

Abstract
Type 2 diabetes and coronary artery disease (CAD) are conditions that cause a substantial public health burden. Since both conditions often coexist in the same individual, it has been hypothesized that they have a common effector. Insulin and hyperglycemia are assumed to play critical roles in this scenario. In recent years, many genetic risk factors for both diabetes and CAD have been discovered, mainly through genome-wide association studies. Genetic aspects of diabetes, diabetic macrovascular complications, and CAD are assumed to have intersections leading to the common effector hypothesis. However, only a few genetic risk factors could be identified that modulate the risk for both conditions. Polymorphisms in TCF7L2 and near the CDKN2A/B genes seem to be of great importance in this regard since they appear to modulate both conditions, and they are not necessarily related to insulinism, or hyperglycemia, for CAD development. Other issues related to this hypothesis, such as the problems of phenotype heterogeneity, are also of interest. Recent studies have contributed to a better understanding of the complex genetics of diabetic macrovascular complications. Much effort is still needed to clarify the associations in the genetics of diabetes, and cardiovascular disease. At present, there is little genetic evidence to support a common effector hypothesis, other than insulin or hyperglycemia, for the association between these conditions.
Keywords: diabetes, coronary artery disease, HLA, polymorphism, genome-wide association study
Abbreviations: ADAMTS9 - disintegrin and metalloproteinase with thrombospondin motifs 9; ADCY5 - adenylate cyclase 5; ADIPOR1 - adiponectin receptor 1; ADRA2A - adrenergic, alpha-2A-, receptor; ANRIL - antisense non-coding RNA in the INK4 locus (also called CDKN2B-AS); ARIC - Atherosclerosis Risk in Communities study; BMI - body mass index; C12orf43 - chromosome 12 open reading frame 43; C2CD4B - C2 calcium-dependent domain containing 4B; CAD - coronary artery disease; CAMK1D - calmodulin-dependent protein kinase 1D; CAPN10 - calpain 10; CDC123 - cell division cycle 123; CDKAL1 - CDK5 regulatory subunit-associated protein 1-like 1; CDKN2A - distal to the genes cyclin-dependent kinase inhibitors 2A; CELSR2 - cadherin EGF LAG seven-pass G-type receptor 2; CRY2 - cryptochrome 2; CVD - cardiovascular disease; CXCL12 - chemokine (C-X-C motif) ligand 12; DCCT - Diabetes Control and Complications Trial; DGKB - diacylglycerol kinase beta 90kDa; DNA - deoxyribonucleic acid; EDIC - Epidemiology of Diabetic Complications and Interventions trial; EGF - epidermal growth factor; ENPP1 - ectonucleotide pyrophosphatase/phosphordi-sterase 1; FADS1 - fatty acid desaturase 1; FOXO1 - forkhead box protein O1; FTO - fat mass and obesity-associated; GCKR - glucokinase regulator; GLIS3 - GLIS family zinc finger 3; GLP-1 - glucagon-like peptide 1; GWAS - genome-wide association studies; HbA1c - glycated hemoglobin; HHEX-IDE - near hematopoietically expressed homeobox and insulin degrading enzyme; HLA - human leukocyte antigen; HNF1A - hepatocyte nuclear factor 1 homeobox A; IGF1 - insulin-like growth factor 1; IGF2BP2 - IGF2 mRNA binding protein 2; JAZF1 - juxtaposed with another zinc finger protein 1; KCNJ11 - potassium inwardly-rectifying channel, subfamily J, member 11; KCNQ1 - potassium voltage-gated channel, KQT-like subfamily, member 1; LADA - latent autoimmune diabetes in adults; LAG - laminin A globular domain; LDL - low-density lipoprotein; LDLR - LDL-receptor; LGR5 - leucine-rich repeat-containing G protein-coupled receptor 5; MADD - MAP kinase-activating death domain; MASS-II - Medicine, Angioplasty, or Surgery Study II; MIA3 - melanoma inhibitory activity family, member 3; MODY - mature onset diabetes of the young; MRAS - Ras-related protein M; MRPS6 - mitochondrial ribosomal protein S6; MTNR1B - melatonin receptor 1B; NF-kappaB - nuclear factor-kappaB; NOTCH2 - notch homolog protein 2; PCSK9 - proprotein convertase subtilisin/kexin type 9; PHACTR1 - phosphatase and actin regulator 1; PPARG - peroxisome proliferator-activated receptor-gamma; PPARGC1A - PPARG coactivator 1-alpha; PROX1 - prospero-homeobox 1; PSRC1 - proline/serine-rich coiled-coil 1; RNA - ribonucleic acid; SGK1 - serum/glucocorticoid regulated kinase 1; SLC30A8 - solute carrier family 30 (zinc transporter), member A8; SNP - single-nucleotide polymorphism; SOD2 - superoxide dismutase 2; SORT1 - sortilin 1; SREBF1 - sterol regulatory element-binding transcription factor 1; T1D - type 1 diabetes; T2D - type 2 diabetes; TCF7L2 - transcription factor 7-like 2; THADA - thyroid adenoma associated; TMEM195 - transmembrane protein 195; TSPAN8 - tetraspanin 8; WDR12 - WD repeat domain 12; WFS1 - wolfram syndrome 1
Introduction
Diabetes is a complex and heterogeneous condition characterized by chronic hyperglycemia. Basically, diabetes can be classified as type 1 diabetes (T1D), type 2 diabetes (T2D), latent autoimmune diabetes in adults (LADA), mature onset diabetes of the young (MODY), gestational diabetes, and secondary diabetes. Of these, MODY is the best example of a condition that has a defined Mendelian genetic pattern (autosomal dominant inheritance). Other monogenic forms of diabetes are also a consequence of rare mutations in a single gene [1]. Although most diabetes cases do not follow a Mendelian inheritance pattern, there is evidence that genetic factors are important in their pathogeneses. For T1D, the major susceptibility locus is related to HLA class II genes at 6p21, which accounts for more than 30%-50% of the genetic risk of T1D. Also, more than 40 non-HLA susceptibility gene markers have been associated with the trait [2].
T2D involves complex genetics. There is an intricate interaction between the environment and genetic background, understood as the contribution of many different genes [1]. In T2D pathophysiology, two conditions are essential: insulin resistance and beta-cell dysfunction (Figure 1). Hyperglycemia is the result of the inability of beta-cells to cover the increased insulin demand caused by reduced insulin sensitivity. Other mechanisms responsible for the pathophysiology of T2D include incretins, particularly glucagon-like peptide (GLP-1), and the deregulation of hepatic glucose disposure due to gluconeogenesis and glycogenolysis, which leads to hyperglycemia. The hepatic glucose production is controlled by hormones, whereas insulin suppresses its production. Glucagon (and catecholamines) stimulates gluconeogenesis and glycogenolysis. Therefore, it is conceivable that increased secretion of glucagon and/or increased hepatic sensitivity to glucagon (i.e. pancreatic alpha-cell dysfunction) contributes to the deregulation of hepatic glucose production, in addition to impaired insulin secretion and reduced hepatic insulin sensitivity [3].
