

Abstract
Phylogenetic trees show a remarkable slowdown in the increase of number of lineages towards the present, a phenomenon which cannot be explained by the standard birth–death model of diversification with constant speciation and extinction rates. The birth–death model instead predicts a constant or accelerating increase in the number of lineages, which has been called the pull of the present. The observed slowdown has been attributed to nonconstancy of the speciation and extinction rates due to some form of diversity dependence (i.e., species-level density dependence), but the mechanisms underlying this are still unclear. Here, we propose an alternative explanation based on the simple concept that speciation takes time to complete. We show that this idea of “protracted” speciation can be incorporated in the standard birth–death model of diversification. The protracted birth–death model predicts a realistic slowdown in the rate of increase of number of lineages in the phylogeny and provides a compelling fit to four bird phylogenies with realistic parameter values. Thus, the effect of recognizing the generally accepted fact that speciation is not an instantaneous event is significant; even if it cannot account for all the observed patterns, it certainly contributes substantially and should therefore be incorporated into future studies.
Keywords: Birth–death model, pull of the present, pull of the recent, protracted speciation, diversity dependence
The temporal pattern of diversification has been of scientific interest for a long time (Yule 1924; Raup et al. 1973). Over the last two decades, methods have been developed to infer diversification rates from phylogenies (Nee et al. 1992; Harvey et al. 1994; Nee et al. 1994a, Nee et al. 1994b; Pybus and Harvey 2000; Nee 2001; Ricklefs 2007). Even though phylogenies may ultimately not be sufficient to accurately estimate speciation and/or extinction rates (Paradis 2003, Paradis 2004: Etienne and Apol 2009: Rabosky 2010), the plot of the number of lineages in the phylogeny versus time, that is, the lineages-through-time (LTT) plot (Nee et al. 1992: Harvey et al. 1994: Pybus and Harvey 2000: Phillimore and Price 2008), often shows a remarkable slowdown towards the present (McPeek 2008; Phillimore and Price 2008; Rabosky and Lovette 2008). In contrast, the standard birth–death model of diversification (Kendall 1948; Raup et al. 1973; Nee et al. 1994a) shows an upward turn towards the present, which has been called the pull of the present (Nee et al. 1994b). To avoid possible confusion, the pull of the present is a phenomenon that is distinct from the pull of the recent (Raup 1979; Jablonski et al. 2003; Nee 2006), which describes the apparently increased rate of diversification seen in the fossil record caused by more complete sampling of recent (and still extant) species. The pull of the present in LTT plots is purely a theoretical phenomenon, a property of the standard birth–death model of diversification. It results from the fact that lineages arising in the recent past are less likely to have become extinct and therefore are more likely to be represented in the phylogeny than lineages arising in the more distant past.
Two explanations of the observed slowdown in LTT plots have been offered. The first is that it is due to a sampling artifact. Two sampling artifacts have been identified. Nee et al. (1994b) showed that taking a small sample from the actual phylogeny produces this slowdown; it transforms the upward turn predicted by the model into a downward turn. More recently, Purvis et al. (2009) argued, on the basis of simulations with the pure birth (i.e., without extinctions, Yule 1924) model, that an apparent slowdown will be observed if there is age dependency in whether nodes are deemed to be speciation events. Sampling effects cannot explain, however, observed slowdowns in (nearly) complete phylogenies (Phillimore and Price 2008). The second explanation is diversity dependence, that is, species-level density dependence (Phillimore and Price 2008), the per species speciation and/or extinction rates are not constant as in the standard birth–death model but decrease with time due to niche filling (Schluter 2000; Ricklefs 2010 ). Although this is certainly a possibility, it has also been argued that in contrast, new species may actually create new niches (Odling-Smee et al. 2003).
