ventricle on day 2 after cisplatin, or the kidney was perfused with normal saline via the left ventricle on days 1 and 2 after cisplatin. When endothelial permeability is increased, the dye extravasates and reaches the subendothelial spaces. Thus, subsequent washing of dye-perfused organs with saline cannot remove the dye. After perfusion, the lung or kidneys were removed, homogenized, and centrifuged. Spectrophotometric analysis of supernatants for Evan's blue dye, at 540 nm, was then performed. The increase in optical density values is indicative of the increase in dye retention, hence, of vascular leakage.
Statistical Analysis
Non-normally distributed data were analyzed by the nonparametric unpaired Mann-Whitney test. Multiple group comparisons are performed using ANOVA, with posttest according to Newman-Keuls. For the survival study, a contingency table for survival on day 4 was analyzed by Fisher's exact test. A P value of <0.05 was considered statistically significant. Values are expressed as means ± SEM.
DISCLOSURES
None.
Acknowledgments
This work was supported by RO1DK056851 and RO1DK056851S1 to CLE, KO8DK069512 to A.J., and R01HL095363 to S.F. Ali Akcay was supported by Fatih University School of Medicine, Department of Nephrology, Ankara, Turkey. Kultigin Turkmen was supported by the Turkish Society of Nephrology. Dong Won Lee was supported by Pusan National University, Pusan, Korea.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
REFERENCES
- 1. Schrier RW: Cancer therapy and renal injury. J Clin Invest 110: 743–745, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Safirstein RL: Renal disease induced by anti-neoplastic agents. In: Diseases of the Kidney and Urinary Tract, 8th ed., edited by Schrier RW, Philadelphia, Lippincott, Williams and Wilkins, 2007, pp. 1068–1081 [Google Scholar]
- 3. Faubel SG, Ljubanovic D, Reznikov LL, Somerset H, Dinarello CA, Edelstein CL: Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int 66: 2202–2213, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Moussion C, Ortega N, Girard JP: The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: A novel “alarmin”? PLoS ONE 3: e3331, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lamkanfi M, Dixit VM: IL-33 raises alarm. Immunity 31: 5–7, 2009 [DOI] [PubMed] [Google Scholar]
- 6. Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, Lavelle EC, Martin SJ: Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31: 84–98, 2009 [DOI] [PubMed] [Google Scholar]
- 7. Cayrol C, Girard JP: The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A 106: 9021–9026, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G: Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem 284: 19420–19426, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akcay A, Nguyen Q, Edelstein CL: Mediators of inflammation in acute kidney injury. Mediators Inflamm. E pub, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu M, Chien CC, Burne-Taney M, Molls RR, Racusen LC, Colvin RB, Rabb H: A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J Am Soc Nephrol 17: 765–774, 2006 [DOI] [PubMed] [Google Scholar]
- 11. Dinarello CA: An IL-1 family member requires caspase-1 processing and signals through the ST2 receptor. Immunity 23: 461–462, 2005 [DOI] [PubMed] [Google Scholar]
- 12. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, Pitman N, Mirchandani A, Rana B, Van Rooijen N, Shepherd M, McSharry C, McInnes IB, Xu D, Liew FY: IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 183: 6469–6477, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY: IL-33 is a chemoattractant for human Th2 cells. European J Immunol 37: 2779–2786, 2007 [DOI] [PubMed] [Google Scholar]
- 14. Liew FY, Pitman N, McInnes IB: Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat Rev Immunol 10: 103–109, 2010 [DOI] [PubMed] [Google Scholar]
- 15. Dursun B, He Z, Somerset H, Oh DJ, Faubel S, Edelstein CL: Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. Am J Physiol Renal Physiol 291: F578–F587, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Sheikh-Hamad D: Cisplatin-induced cytoxicity: Is the nucleus relevant? Am J Physiol Renal Physiol 295: F42–43, 2008 [DOI] [PubMed] [Google Scholar]
- 17. Sartori S, Nielsen I, Maestri A, Beltrami D, Trevisani L, Pazzi P: Acute gastroduodenal mucosal injury after cisplatin plus etoposide chemotherapy. Clinical and endoscopic study. Oncology 48: 356–361, 1991 [DOI] [PubMed] [Google Scholar]
- 18. Wang J, He D, Zhang Q, Han Y, Jin S, Qi F: Resveratrol protects against Cisplatin-induced cardiotoxicity by alleviating oxidative damage. Cancer Biother Radiopharm 24: 675–680, 2009 [DOI] [PubMed] [Google Scholar]
- 19. Leo F, Pelosi G, Sonzogni A, Chilosi M, Bonomo G, Spaggiari L: Structural lung damage after chemotherapy fact or fiction? Lung Cancer 67: 306–310, 2010 [DOI] [PubMed] [Google Scholar]
- 20. Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, Edelstein CL: Cisplatin-induced ARF is associated with an increase in the cytokines IL-1β, IL-18, IL-6 and neutrophil infiltration in the kidney. J Pharmacol Exp Ther 322: 8–15, 2007 [DOI] [PubMed] [Google Scholar]