Figure 1. Possible mechanisms of confirmed and potential risk SNPs in type 2 diabetes.
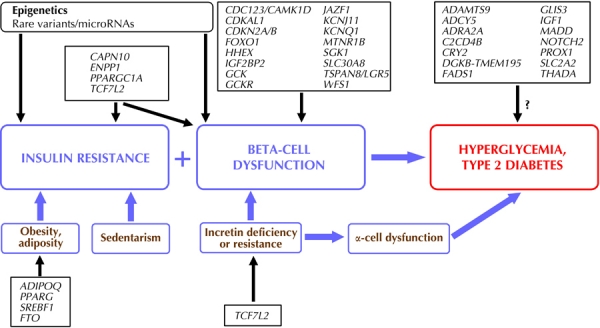
Many single-nucleotide polymorphisms (SNPs) affect pancreatic beta-cell function. Gene symbols represent SNPs in or near these gene loci. Likely, epigenetic alterations, microRNAs, and/or rare genetic variants also have a critical role. The mechanisms by which some genes increase the risk of diabetes are not yet known.
The increasing prevalence of diabetes and the chronic complications related to this condition makes it a major global health problem. Basically, the complications are classified as microvascular (diabetic retinopathy, neuropathy, and nephropathy) and macrovascular (stroke, acute coronary syndrome, and chronic peripheral arterial disease) complications. Of these, cardiovascular disease (CVD) causes the most deaths in these individuals [4]. Both T2D and CVD are common, complex conditions with a substantial public health burden [5]. Also, both conditions have a quantifiable genetic component in their pathophysiology. In most affected individuals, CVD coexists with metabolic risk factors, including diabetes. It has been well known for decades that diabetes increases the risk of cardiovascular disease. Indeed approximately 65% of deaths in T2D patients are related to coronary artery disease (CAD) or stroke [6]. CAD is clearly associated with hyperglycemia and with non-glycemic factors related to the metabolic syndrome, including hypertension, dyslipidemia, hypercoagulability, and chronic inflammation. Therefore it is conceivable that the same genetic risk factors may play a role in diabetes and in diabetic macrovascular complications.
Despite proven association between diabetes and atherosclerosis, the underlying mechanisms linking these two conditions are not fully understood [4]. Classically, it has been taught that this clear epidemiologic association is due to high insulin levels and/or hyperglycemia. However, this view was based on limited understanding of how common genetic variants modulate T2D and CVD. Candidate gene studies and linkage analyses identified only a few susceptibility loci that could be replicated consistently in large-scale studies. Nonetheless, in the past decade, genome-wide association studies (GWAS) have dramatically increased the number of common single-nucleotide polymorphisms (SNPs) with confirmed association with T2D, or cardiovascular traits [5]. As GWAS offer an image of the entire genome variation, the approach could be used to empirically test the existence of specific and pleiotropic determinants of both conditions, i.e., common effectors other than hyperglycemia and/or hyperinsulinism.
In this review, we highlight the findings from recent studies identifying risk loci in T2D, diabetic macrovascular complications, and CAD. We pay special attention to the intersections between the genetics of these conditions, as a way to explore the common effectors of both conditions. apart from the usual insulin/hyperglycemia hypothesis.
Genetics of type 2 diabetes
There is strong evidence for a genetic component of T2D risk. First, the observation of a wide range of diabetes prevalence in different ethnic groups, from very low levels of around 1% in some populations, such as tribes of Mapuche Indians or Chinese that live in rural areas, to extremely high levels, as found in Nauru and Pima Indians in Arizona [7]. A part of this ethnic variability can be attributed to non-genetic environmental and cultural factors. However, the observation that the disease prevalence varies substantially among ethnic groups who share the same environment, supports the hypothesis that genetic factors contribute to disease predisposition [8]. Familial aggregation studies that compared the disease prevalence within family members of a proband according to that expected in the general population showed the importance of the genetic factors. A greater prevalence in family members is thought to be due to an increased number of genes shared among them, including genes that play a role in disease predisposition [8]. T2D occurs more frequently among individuals who have first-degree relatives with diabetes: data from the Framingham Offspring Study reveal that children of one parent with T2D have a 3.5 times greater risk of developing the disease compared with an individual from the general population, and 6.1 times, when both parents have T2D [9]. The Isfahan Diabetes Prevention Study found a 10.3% higher diabetes prevalence among first-degree relatives of T2D patients compared with 6.0% for a control population of the same age [10]. Also, there is significant concordance in twin studies [11]. Monozygotic twins are genetically identical, while dizygotic twins share only half their genes on average. Considering that both kinds of twins tend to share much of their environment (known as the "equal environment assumption"), increased concordance rates for disease in monozygotic twins compared with dizygotic twins are indicative of genetic factors contributing to disease predisposition [8].
However, the inheritance pattern in T2D is complex. It is hypothesized that many genes affect disease predisposition and both gene-gene and gene-environment interactions impact disease risk. This proposed scenario led to initial difficulties in identifying genetic risk factors for T2D. Only recently, with the advent of GWAS, it was discovered that an avalanche of genes is associated with T2D, and other related complex phenotypes.
Most association studies employ the traditional case-control design in which the prevalence of a putative disease marker is compared between persons with a disorder (cases), and persons without the disorder (controls). This design is also used in GWAS, but it can simultaneously assess hundreds of thousands of markers using arrays of high-density SNPs. The SNPs are exchanges of single base pairs and represent approximately 90% of the sequence variation within the human genome. Arrays commonly used in these studies are able to assess 500,000 to 1,000,000 SNPs simultaneously [12].
The first GWA studies for T2D susceptibility loci were conducted in populations of European ancestry [13-17]. These studies found replicated evidence for association of the following genes:
1. transcription factor 7-like 2 (TCF7L2),
2. peroxisome proliferator-activated receptor-g (PPARG), and
3. potassium inwardly-rectifying channel, subfamily J, member 11 (KCNJ11).