Here, we offer an alternative explanation of the slowdown which is an extension of the standard birth–death model in which speciation is assumed not to take place instantaneously but is allowed to take time. There is general agreement that speciation takes time (Avise 1999). Speciation requires reproductive isolation, which could be either prezygotic (e.g., due to mate choice) or postzygotic (e.g., due to reduced hybrid fitness). Both prezygotic and postzygotic isolations are correlated with genetic distance between pairs of species, which is a strong indicator of time since divergence between the species (Coyne and Orr 2004). There is clear evidence that a considerable amount of time may be needed to create the genetic distance required to distinguish two `good' species. For example, fertile and viable hybrids can exist for millions of years since the initial divergence (Coyne and Orr 2004), and old populations on islands are much more likely to be recognized as taxonomically distinct than young populations (Price et al. 2010). Avise (1999) provides various estimates of upper and lower bounds to the duration of speciation (the upper bound being set by the divergence time of sister species, see also Rosindell et al. (2010), and the lower bound being set by the divergence time of phylogroups). For birds and mammals, he reports values between 1 and 3 Myr. In fish and herpetofauna, the reported rates are similar to those of birds and mammals but could in reality be much larger due to slower mitochondrial DNA clocks. Examples of 5 Myr exist in salamanders. Only in exceptional cases, for example, polyploidy in plants, can speciation occur instantaneously. Most detailed genetic models of speciation also predict that speciation takes time (Gavrilets 2004). Here, in order to preserve generality, we deliberately do not assume a specific mechanism for speciation but only recognize the simple fact that it is gradual rather than instantaneous. This form of speciation, termed “protracted speciation” by Rosindell et al. (2010), thus implicitly captures the outcome of what in reality are complex, ecological, and genetic processes, which given enough time lead to the birth of a new species (Schluter 2009).
Protracted speciation has been shown to resolve problems with the predictions of the neutral theory of biodiversity on speciation rate and species longevities (Hubbell 2001; Rosindell et al. 2010). Here, we show how it explains the slowdown in LTT plots, in general, and when applied to four bird phylogenies. Moreover, we show that it can predict more imbalanced phylogenies than the standard birth–death model. We first study the pure birth model (i.e., no extinctions) with protracted speciation because it allows analytical treatment, which elegantly proves our point mathematically. We then explore the birth–death model with protracted speciation by simulation.
RESULTS
Model Predictions
Pure birth model
We start with the pure birth model or Yule (1924) model of diversification. We denote the number of species with Ng where the subscript g will become clear later. At a constant rate λ1, species produce new species. There is no extinction in this model. Figure 1A shows the pure birth process. The probability that at time t there are Ng species is given by the following master equation:
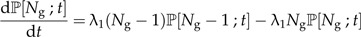 |
(1a) |
with initial condition
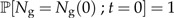 |
(1b) |
This equation can be completely solved analytically, but here, we are only interested in the expected number of species at time t which obeys the ODE
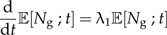 |
(2a) |
with initial condition
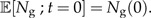 |
(2b) |
The solution is straightforward:
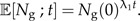 |
(3) |
Because all species survive (no extinction), the expected number of ancestral lineages in the phylogeny, L, at time t for the good species that are extant the present time T is simply given by
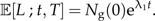 |
(4) |
The present time T is irrelevant for this model but will be relevant for the protracted form of this model. From Equation (4), we see that the logarithm of the number of lineages increases linearly with time t:
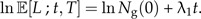 |
(5) |
Figure 1.

The pure birth model a) with and b) without protracted speciation. Dotted lines indicate an incipient species and solid lines are good species. c) Phylogeny of the protracted pure birth process of panel b: only those lineages that have completed speciation before the present will show up in the phylogeny. Note that the branching points are at the times that the incipient species are produced, not at the times that they become good species.