- 21. Bizzarri C, Beccari AR, Bertini R, Cavicchia MR, Giorgini S, Allegretti M: ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol Therap 112: 139–149, 2006 [DOI] [PubMed] [Google Scholar]
- 22. Stillie R, Farooq SM, Gordon JR, Stadnyk AW: The functional significance behind expressing two IL-8 receptor types on PMN. Journal of Leukoc Biol 86: 529–543, 2009 [DOI] [PubMed] [Google Scholar]
- 23. He Z, Altmann C, Hoke TS, Ljubanovic D, Jani A, Dinarello CA, Faubel S, Edelstein CL: Interleukin-18 (IL-18) binding protein transgenic mice are protected against ischemic AKI. Am J Physiol Renal Physiol 295: F1414–F1421, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kakkar R, Lee RT: The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat Rev Drug Discov 7: 827–840, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, Baker AH, McInnes IB, Liew FY: IL-33 reduces the development of atherosclerosis. J Exp Med 339–346, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimpo M, Morrow DA, Weinberg EO, Sabatine MS, Murphy SA, Antman EM, Lee RT: Serum levels of the interleukin-1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. Circulation 109: 2186–2190, 2004 [DOI] [PubMed] [Google Scholar]
- 27. Weinberg EO, Shimpo M, Hurwitz S, Tominaga S, Rouleau JL, Lee RT: Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation 107: 721–726, 2003 [DOI] [PubMed] [Google Scholar]
- 28. Matsuyama Y, Okazaki H, Tamemoto H, Kimura H, Kamata Y, Nagatani K, Nagashima T, Hayakawa M, Iwamoto M, Yoshio T, Tominaga S, Minota S: Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J Rheum 37: 18–25, 2010 [DOI] [PubMed] [Google Scholar]
- 29. Lu L, Oh DJ, Dursun B, He Z, Hoke TS, Faubel S, Edelstein CL: Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J Pharmacol Exp Ther 324: 111–117, 2007 [DOI] [PubMed] [Google Scholar]
- 30. Jackson AM, Rose BD, Graff LG, Jacobs JB, Schwartz JH, Strauss GM, Yang JP, Rudnick MR, Elfenbein IB, Narins RG: Thrombotic microangiopathy and renal failure associated with antineoplastic chemotherapy. Ann Intern Med 101: 41–44, 1984 [DOI] [PubMed] [Google Scholar]
- 31. Fields SM, Lindley CM: Thrombotic microangiopathy associated with chemotherapy: case report and review of the literature. DICP 23: 582–588, 1989 [DOI] [PubMed] [Google Scholar]
- 32. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP: IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A 104: 282–287, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayakawa M, Hayakawa H, Matsuyama Y, Tamemoto H, Okazaki H, Tominaga S: Mature IL-33 is produced by calpain-mediated cleavage in vivo. Biochem Biophys Res Comm 387: 218–222, 2009 [DOI] [PubMed] [Google Scholar]
- 34. Smith DE: IL-33: A tissue derived cytokine pathway in allergic inflammation and asthma. Clin Exp Allergy 40: 200–208, 2009 [DOI] [PubMed] [Google Scholar]
- 35. Lee H, Nho D, Chung HS, Lee H, Shin MK, Kim SH, Bae H: CD4+CD25+ regulatory T cells attenuate cisplatin-induced nephrotoxicity in mice. Kidney Int 78: 1100–1109, 2010 [DOI] [PubMed] [Google Scholar]
- 36. Ramesh G, Reeves WB: TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramesh G, Reeves WB: TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 285: F610–F618, 2003 [DOI] [PubMed] [Google Scholar]
- 38. Franchi L, Eigenbrod T, Nunez G: Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183: 792–796, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bolisetty S, Agarwal A: Neutrophils in acute kidney injury: Not neutral any more. Kidney Int 75: 674–676, 2009 [DOI] [PubMed] [Google Scholar]
- 40. Gangadharan C, Thoh M, Manna SK: Late phase activation of NFKB by doxorubicin is mediated by IL-8 and induction of apoptosis via FasL. Breast Cancer Res Treat 120: 671–683, 2010 [DOI] [PubMed] [Google Scholar]
- 41. Gobe G, Zhang XJ, Willgoss DA, Schoch E, Hogg NA, Endre ZH: Relationship between expression of Bcl-2 genes and growth factors in ischemic acute renal failure in the rat. J Am Soc Nephrol 11: 454–467, 2000 [DOI] [PubMed] [Google Scholar]
- 42. Faubel SG, Ljubanovic D, Poole B, Dursun B, Cushing S, He Z, Gill RG, Edelstein CL: Peripheral CD4 T cell depletion is not sufficient to prevent ischemic acute renal failure. Transplant 80: 643–649, 2005 [DOI] [PubMed] [Google Scholar]
- 43. Dialynas DP, Quan ZS, Wall KA, Pierres A, Quintans J, Loken MR, Pierres M, Fitch FW: Characterization of the murine T cell surface molecule, designated L3T4, identified by monoclonal antibody GK1.5: Similarity of L3T4 to the human Leu-3/T4 molecule. J Immunol 131: 2445–2451, 1983 [PubMed] [Google Scholar]
- 44. Vielhauer V, Anders HJ, Perez dL, Luckow B, Schlondorff D, Mack M: Phenotyping renal leukocyte subsets by four-color flow cytometry: Characterization of chemokine receptor expression. Nephron Exp Nephrol 93: e63, 2003 [DOI] [PubMed] [Google Scholar]
- 45. Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM: Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol 172: 2853–2860, 2004 [DOI] [PubMed] [Google Scholar]
- 46. Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW: Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science 265: 682–684, 1994 [DOI] [PubMed] [Google Scholar]