Six additional loci have been identified:
1. solute carrier family 30 (zinc transporter), member 8 (SLC30A8),
2. insulin-like growth factor 2 mRNA binding protein 2 (IGF2BP2),
3. fat mass and obesity-associated (FTO),
4. near hematopoietically expressed homeobox and insulin degrading enzyme (HHEX-IDE),
5. CDK5 regulatory subunit-associated protein 1-like 1 (CDKAL1), and
6. distal to the genes cyclin-dependent kinase inhibitors 2A and 2B (CDKN2A-CDKN2B).
These genes have the highest strength of association between the risk genes for T2D. However, with increasing sample size and wider coverage of the genome, it has been possible to identify other susceptibility loci for T2D, namely:
1. calpain 10 (CAPN10),
2. ectonucleotide pyrophosphatase/phosphor-disterase 1 (ENPP1),
3. forkhead box protein O1 (FOXO1),
4. potassium voltage-gated channel, KQT-like subfamily, member 1 (KCNQ1),
5. melatonin receptor 1B (MTNR1B),
6. peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGC1A),
7. sterol regulatory element-binding transcription factor 1 (SREBF1),
8. serum/glucocorticoid regulated kinase 1 (SGK1), and
9. wolfram syndrome 1 (WFS1).
Later meta-analysis comprising thousands of individuals, and millions of SNPs, have detected previously unknown loci with robust evidence for association (p less than 5x10-8), including:
1. juxtaposed with another zinc finger protein 1 (JAZF1),
2. cell division cycle 123 / calcium / calmodulin-dependent protein kinase 1D (CDC123-CAMK1D),
3. tetraspanin 8 / leucine-rich repeat-containing G protein-coupled receptor 5 (TSPAN8-LGR5),
4. thyroid adenoma associated (THADA),
5. disintegrin and metalloproteinase with thrombospondin motifs 9 (ADAMTS9), and
6. neurogenic locus notch homolog protein 2 (NOTCH2) genes [18].
Also, loci have been found in or near:
1. adenylate cyclase 5 (ADCY5),
2. MAP kinase-activating death domain (MADD),
3. adrenergic, alpha-2A-, receptor (ADRA2A),
4. cryptochrome 2 (CRY2),
5. fatty acid desaturase 1 (FADS1),
6. GLIS family zinc finger 3 (GLIS3),
7. solute carrier family 2 (SLC2A2),
8. prospero-homeobox 1 (PROX1),
9. C2 calcium-dependent domain containing 4B (C2CD4B) insulin-like growth factor 1 (IGF1),
10. glucokinase (GCK),
11. glucokinase regulator (GCKR), and
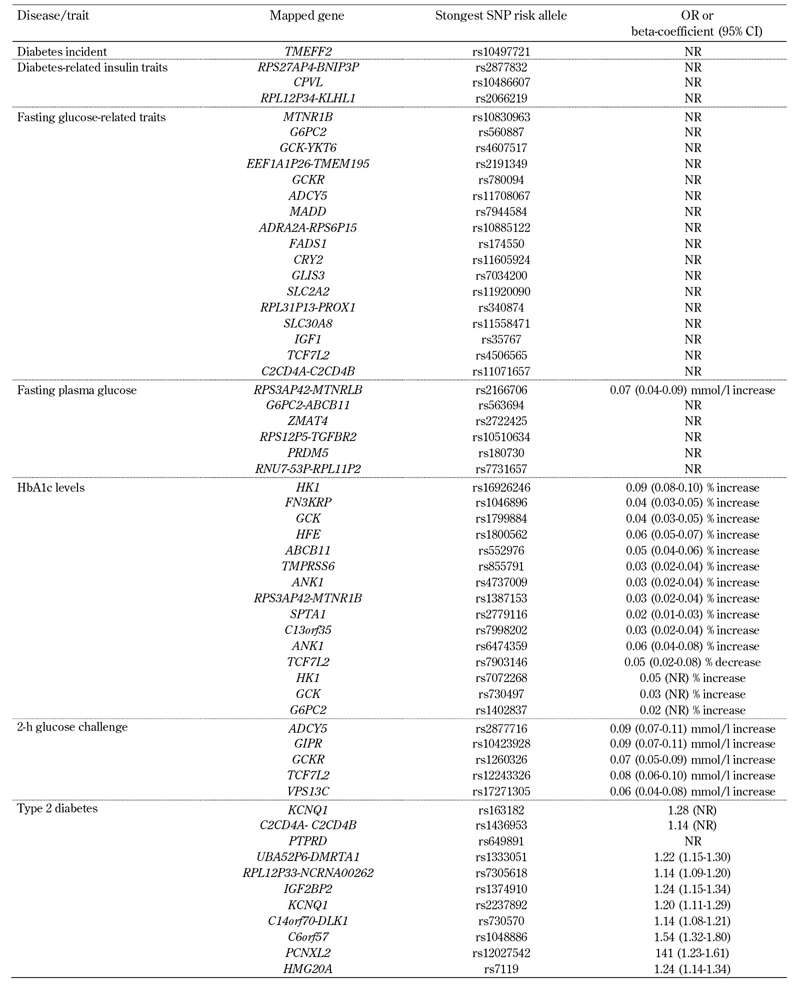
12. diacylglycerol kinase, beta 90kDa / transmembrane protein 195 (DGKB-TMEM195) (Table 1) [19].
Table 1. Single-nucleotide polymorphisms associated with type 2 diabetes and/or diabetes-related phenotypes in GWAS.
Genetics of coronary artery disease
Several GWAS for CAD have been conducted in the general population [20]. In GWAS analyzing CAD and myocardial infarction (MI), fewer genetic variants meet the established threshold of p < 5.10-8 compared with T2D [5]. The most studied and replicated locus associated with MI and CAD is located on chromosome 9p21.3 [21]. This locus is near the CDKN2A and CDKN2B genes, contains no annotated genes, and is not associated with established CVD risk factors such as plasma lipoproteins, or hypertension. Interestingly, it has also been associated with diabetes [22]. A number of subsequent GWAS analyzing CAD have found additional loci associated with MI or CAD risk in the general population, with genome-wide significance [23-25]. Most of these variants are associated with a relatively small increase in cardiovascular risk. Allelic odds ratios do not exceed 1.3 (Figure 2).
Figure 2. Classic risk factors in the formation and progression of atherosclerotic plaque, and possible pathogenic mechanisms of coronary artery disease risk genes.