PROTRACTED PURE BIRTH MODEL
Now we make speciation a protracted process. That is, each extant species still produces new species at a rate λ1, but these new species are not yet good species. Instead, they are incipient species which become good species at a rate λ2. This means that the time needed to complete speciation is exponentially distributed with parameter λ2, so the mean time it will take to complete speciation is . Note that this protracted speciation model differs slightly from that analyzed by Rosindell et al. (2010) who assumed a fixed time to complete speciation. If anything, a stochastically varying time to complete speciation seems more realistic. Incipient species give rise to new incipient species at rate λ3 while they are incipient. Figure 1B shows this protracted pure birth process. Again, we can write down a master equation, but now for the probability ℙ[Ng,Ni;t] that at time t there are Ng good (hence the subscript g) species and Ni incipient species:
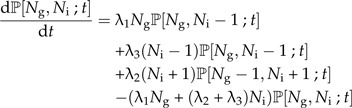 |
(6a) |
with initial condition
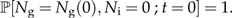 |
(6b) |
This model cannot be solved analytically for ℙ[Ng,Ni;t], but we can write down expressions for the expected number of good and incipient species at time t (see Supplementary Data) :
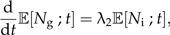 |
(7a) |
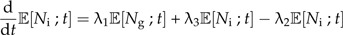 |
(7b) |
with initial condition
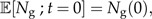 |
(7c) |
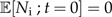 |
(7d) |
This can be solved in general (see Supplementary Data), but here, we look at the special case where incipient species gives rise to new incipient species at the same rate as good species do (λ3 = λ1) in which case, we have (see Supplementary Data):
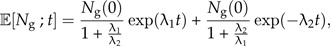 |
(8a) |
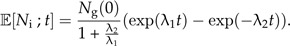 |
(8b) |
The expected number of ancestral lineages L at time t for the good species extant at time T is the sum of the expected number of good species and the expected number of incipient species which have at least one good descendant species before time T:
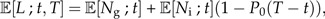 |
(9) |
where P0(T − t) is the probability that none of the descendants becomes a good species. Figure 1C illustrates that not all incipient species contribute to L because not all incipient species leave good descendant species before T. In online Supplementary Data, we derive an analytical expression for P0(t):
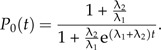 |
(10) |
Inserting this expression and Equation (8) in Equation (9) yields
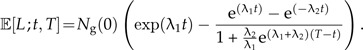 |
(11) |
We observe that the number of lineages increases less with t than in the pure birth model without protracted speciation (λ2 = ∞), because the second term on the right hand side increases with t so the closer to the present, the more 𝔼[L;t,T] will differ from the pure birth model. Figure 2 illustrates how the slowdown in increase of the number of lineages depends on the parameter λ2.
Figure 2.

Expected LTT plot for the protracted pure birth model (i.e., no extinction) for various values of the speciation completion rate λ2. The value of the speciation initiation rate λ1 is 0.5. The curve for λ2 = ∞ is barely visible, as it almost coincides with the curve for λ2 = 10.
The formulas above assume that we start with Ng(0) good species, but in practice, we look at a phylogeny starting with two lineages at crown age at which point at least one of the two lineages is incipient (because it originates from the other at this point). In online Supplementary Data, we derive analogous expressions for this initial condition.
BIRTH–DEATH MODEL
Starting from the pure birth model, but allowing for extinctions, one obtains the well-known birth–death model (Kendall 1948) for which the master equation reads:
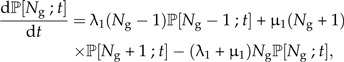 |
(12a) |
where μ1 is the extinction rate and the initial conditions are given by Equation (1b). See Figure 3, panels A and B, for an illustration of the birth–death process. The solution for this model can be obtained analytically with the method of characteristics. Here, we are interested in the expected number of species at time t, which is given by
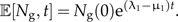 |
(12b) |
To compute the number of ancestral lineages L at time t of the species extant at time T, we first need the probability, ℙt1,t2, that there are surviving lineages at time t2 for a process starting at t1 with a single individual. This probability is given by (Kendall 1948; Nee et al. 1994a)
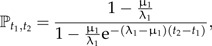 |
(13) |
assuming that λ1 = /μ1. The number of ancestral lineages at time t conditional on survival of the clade until the present time T is given by (Nee et al. 1994a, see also online Supplementary Data)
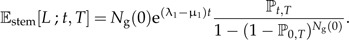 |
(14) |
This expression is valid for the expected number of lineages when starting with Ng(0) species at the stem age t = 0. In practice, we usually have data on crown age, the branching point of the first two ancestral lineages. To produce the model's expectations for a phylogeny with a prescribed crown age, we must require that both ancestral lineages survive because if only one survives, there may still be a phylogeny, but it does not have the prescribed crown age. Mathematically, starting with two lineages at the crown age t = 0 implies that we can simply take twice Equation (14) with Ng(0) = 1:
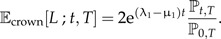 |
(15) |
Regardless of whether we use the stem age–based Equation (14) or crown-age based Equation (15), the logarithm of the number of lineages increases more than linearly with time:
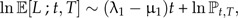 |
(16) |
where we ignored all terms that do not depend on t because ℙt,T increases more than linearly with t. This means that the model predicts an upturn in number of lineages near the present. This phenomenon is called the pull of the present (Nee et al. 1994b) and can be seen in Figure 4. The verbal explanation is that recently arisen species have not had the time to become extinct causing an apparent acceleration of diversification near the present.