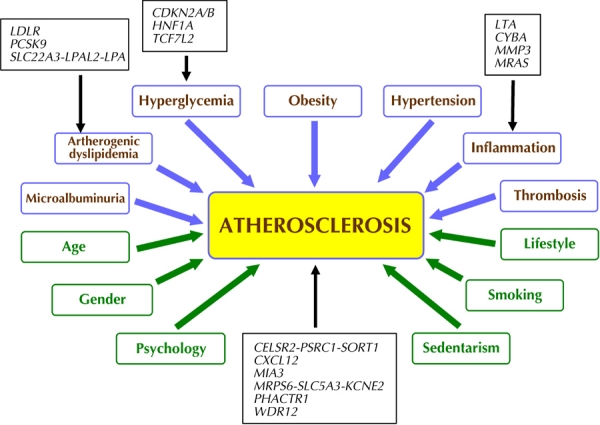
Blue: risk factors associated with metabolic syndrome (MS) and type 2 diabetes (T2D). Green: risk factor not directly associated with MS and/or T2D. With respect to diabetes, some mechanisms by which genes modulate the risk of atherosclerosis are not yet known.
Similar to other complex disorders, most of the CAD-associated variants are located in non-coding regions, suggesting an effect on regulatory elements and gene expression, rather than on the amino acid sequence [20]. Other than the SNPs mentioned above, SNPs from the following genes also reached the significance levels required in GWAS:
1. proprotein convertase subtilisin/kexin type 9 (PCSK9),
2. WD repeat domain 12 (WDR12),
3. phosphatase and actin regulator 1 (PHACTR1),
4. LDL-receptor (LDLR),
5. mitochondrial ribosomal protein S6 / solute carrier family 5 / potassium voltage-gated channel subfamily E member 2 (MRPS6-SLC5A3-KCNE2) [24],
6. cadherin EGF LAG seven-pass G-type receptor 2 / praline / serine-rich coiled-coil 1 / sortilin 1 (CELSR2-PSRC1-SORT1),
7. melanoma inhibitory activity family, member 3 (MIA3),
8. chemokine (C-X-C motif) ligand 12 (CXCL12) [13, 24, 25],
9. hepatocyte nuclear factor 1 homeobox A / chromosome 12 open reading frame 43 (HNF1A-C12orf43),
10. Ras-related protein M (MRAS) (Table 2) [23].
Table 2. Single-nucleotide polymorphisms associated with CAD and coronary atherosclerosis-related phenotypes in GWAS.

Also, two haplotypes from the SLC22A3-LPAL2-LPA gene cluster are associated to CAD [26]. Interestingly, most genes discovered in GWAS, and that appear to be involved with CAD, were not previously implicated in the etiology of atherosclerosis (Figure 2). Notable exceptions are:
1. LDLR, which codes for the LDL receptor,
2. PCSK9, which codes for a serine protease that is mutated in Mendelian forms of hypercholesterolemia [13], and
3. the SLC22A3-LPAL2-LPA cluster, which includes the gene for the atherogenic lipoprotein Lp(a).
Mutations in the gene HNF1A are involved in the pathogenesis of MODY. Clearly, further functional experiments are required to assess the molecular mechanisms underlying CAD/MI genetic risk factors.
Overlap between the genetics of T2D and CAD, and the genetics of diabetic macrovascular complications
Results from genome-wide association studies
As mentioned previously, T2D and CAD have many intersections and coexist in many cases. Therefore, genetic risk factors can exist and coincide in these two conditions. Indeed, 40-50% of the variance in two indices for assessing the extent of atherosclerosis (i.e. coronary calcium and carotid intima-media thickness) can be attributed to familial factors among individuals with T2D [27-29]. This effect persists after adjustment for known cardiovascular risk factors, suggesting that there are unidentified pathways related to both diabetes development and atherogenesis [20].
Currently, results from GWAS for CAD specifically conducted in diabetic patients are not available. However, GWAS for CAD have been performed in the general population. They have found relevant results in diabetic subjects [20]. Independent GWAS for CAD or MI have identified strong evidence for the association in the locus on chromosome 9p21, near the CDKN2A and CDKN2B genes [13, 21, 30]. These studies reported SNPs in strong linkage disequilibrium (LD) with each other in populations of European ancestry and secondary signals in the adjacent LD block [5, 21]. This LD block is devoid of protein-coding genes, but includes the most 3' exons of a non-coding gene known as ANRIL (a.k.a. CDKN2B-AS), which is expressed in tissues involved in atherosclerosis [22]. Prime ANRIL target candidates are the adjacent cell-cycle genes CDKN2A and CDKN2B involved in the control of cell proliferation, cell aging, and apoptosis. We recall that polymorphisms located in the same region are also associated with T2D (Figure 2, Table 3). In a replication study, the associations between these SNPs with both CAD and T2D were independent of each other. Also, these SNPs were not associated with changes in cholesterol, fibrinogen, albumin, uric acid, bilirubin, or homocysteine [22].
Table 3. Genes with single-nucleotide polymorphisms associated with both type 2 diabetes and coronary artery disease in GWAS.
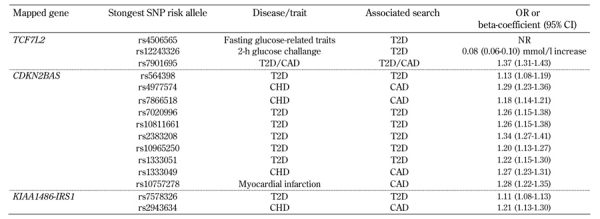
Legend: Data are OR or beta-coefficient and 95% CI. SNP: single-nucleotide polymorphism. OR: odds ratio. CI: Confidence interval. NR: not reported. T2D: type 2 diabetes. CAD: coronary artery disease. CHD: coronary heart disease.
The contribution of glycemia and genotype to CAD risk
Nevertheless, data from the Joslin Heart Study suggest that this locus may have a larger effect on CAD risk among T2D patients than in healthy controls because of an interaction with the diabetic milieu. This study compared cases of T2D and angiographic evidence of CAD with type 2 diabetic controls having a negative cardiovascular history of CAD and a normal exercise treadmill test. Odds ratios were 1.45 for heterozygotes and 2.37 for homozygotes. These values were consistent with an additive mode of inheritance, even after adjustment for cardiovascular risk factors [31]. The effects of the risk genotype were significantly increased by poor glycemic control. Therefore, it may be hypothesized that the association between these risk alleles, T2D, and cardiovascular disease occurs because of elevations in glycemia. However, the influence of other non-glycemic factors must not be disregarded. In fact, the current understanding of the effects of this locus on T2D and CAD suggest that there are two risk alleles affecting CAD and T2D independently. But it is important to develop new functional studies dissecting these extremely robust genotype-phenotype associations.