FIGURE 3.

The birth–death model with and without protracted speciation. a) A birth–death process that is extinct before the present time T, an eventuality that most analyses are conditioned against. b) A birth–death process that survives up to the present time T. c) The birth–death process of b where speciation is protracted. Dotted lines indicate an incipient species and solid lines are good species. d) Phylogeny of the protracted birth–death process of panel c. Only those lineages that have completed speciation or incipient lineages whose parent species has gone extinct before the present will show up in the phylogeny.
FIGURE 4.

Expected LTT plots for the protracted birth–death model for various values of the extinction rate μ1 = μ2 (upper panels) and the corresponding histograms of the slowdown statistic r (bottom panels). The lines are for different speciation completion rates λ2. The value of the speciation initiation rate λ1 is set at 0.5. The curve for λ2 = ∞ is barely visible, as it almost coincides with the curve for λ2 = 10.
PROTRACTED BIRTH–DEATH MODEL
Making speciation protracted changes the master equation to
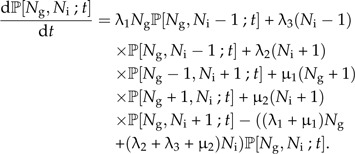 |
(17) |
See Figure 3C for an illustration of the process. As in the protracted pure birth model, the master equation cannot be solved analytically. Nevertheless, it is again possible to obtain analytical expressions for the expected number of good and incipient species at time t. We will not write out these expressions and their solutions explicitly because they are cumbersome to write, and, more importantly, they cannot easily be used to obtain an expression for 𝔼[L;t,T]. First of all, conditioning on survival to the present, as in Equation (14), is no longer trivial because this requires (a function of) ℙ[Ng,Ni;t] for which an analytical solution is lacking. Furthermore, the addition of the expectations of Ng and Ni with a correction for the latter, as in Equation (9), no longer holds, because of the complicating factor that even if an incipient species has not become a good species by time T, it will be counted as a good species if its immediate ancestor was good but has become extinct (it simply replaces this extinct ancestor, because as long as it has not completed speciation, it will be considered identical to the ancestor species). Figure 3D explains this.
Because further analytical treatment seems extremely challenging, we simulated the process in order to gain insight in the behavior of the protracted birth–death model. Simulations also have the advantage that we can produce the (expected) number of lineages through time starting from the crown age, rather than the stem age, which require that conditioning should be done on the survival to the present of at least two lineages.
The simulation procedure of the protracted birth–death model is straightforward: We used the Gillespie (1976) algorithm to simulate the master Equation (17). We started with one good species and one incipient species because we wanted to look at the results for crown age. Alternatively, one could start with two incipient species, but this does not affect the results qualitatively. For every newly arisen incipient species, we recorded, during the simulation, the exact time when it arose (time of birth), from which species it arose (parent species), when it completes speciation (time of maturation), and when it becomes extinct (time of death), noting that speciation completion and extinction need not occur. At the end of the simulation (at T = 15 Myr in our simulations), we constructed the phylogeny from this information to check whether the phylogeny had the initial good and incipient species as common ancestors (the common ancestors could be younger which must be ruled out because all iterations of the simulation must have the same crown age so that they can be averaged). If not, the phylogeny was ignored and the whole procedure was repeated.
Constructing this phylogeny is less straightforward than running the simulation. To obtain the phylogeny, we counted all extant species regarded as good species at the last event in the simulation (time t). Of course, good species qualify to be regarded as good, but there is a special case where an incipient species must also be regarded as good: when it arose from a good species that has since become extinct (Fig. 3C). However, when several incipient species all arose from a now extinct good species, only one should be counted as a good species; here, we adopted the convention of always choosing the youngest orphaned incipient species. To find the branching times of the phylogeny, the lineages considered good at time t were followed backwards in time until the birth event of one of these lineages (as an incipient species) was encountered. We define the age of a species as the time that passes between birth and death (extinction), not the time between maturation and death (extinction). This definition most closely matches the way real phylogenies are constructed. We continued the backwards search for birth events until one lineage remained at the crown age: The point just before the birth event of the second still extant lineage. In all our simulations, we assumed that incipient species produce new incipient species at the same rate as good species do (λ3 = λ1), but our code allows for different values for these rates (λ3 = /λ1). Online Supplementary Data contains a pseudo-code for the simulations which explains the construction of the phylogeny in more detail. A Matlab code is available upon request from the corresponding author.
Figure 4 shows the LTT plots for various extinction rates (where extinction rates of incipient and good species are identical). Furthermore, it shows histograms of the slowdown statistic Δr which is calculated as (Pigot et al. 2010)
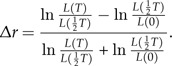 |
(18) |
This is a better statistic than the often-used γ-statistic which depends on the size of the tree. It is clear that, as in the protracted pure birth model, making speciation protracted (lower values of λ2) causes a slowdown in the increase of the number of lineages through time. Remarkably, not only does the mean of the measure of slowdown Δr shift to the left but also the variance of Δr becomes larger for smaller values of λ2 (longer mean time to complete speciation).
In Figure 4, the extinction rates of incipient and good species are set equal. This may not be realistic (see Discussion section). Figure 5 shows the effect of different extinction rates for incipient and good species. When the incipient species extinction rate is high, protracted speciation no longer causes a slowdown in the LTT plot. The reason is simple: When incipient species are highly likely to go extinct, incipient species that manage to become good species will necessarily have to do so quickly, that is, they should take a short time to complete speciation. The situation is then similar to the birth–death process without protracted speciation, with the corresponding pull of the present.
FIGURE 5.

Expected LTT plots for the protracted birth–death model for various values of the incipient species extinction rate μ2, where the extinction rate of good species is set at μ1 = 0.4. The lines are for different speciation completion rates λ2. The value of the speciation initiation rate λ1 is set at 0.5.
SPECIATION COMPLETION TIME AND DURATION OF SPECIATION
The parameter λ2 measures the rate of incipient species completing speciation, and therefore, its inverse, , measures the time to complete speciation. However, this is not the same as the duration of speciation that is measured from speciation events that have actually occurred because some incipient species may never become good species because they go extinct (at rate μ2 in our model) and because each incipient species may produce incipient species itself (at rate λ3 in our model) that complete speciation before their parent does. One can derive an expression for the mean duration of speciation τ in terms of the model parameters (see online Supplementary Data):
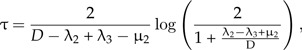 |
(19) |
where
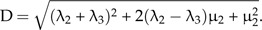 |
(20) |
If λ3 = 0, this expression simplifies to (see online Supplementary Data)
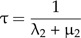 |
(21a) |
and for μ2 = 0, we have
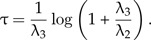 |
(21b) |
Except for this last case, the distribution of the mean duration of speciation is no longer exponential and may have an interior mode (see online Supplementary Data).
MODEL FIT TO DATA
An exact expression for the likelihood of a phylogeny (and therefore an LTT) can be derived in the special case λ3 = λ1 and μ1 = μ2 = 0 (see online Supplementary Data):
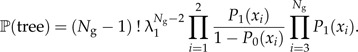 |
(22) |
where the xi are the branching times and
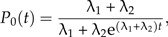 |
(23a) |
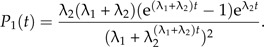 |
(23b) |
Figure 6 shows the fit of the protracted birth model (in green) to four bird phylogenies (Acanthiza, Cracidae, Myiborus, and Toxostoma) using this likelihood. The phylogenies were selected from the bird phylogenies of Phillimore and Price (2008) with the criteria that a slowdown must be clearly visible in the data and that there are no missing species. Table 1 contains the parameter estimates.
FIGURE 6.

LTT plots (stars) of four bird phylogenies, selected from Phillimore & Price (2008) (see text for selection criteria) and the fits of the protracted birth model (gray) and the protracted birth–death model (black). The former is obtained through maximum likelihood, the latter by least squares (see text). We assumed that μ2 = μ1 and λ3 = λ1, so the fitted parameters are λ1, λ2, and μ1.