Candidate genes for CAD risk in the adiponectin pathway
Interesting results concerning candidate genes for diabetic cardiovascular complications have arisen from studies on the adiponectin pathway. Adiponectin is a proteic hormone produced and secreted exclusively by adipocytes. It regulates the metabolism of lipids and glucose by insulin-sensitizing effects. Adiponectin also has anti-inflammatory and direct anti-atherogenic effects by inhibiting monocyte adhesion to the endothelium, smooth muscle cell proliferation, and foam cell formation in the arterial wall [32]. In a meta-analysis of 827 CVD cases, and 1,887 CVD-free control subjects, the polymorphism +276G→T in the adiponectin gene was significantly associated with approximately 45% decreased CVD risk under a recessive inheritance model in diabetic patients [33].
Also, three SNPs located in the adiponectin receptor 1 (ADIPOR1) gene were all significantly associated with CAD among individuals with T2D. In the derivation population, the allelic odds ratios were in the range of 1.3-1.4. A similar, but not significant, trend was found in the replication sample [34]. This effect appears to be related to lower ADIPOR1 mRNA levels in carriers of the risk genotypes. In the same line of prior studies, there is an association between the adiponectin rs266729 promoter gene variant (-11377C > G) and plasma markers of oxidative stress in diabetic subjects, including molecules of oxidized-LDL [35]. Another study of interest in the association of oxidative stress with macrovascular complications of diabetes was performed by Jones et al. [36]. The authors found an association between risk genotype TT of mitochondrial superoxide dismutase 2 (SOD2), which codes an endogenous antioxidant enzyme, and the prevalence of CAD (odds ratio = 2.2) in diabetic females.
Single nucleotide polymorphisms as risk factors for T2D and cardiac artery disease
Considering the close relationship between T2D and cardiovascular disease, an intriguing question is whether there is an association between CAD and SNPs that are known risk factors for T2D. In this context, Pechlivanis et al. have evaluated the association between coronary artery calcification and 11 validated risk SNPs for diabetes in the Heinz Nixdorf recall cohort. They have shown that only the genetic variant near CDKN2A/2B was associated with quantitative coronary artery calcification. However, as previously discussed, one cannot exclude that the effect of another 9p21 gene was captured in the study [37].
There is also evidence for a strong interplay between TCF7L2 and CAD. TCF7L2 gene polymorphisms are the major known genetic risk factors for T2D. Common variants of this gene were significantly associated with diabetes, and they were consistently replicated in different populations worldwide. Different studies have found the highest odds ratios for diabetes in TCF7L2 variants, among all known variants [38]. In this context, Bielinski et al. have tested the TCF7L2 SNPs, rs7903146, rs12255372, rs7901695, rs11196205, and rs7895340, for associations with cardiovascular events in more than 13,000 individuals from the ARIC population (Atherosclerosis Risk in Communities). The authors did not find any significant association between TCF7L2 SNPs and the incidence of CAD, ischemic stroke, prevalence of peripheral arterial disease, or mortality from any of the causes in the full cohort. Stratifying the population by race or diagnosis of diabetes did not change this finding [39]. However, in a later study of about nine hundred subjects referred for cardiac catheterization for CAD diagnosis, our group has found a significant association between the TCF7L2 rs7903146 polymorphism and the prevalence and severity of CAD. Using an independent sample from the MASS-II Trial, prospectively followed-up for 5 years and assessed for major cardiovascular events incidence, we found no association between the TCF7L2 genotype and coronary lesions in diabetic subjects, although the diabetic patients did have a higher prevalence of coronary lesions. However, non-diabetic individuals carrying the risk allele were associated with a significantly higher frequency of coronary lesions than non-diabetics without the risk allele (adjusted odds ratio = 2.32 per T allele, 95%CI 1.27-4.24).
The presence of multi-vessel CAD was associated with the risk genotype in non-diabetics. Similarly, based on the prospective sample analysis, non-diabetics carrying the risk genotype had significantly more cardiovascular end-point events than non-risk carriers, mainly due to an increased incidence of death [40]. The results of these two studies are divergent, but there are major methodological differences between them. In the ARIC study, all patients with prevalent CAD were excluded so that all subjects were free of prior CVD at baseline. Whereas, our study included patients with CAD. These methodological differences, and the a priori risk of cardiovascular events in these populations, can explain the contradictory results.
Recently, a cross-sectional study assessed the TCF7L2 variants, rs7903146, rs12255372, and rs11196205, in 1,650 patients undergoing coronary angiography for the evaluation of established or suspected stable CAD. Interestingly, the variant rs7903146 was significantly associated with CAD in the total study cohort (adjusted additive odds ratio = 1.29, 95%CI 1.09-1.53). However, this association was stronger in T2D patients than in non-diabetic subjects even after adjustment by glycemia and HbA1c levels [41]. The reasons for these contradictory results in diabetic and non-diabetic subjects are not known. However, there are differences between the two studies concerning subject characteristics (e.g. mean age, prevalence of male gender and hypertension, and plasma levels of cholesterol). Also, there are differences in the model of inheritance used in the analysis, and the genetic background. Any, or all, of these differences could have contributed to the apparent divergent findings.
Finally, another study to assess the combined risk of several polymorphisms for T2D found similar results. It showed an association between cardiovascular events and combined genotype risk in non-diabetics, even after exclusion of TCF7L2 data [42]. The importance of TCF7L2 for CAD was confirmed in a study that reviewed data from the ARIC study. This study showed a significant increase in the risk of CAD only among lean individuals homozygous for the risk allele of the TCF7L2 rs7903146 gene risk variant (hazard ratio = 1.42, 95%CI 1.03,1.97) [43].
The mechanisms by which the carriers of the TCF7L2 risk allele have a higher risk of CAD are still unknown. The simplest explanation is the association of TCF7L2 with diabetes and hyperglycemia, at diabetic or non-diabetic levels. However, beyond hyperglycemia and diabetes, some studies have observed that the activation of TCF-4 transcription, encoded by TCF7L2, is related to the signaling pathway of nuclear factor-κB (NF-κB), which regulates inflammatory signaling pathways [44]. Therefore, NF-κB might be involved in an additional epistatic mechanism operating in the vascular wall. Also, other effects beyond carbohydrate metabolism, such as plasma lipid concentrations, blood pressure, markers of coagulation/inflammation [45], and enhanced sympathetic nervous system activity [46] may play a role for explaining these data.
Understanding the molecular mechanisms of risk association
There is increasing evidence to suggest that genetic factors related to T2D have more implications for the dysfunction of pancreatic beta-cells than for insulin resistance. Most of the genes associated with T2D risk are confirmedly, or potentially, related to pancreatic beta-cell function (Figure 1). In T2D pathophysiology, insulin resistance causes beta-cells to increase insulin secretion for the maintenance of normoglycemia. Therefore, genetically determined defects would only become evident in the presence of insulin resistance. However, T2D has an important environmental component. Each polymorphism associated with T2D only makes a small contribution to the risk of developing the disease, as observed by the relatively low OR values found in GWAS (unlike other clinical risk factors). Therefore, it is assumed that environmental conditions increase T2D risk, especially (but not only) by impairing insulin sensitivity because of the relationship between visceral obesity, sedentary lifestyle, and insulin resistance. If insulin secretion capacity by beta-cells is sufficient, then secretion can increase to compensate impaired insulin action, and hyperglycemia would not occur.