TABLE 1.
Parameter estimates of four bird phylogenies for the protracted birth model (pb, using likelihood) and the protracted birth–death model (pbd, using least squares)
| Data set | Model | λ 1 = λ3 (Myr −1) | λ 2 (Myr −1) | μ 1 = μ2 (Myr −1) | GOF | τ (Myr ) |
| Acanthiza | pb | 0.47 | 0.04 | 0 | – 7.89 | 5.16 |
| pbd | 0.66 | 0.07 | 0.3 | 0.35 | 3.81 | |
| Cracidae | pb | 0.96 | 0.12 | 0 | 1.13 | 2.31 |
| pbd | 1.09 | 0.16 | 0.3 | 0.47 | 1.95 | |
| Myiborus | pb | 0.48 | 0.89 | 0 | – 2.71 | 0.89 |
| pbd | 0.81 | 0.39 | 0.3 | 0.41 | 1.29 | |
| Toxostoma | pb | 0.43 | 0.05 | 0 | – 8.34 | 5.19 |
| pbd | 0.65 | 0.06 | 0.3 | 0.24 | 3.98 |
Because the extinction rate cannot be estimated reliably (the goodness of fit [GOF] did not change over a wide range of extinction values), the reported value of 0.3 was set beforehand. Also shown are the corresponding values of the GOF statistic, i.e. maximum likelihood and least-ssquared distance, and the estimated average duration of speciation (τ).
We have not yet been able to find an expression for the likelihood for the protracted model with extinction because of difficulties defining good species (incipient species with extinct good parents must be considered good). Therefore, we employed a different fitting method to fit the model to four bird phylogenies. The fitting method is a least-squares fit of the full LTT plot: Using a simplex optimization algorithm, we searched for the parameters that minimize the distance between the observed LTT plot and the expected LTT plot, where the latter was obtained by simulation (10,000 iterations with a different seed for each iteration and the seeds fixed during the optimization to minimize noise). We found that we could not estimate the parameters reliably because the fit did not show an observable change across a wide range of extinction rates, but the speciation initiation and speciation completion rates did depend on the value of the extinction rate. Figure 6 shows the fit for an extinction rate of 0.3 Myr − 1, whereas Table shows the corresponding parameter estimates of speciation initiation and speciation completion rates and the durations of speciation calculated from these rates with Equation (19). The durations of speciation are definitely in the right ball park, but because of the difficulty in accurately estimating the parameters, they should be interpreted with care.
It may seem that the fit of the protracted birth model, using maximum likelihood, is not as good as the fit of the protracted birth–death model, using least squares, but this is only apparent. Maximizing the likelihood finds the parameters that make the observed data set the mode of the probability distribution of phylogenies, whereas the least-squares approach finds the parameters that make it the mean. Expected LTT plot are, by definition, based on the mean.
DISCUSSION
We have shown that protracted speciation, which only assumes that speciation takes time rather than occurs instantaneously, can explain the observed slowdown in LTT plots. The verbal argument is simple: Speciation events that initiated in the recent past may not have completed yet, so they do not count towards the total number of extant species at the present. Stated otherwise, to find branching points in the phylogenetic tree, one has to look back into the past at least as far as the time needed for speciation to complete, which is on average . This causes many recent branching points to disappear, but deeper branching points will be counted as producing good lineages because they almost always leave enough time for speciation to complete.
Not all real phylogenies show a slowdown in diversification. This may be considered to be at odds with our results, but they are actually fully consistent with them. Figure 4 shows the distribution of the slowdown statistic Δr across 10,000 phylogenies simulated with the same parameter set. Whereas the bulk of the simulations show negative values of the slowdown statistic, some of them have slowdown parameters larger than 0. Also, the protractedness parameter λ2 need not be the same for all clades because the speciation process may be different across different clades.