The underlying mechanisms that explain how these common genetic variants affect the function of pancreatic beta-cells have been proposed only for a few genes, after in vitro and in vivo studies [12]. Recently, efforts have been made to find pathophysiological explanations [47]. In a meta-analysis, Ingelsson et al. found statistical significance (p = 2.1x10-71) for the relationship between glucose-raising allele carriers and abnormal insulin processing and/or insulin secretion [48]. Nevertheless, progress towards understanding the disease mechanisms has been slowed by the modest effect sizes, and the fact that most GWAS signals map away from coding sequences [47].
In fact, there is an apparent difficulty in establishing a causal link between an associated allele and the molecular aspects of disease development. This impairs the understanding of how T2D risk alleles impact the prediction of cardiovascular events in diabetic and non-diabetic individuals.
Difficulties in determining the phenotypes
The characterization of the type of diabetes, or even the correct classification in T2D, for example, is a major concern. Misclassification may have significant consequences on the results of genetic association studies. At the molecular level, diabetes represents many diseases with a common phenotype, i.e. hyperglycemia, and the definition of disease can vary between studies. In many cases, these different molecular disorders are all grouped together in the same study with consequent loss of statistical power due to increased phenotypic heterogeneity [8]. Clinically, it has been difficult to distinguish each of these different diabetes categories. It is hoped that genetic studies can help to provide greater clarity in this regard.
The same applies to the determinination of CAD phenotypes. In this case, the tools for evaluation and confirmation are even more heterogeneous. Several studies were published with conflicting findings, which were due to either false positives from marginal p-values, or from false negatives because of the lack of statistical power. Another explanation may be that different phenotypes were investigated in different studies e.g., clinically significant CAD vs. coronary artery calcium [20]. At least in part, these problems explain the difficulty in establishing an association between polymorphisms and CAD, and lead to the following questions:
1. Is it possible to extrapolate the results of GWAS performed in Caucasians to African or South-American populations?
2. Is the preponderance of genes related to insulin secretion rather than insulin resistance at the genetic risk of T2D due to a weight limit in GWAS?
3. What is the best method for defining CAD?
The combination of studies using different phenotypic criteria should be avoided, or should only be used with limitations. This concept is particularly important, since it limits the comparison of GWAS results for both T2D and CAD as a test for a common molecular defect hypothesis.
At the moment, it is likely that phenotype misclassification is distorting results of genetic association studies, and impacts on the ability of genetic association studies to define a particular hyperglycemic or atherosclerotic phenotype. It is possible that the use of inclusion criteria such as age, body mass index (BMI), or ethnicity is biasing the observed results. Establishing the correct phenotype is very important for genetic association studies. This is evidenced in other polygenic diseases where the phenotypic diagnosis is less variable. For example, GWAS of rheumatoid arthritis have more consistent results, despite similar degrees of heritability [49].
Is the "lacking heritability" hiding potential common effectors?
It is clear that there are problems with GWAS. However, despite the limitations, GWAS offer an image of the entire genome for genes affecting susceptibility to diabetic complications [20]. In the last decade, this advantage has led to a boom in the investigation of genetic risk factors for complex diseases. Nevertheless, the discovery of risk genes for complex diseases through GWAS cannot explain all the heritability aspects of these diseases. The heritability of many complex diseases, such as T2D and CAD, have been estimated from twin and family studies. There are estimates of how much the identified genes explain the total genetic variance of a given complex disease [50]. Most variants identified from GWAS confer relatively small increments in risk, and explain only a small proportion of familial clustering. This leads to the question how the "lacking" heritability can be explained [51].
According to the common variant disease hypothesis, complex diseases such as T2D and CAD are caused by the simultaneous occurrence of common DNA sequence variations (minor allele frequencies of more than 5%) in many genes [12]. Each of these DNA alterations is supposed to exert only moderate effects on the affected genes' function and/or expression; but in sum, these variations would confer an increased susceptibility to disease. This idea initiated the search for a set of SNPs associated with complex diseases. Then, the combination of information from each polymorphism would sum up the total heritability.
However, the known genetic risk factors for both T2D and CAD only explain one quarter of the predicted heritability, at best. A recent study assessed 25 risk SNPs for T2D, with a heritability of approximately 42%. These SNPs explained approximately 28% [50]. Similarly, the heritability of CAD (including myocardial infarction) is about 49%, with 12 studied loci explaining about 25% of heritability. This confirms that a considerable proportion of heritability is still "lacking".
With a larger sample size and increased genome coverage, more loci are implicated in pathophysiological mechanisms of polygenic diseases. However, it is expected that the strength of association between the recently discovered SNPs and complex diseases is even lower than the association found in the first SNPs. Therefore, according to this hypothesis, many new SNPs associated with T2D or CAD need to be discovered so that we can learn about the heritability. However, it is difficult to conceive that unknown variants with small effects can account for the unidentified common effectors of both diseases.
Beside the purely genetic association, gene-gene and gene-environment interactions, epigenetics, and the role of microRNAs are also not fully understood [52]. Some environmental disturbances, such as low birth weight, obesity, sedentary lifestyle, and aging may lead to T2D by affecting the expression of genes through epigenetic modifications. These epigenetic modifications of the genome provide mechanisms for stable propagation of the expression of some genes from one cellular generation to another. This is achieved either by modification in histones or DNA methylation [53]. Indeed, subsequent to a period of prior hyperglycemic exposure, some individuals with diabetes experience a continued progression of vascular complications even after glycemic control. This phenomenon is termed "metabolic memory" [54]. Classically, it has been demonstrated in clinical trials such as the Diabetes Control and Complications Trial (DCCT), and the follow-up Epidemiology of Diabetic Complications and Interventions Trial (EDIC) in type 1 diabetic subjects [55], and later in other clinical trials with T2D patients [54].