In our model, we have assumed the simplest form of protracted speciation, where good species give rise to incipient species at a constant rate λ1 and incipient species complete their speciation at a constant rate λ2. The latter means that the time to complete speciation is exponentially distributed. Naturally, speciation is a much more intricate process than these simple assumptions suggest. An incipient species may have to go through several stages before it is considered a good species, for example, if speciation requires an accumulation of mutations (Gavrilets 2004). This can be modeled by assuming higher order incipient species Ni,j (where j is the order) that transform into one another with constant rates λ2,j. We then look at the dynamics of ℙ[Ng,Ni,1,…,Ni,n,t] when the highest order is n. The time spent in each of these incipient states is still exponentially distributed with parameter λ2,j, but the total time to complete speciation, which is the sum of the times spent in each incipient state, is no longer exponentially distributed. For example, when all rates are identical (λ2,j = λ2 for all j = 1,…,n), then the total time to complete speciation is gamma distributed (Akkouchi 2008):
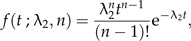 |
(24) |
whereas for different parameters λ2,j, the probability distribution is
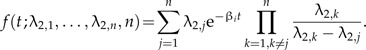 |
(25) |
These distributions, unlike the exponential distribution, are hump shaped with their mode at time larger than 0 and very flexible, so a wide variety of speciation modes could be incorporated. Such alternative distributions for the time for speciation to complete will have consequences for the quantitative shape of the LTT plot, but we expect that it will not change our results qualitatively: There will still be a slowdown of the increase of the number of lineages. For example, consider a model with n incipient states with identical rates nλ2. We expect the slowdown to be more pronounced because while the mean time to complete speciation is still , the modal time to complete speciation, , is larger than 0. Furthermore, note that for this model, the larger the number of incipient stages, the more peaked is the distribution of the speciation-completion time. In the limit of an infinite number of incipient stages, the speciation completion time becomes fixed. This is the case considered in Rosindell et al. (2010) in the context of the neutral theory of biodiversity.
The parameter μ2 is the extinction rate of incipient species. It can be argued that incipient species are more likely to become extinct (and hence have higher μ2 values) because of smaller population size or because gene flow causes the incipient species to merge with its parent (McPeek 2008). However, one could also argue that incipient species have a lower extinction rate because they are likely to fill a new niche in which there is less competition. In reality, both processes may play a role. A possible scenario is that initially the extinction rate is high, but this extinction rate decreases as time goes by. This can all be incorporated in the protracted speciation model with multiple incipient states. For example, in a model with two incipient states, the first state has a higher extinction rate than the second.
Not only can protracted speciation explain slowdown in LTT plots, it can also predict more imbalanced trees than the standard birth–death model, in line with observations (Blum and Francois 2006; Phillimore and Price 2008), if λ3 < λ1 and λ2 is small ( < 1). This is not due to protractedness per se but due to the fact that it takes longer for a newly arisen incipient species to speciate further than for a good species when λ3 < λ1. This induces a time lag which has been shown to change tree imbalance in simulations (Losos and Adler 1995; Rogers 1996) and analytically (Pinelis 2003). We show this result for λ3 = 0 in online Supplementary Data.
The protracted speciation model is a parsimonious explanation for the slowdown in diversification that has, to the best of our knowledge, gone unnoticed in the literature. Only Weir and Schluter (2007) constructed a model which, in hindsight, can be interpreted as an early protracted speciation model, but it was not used to explain slowdowns in clade diversification. In fact, our model now allows for a rigorous reanalysis of their pioneering work, relating the latitudinal diversity gradient to a latitudinal trend in the duration of speciation. Our protracted speciation model bears some resemblance to the explanations in terms of diversity dependence as well as age dependence of nodes. Diversity dependence induces a nonconstant rate of speciation, and, likewise, protracted speciation induces nonconstant rate of speciation where the speciation rate decreases near the present day to discount any speciation events that have started, but not yet completed. The main difference between our model and the diversity-dependence model is that in the latter, the speciation rate depends on the number of extant species which naturally increases with time. Diversity dependence means that the species are no longer independent from one another, and hence, the analytical treatment of the diversity-dependent birth–death model is challenging; recent work in the field (Rabosky and Lovette 2008) has been criticized for making unrealistic assumptions for the sake of tractability (Bokma 2009). Analytical treatment of the protracted speciation model interpreted as a time-dependent birth–death model is also not trivial because the distribution of speciation completion times cannot be simply translated into a time-dependent speciation rate, but perhaps, this interpretation provides good approximations and the idea merits further study. The second explanation, of age dependence of nodes (Purvis et al. 2009), is actually an emergent property of protracted speciation. Missing species are not interpreted as sampling effects but as unfinished speciation processes. In fact, all explanations for the slowdown in diversification must involve a mechanism for lowering the speciation rate at times near the present. That diversification rates vary in time is generally accepted (Bokma 2003), but the question is why they should mostly be decreasing right now. The answer is inherent in the explanations in terms of sampling effects and protracted speciation, but it is a separate assumption in the explanation in terms of diversity dependence. Diversity dependence certainly plays a role, but it may be confined to specific cases, for example, Rabosky and Lovette 2008, whereas protracted speciation seems a more general phenomenon. The only way to find out is to construct a model that contains both protracted speciation and diversity dependence and compare it to a model with just protracted speciation.