Although the role of DNA methylation in the pathogenesis of cardiovascular diseases is not completely understood, atherosclerosis was associated with global hypomethylation in smooth muscle cells of atherosclerotic lesions from humans and animals. Altered DNA methylation of several candidate genes linked to atherosclerosis was identified in both vascular smooth muscle cells and endothelial cells in mouse models [54]. Also, other CAD risk factors such as hyperhomocysteinemia, hypercholesterolemia, and inflammation have been associated with atherosclerosis, by altering DNA methylation [56]. The role of epigenic factors is particularly important in the current discussion on the common effector hypothesis since genetic risk factors for diabetes may be overrepresented in GWAS on CAD, even if the standard adjustments for BMI, diabetes status, and glucose control have been made. However, as previously discussed, such overrepresentation has not been observed.
Final considerations
Considering the epidemiological importance of T2D and CAD, it is worth working towards a better understanding of their genetic basis. There are many challenges remaining in the identification of genetic variants that influence the risk of T2D, diabetic macrovascular complications, and CAD. Large clinical studies are needed to discover additional genetic loci. However, if cohorts are combined, heterogeneity across studies may become a problem.
Most of the GWAS published are from samples of European ancestry populations. It will be important to reassess the recognized variants in other populations. A few GWAS have evaluated structural variants or rare alleles, which are likely to be responsible for a segment of the "lacking heritability", and are important targets of resequencing. Additional biological mechanisms of complex diseases may be discovered through studies of gene-gene and gene-environment interactions, especially if there are interactions with dietary and physical activity factors. These studies should aim to elucidate the mechanisms of inter-individual variability. All this together could clarify important aspects of the genetics of both T2D and CAD. A better understanding of the complex mechanisms will certainly contribute to finding preventative therapies for these two conditions.
At present, there is little genetic evidence to support a common effector hypothesis other than insulin or hyperglycemia, for these conditions. Most probably, genetic risk factors for diabetes do indeed confer risk for CAD. This is not caused by intrinsic and pleiotropic atherosclerosis-promoting mechanisms, but more likely by the hyperglycemic/insulinic diabetic environment.
Disclosures: The authors report no conflict of interests.
References
- 1.Malecki MT. Genetics of type 2 diabetes mellitus. Diabetes Res Clin Pract. 2005;68(Suppl 1):S10–S21. doi: 10.1016/j.diabres.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Steck AK, Rewers MJ. Genetics of type 1 diabetes. Clin Chem. 2011;57(2):176–185. doi: 10.1373/clinchem.2010.148221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burcelin R, Knauf C, Cani PD. Pancreatic alpha-cell dysfunction in diabetes. Diabetes Metab. 2008;34(Suppl 2):S49–S55. doi: 10.1016/S1262-3636(08)73395-0. [DOI] [PubMed] [Google Scholar]
- 4.Milicevic Z, Raz I, Beattie SD, Campaigne BN, Sarwat S, Gromniak E, Kowalska I, Galic E, Tan M, Hanefeld M. Natural history of cardiovascular disease in patients with diabetes: role of hyperglycemia. Diabetes Care. 2008;31(Suppl 2):S155–S160. doi: 10.2337/dc08-s240. [DOI] [PubMed] [Google Scholar]
- 5.Mohlke KL, Boehnke M, Abecasis GR. Metabolic and cardiovascular traits: an abundance of recently identified common genetic variants. Hum Mol Genet. 2008;17(R2):R102–R108. doi: 10.1093/hmg/ddn275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrish NJ, Wang SL, Stevens LK, Fuller JH, Keen H. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia. 2001;44(Suppl 2):S14–S21. doi: 10.1007/pl00002934. [DOI] [PubMed] [Google Scholar]
- 7.King H, Rewers M. Global estimates for prevalence of diabetes mellitus and impaired glucose tolerance in adults. WHO Ad Hoc Diabetes Reporting Group. Diabetes Care. 1993;16(1):157–177. doi: 10.2337/diacare.16.1.157. [DOI] [PubMed] [Google Scholar]
- 8.Barroso I. Genetics of type 2 diabetes. Diabet Med. 2005;22(5):517–535. doi: 10.1111/j.1464-5491.2005.01550.x. [DOI] [PubMed] [Google Scholar]
- 9.Meigs JB, Cupples LA, Wilson PW. Parental transmission of type 2 diabetes: the Framingham Offspring Study. Diabetes. 2000;49(12):2201–2207. doi: 10.2337/diabetes.49.12.2201. [DOI] [PubMed] [Google Scholar]
- 10.Amini M, Janghorbani M. Diabetes and impaired glucose regulation in first-degree relatives of patients with type 2 diabetes in Isfahan, Iran: prevalence and risk factors. Rev Diabet Stud. 2007;4(3):169–176. doi: 10.1900/RDS.2007.4.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance - a population-based twin study. Diabetologia. 1999;42(2):139–145. doi: 10.1007/s001250051131. [DOI] [PubMed] [Google Scholar]
- 12.Staiger H, Machicao F, Fritsche A, Haring HU. Pathomechanisms of type 2 diabetes genes. Endocr Rev. 2009;30(6):557–585. doi: 10.1210/er.2009-0017. [DOI] [PubMed] [Google Scholar]
- 13.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ. et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 15.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU. et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316(5829):1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S. et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445(7130):881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 17.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S. et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39(6):770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 18.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40(5):638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL. et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet;42(2):105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doria A. Genetics of diabetes complications. Curr Diab Rep. 2010;10(6):467–475. doi: 10.1007/s11892-010-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR. et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, Green F, Clarke R, Collins R, Franzosi MG, Tognoni G. et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17(6):806–814. doi: 10.1093/hmg/ddm352. [DOI] [PubMed] [Google Scholar]
- 23.Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, Linsel-Nitschke P, Kathiresan S, Wright B, Tregouet DA. et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41(3):280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H. et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41(3):334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE. et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel-Nitschke P, Perret C, DeSuremain M. et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41(3):283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 27.Bowden DW, Cox AJ, Freedman BI, Hugenschimdt CE, Wagenknecht LE, Herrington D, Agarwal S, Register TD, Maldjian JA, Ng MC. et al. Review of the Diabetes Heart Study (DHS) family of studies: a comprehensively examined sample for genetic and epidemiological studies of type 2 diabetes and its complications. Rev Diabet Stud. 2010;7(3):188–201. doi: 10.1900/RDS.2010.7.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke. 2002;33(7):1876–1881. doi: 10.1161/01.str.0000019909.71547.aa. [DOI] [PubMed] [Google Scholar]
- 29.Wagenknecht LE, Bowden DW, Carr JJ, Langefeld CD, Freedman BI, Rich SS. Familial aggregation of coronary artery calcium in families with type 2 diabetes. Diabetes. 2001;50(4):861–866. doi: 10.2337/diabetes.50.4.861. [DOI] [PubMed] [Google Scholar]
- 30.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G. et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 31.Doria A, Wojcik J, Xu R, Gervino EV, Hauser TH, Johnstone MT, Nolan D, Hu FB, Warram JH. Interaction between poor glycemic control and 9p21 locus on risk of coronary artery disease in type 2 diabetes. JAMA. 2008;300(20):2389–2397. doi: 10.1001/jama.2008.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26(3):439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 33.Qi L, Doria A, Manson JE, Meigs JB, Hunter D, Mantzoros CS, Hu FB. Adiponectin genetic variability, plasma adiponectin, and cardiovascular risk in patients with type 2 diabetes. Diabetes. 2006;55(5):1512–1516. doi: 10.2337/db05-1520. [DOI] [PubMed] [Google Scholar]
- 34.Soccio T, Zhang YY, Bacci S, Mlynarski W, Placha G, Raggio G, Di Paola R, Marucci A, Johnstone MT, Gervino EV. et al. Common haplotypes at the adiponectin receptor 1 (ADIPOR1) locus are associated with increased risk of coronary artery disease in type 2 diabetes. Diabetes. 2006;55(10):2763–2770. doi: 10.2337/db06-0613. [DOI] [PubMed] [Google Scholar]
- 35.Prior SL, Gable DR, Cooper JA, Bain SC, Hurel SJ, Humphries SE, Stephens JW. Association between the adiponectin promoter rs266729 gene variant and oxidative stress in patients with diabetes mellitus. Eur Heart J. 2009;30(10):1263–1269. doi: 10.1093/eurheartj/ehp090. [DOI] [PubMed] [Google Scholar]
- 36.Jones DA, Prior SL, Tang TS, Bain SC, Hurel SJ, Humphries SE, Stephens JW. Association between the rs4880 superoxide dismutase 2 (C>T) gene variant and coronary heart disease in diabetes mellitus. Diabetes Res Clin Pract. 2010;90(2):196–201. doi: 10.1016/j.diabres.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 37.Pechlivanis S, Scherag A, Muhleisen TW, Mohlenkamp S, Horsthemke B, Boes T, Brocker-Preuss M, Mann K, Erbel R, Jockel KH, Nothen MM, Moebus S. Coronary artery calcification and its relationship to validated genetic variants for diabetes mellitus assessed in the Heinz Nixdorf recall cohort. Arterioscler Thromb Vasc. Biol;30(9):1867–1872. doi: 10.1161/ATVBAHA.110.208496. [DOI] [PubMed] [Google Scholar]
- 38.Perry JR, Frayling TM. New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr Opin Clin Nutr Metab Care. 2008;11(4):371–377. doi: 10.1097/MCO.0b013e32830349a1. [DOI] [PubMed] [Google Scholar]
- 39.Bielinski SJ, Pankow JS, Folsom AR, North KE, Boerwinkle E. TCF7L2 single nucleotide polymorphisms, cardiovascular disease and all-cause mortality: the Atherosclerosis Risk in Communities (ARIC) study. Diabetologia. 2008;51(6):968–970. doi: 10.1007/s00125-008-1004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sousa AG, Marquezine GF, Lemos PA, Martinez E, Lopes N, Hueb WA, Krieger JE, Pereira AC. TCF7L2 polymorphism rs7903146 is associated with coronary artery disease severity and mortality. PLoS One. 2009;4(11):e7697. doi: 10.1371/journal.pone.0007697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muendlein A, Saely CH, Geller-Rhomberg S, Sonderegger G, Rein P, Winder T, Beer S, Vonbank A, Drexel H. Single nucleotide polymorphisms of TCF7L2 are linked to diabetic coronary atherosclerosis. PLoS One. 2011;6(3):e17978. doi: 10.1371/journal.pone.0017978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sousa AG, Lopes NH, Hueb WA, Krieger JE, Pereira AC. Genetic variants of diabetes risk and incident cardiovascular events in chronic coronary artery disease. PLoS One. 2011;6(1):e16341. doi: 10.1371/journal.pone.0016341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kucharska-Newton AM, Monda KL, Bielinski SJ, Boerwinkle E, Rea TD, Rosamond WD, Pankow JS, Kottgen A, Heiss G, North KE. Role of BMI in the Association of the TCF7L2 rs7903146 Variant with Coronary Heart Disease: The Atherosclerosis Risk in Communities (ARIC) Study. J Obes. 2010;2010: 651903. doi: 10.1155/2010/651903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, Hall JL. A role for the beta-catenin/T-cell factor signaling cascade in vascular remodeling. Circ Res. 2002;90(3):340–347. doi: 10.1161/hh0302.104466. [DOI] [PubMed] [Google Scholar]
- 45.Delgado-Lista J, Perez-Martinez P, Garcia-Rios A, Phillips CM, Williams CM, Gulseth HL, Helal O, Blaak EE, Kiec-Wilk B, Basu S. et al. Pleiotropic effects of TCF7L2 gene variants and its modulation in the metabolic syndrome: from the LIPGENE study. Atherosclerosis. 2011;214(1):110–116. doi: 10.1016/j.atherosclerosis.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 46.Boccardi V, Ambrosino I, Papa M, Fiore D, Rizzo MR, Paolisso G, Barbieri M. Potential role of TCF7L2 gene variants on cardiac sympathetic/parasympathetic activity. Eur J Hum Genet. 2010;18(12):1333–1338. doi: 10.1038/ejhg.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van de Bunt M, Gloyn AL. From genetic association to molecular mechanism. Curr Diab Rep. 2010;10(6):452–466. doi: 10.1007/s11892-010-0150-2. [DOI] [PubMed] [Google Scholar]
- 48.Ingelsson E, Langenberg C, Hivert MF, Prokopenko I, Lyssenko V, Dupuis J, Magi R, Sharp S, Jackson AU, Assimes TL. et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes. 2010;59(5):1266–1275. doi: 10.2337/db09-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A. et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42(6):508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.So HC, Gui AH, Cherny SS, Sham PC. Evaluating the heritability explained by known susceptibility variants: a survey of ten complex diseases. Genet Epidemiol. 2011;35(5):310–317. doi: 10.1002/gepi.20579. [DOI] [PubMed] [Google Scholar]
- 51.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A. et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guay C, Roggli E, Nesca V, Jacovetti C, Regazzi R. Diabetes mellitus, a microRNA-related disease? Transl Res. 2011;157(4):253–264. doi: 10.1016/j.trsl.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Pinney SE, Simmons RA. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab. 2010;21(4):223–229. doi: 10.1016/j.tem.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90(3):421–429. doi: 10.1093/cvr/cvr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ, Raskin P, Zinman B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353(25):2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, Heimburger O, Barany P, Alvestrand A, Nordfors L. et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? J Intern Med. 2007;261(5):488–499. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]