We used a least-squares approach to estimate model parameters in the protracted birth–death model because we have not yet been able to derive a full likelihood formula for this model when extinction is nonzero. Such a likelihood formula is useful for a statistically sound estimation of parameters from given phylogenies and comparison of the performance of different models (although maximum-likelihood–based estimated tend to be biased). Nevertheless, the simplicity of protracted speciation leaves us hopeful that such a likelihood formula will become available for our model or a variant that also contains the essence of protracted speciation. However, even if the likelihood can be computed, the estimates are unlikely to be very accurate because we expect the likelihood surface to be fairly flat across a wide range of extinction rates. This expectation is based on results with the least-squares approach where the least-squares distance between model and data showed no observable change across a wide range of extinction rates. Our inability to accurately estimate parameters will be even more pronounced in more realistic models with λ1 = /λ3 and μ1 = /μ2 or a more realistic distribution of speciation completion times because of the higher number of df. Consequently, the phylogeny alone might not allow accurate estimation of the key parameters and the relative contributions of protracted speciation and other mechanisms such as diversity dependence in explaining the slowdown. This may require development of methods employing additional data such as the fossil record (Paradis 2003, Paradis 2004; Etienne and Apol 2009; Purvis et al. 2009).
We have shown that the observed slowdowns in diversification can be explained with the simple assumption that speciation takes time. In no way do we exclude the possibility or importance of diversity-dependent diversification; rather, we regard protracted speciation as an alternative, possibly complementary parsimonious explanation that cannot be ignored in future models of diversification.
SUPPLEMENTARY MATERIAL
FUNDING
This work was financially supported by the EPSRC (grant EP/F043112/1) and the Netherlands Organization for Scientifi c Research (NWO).
Supplementary Material
Acknowledgments
We are most grateful to Albert Phillimore and Bart Haegeman for very stimulating discussions and comments on the manuscript and the former for providing the bird phylogenies. J.R. thanks the EPSRC (grant EP/F043112/1) and R.S.E. thanks the Netherlands Organization for Scientific Research (NWO) for financial support.
References
- Akkouchi M. On the convolution of exponential distributions. J. Chungcheong Math. Soc. 2008;21:501–510. [Google Scholar]
- Avise JC. Cambridge (MA): Harvard University Press; 1999. Phylogeography: the history and formation of species. [Google Scholar]
- Blum MGB, Francois O. Which random processes describe the tree of life? A large-scale study of phylogenetic tree imbalance. Sys. Biol. 2006;55:685–691. doi: 10.1080/10635150600889625. [DOI] [PubMed] [Google Scholar]
- Bokma F. Testing for equal rates of cladogenesis in diverse taxa. Evolution. 2003;57:2469–2474. doi: 10.1111/j.0014-3820.2003.tb01492.x. [DOI] [PubMed] [Google Scholar]
- Bokma F. Problems detecting density-dependent diversification on phylogenies. Proc. R. Soc. Lond. B. 2009;276:993–994. doi: 10.1098/rspb.2008.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Sunderland (MA): Sinauer; 2004. Speciation. [Google Scholar]
- Etienne RS, Apol MEF. Estimating speciation and extinction rates from diversity data and the fossil record. Evolution. 2009;63:244–255. doi: 10.1111/j.1558-5646.2008.00537.x. [DOI] [PubMed] [Google Scholar]
- Gavrilets S. Princeton (NJ): Princeton University Press; 2004. Fitness landscapes and the origin of species. [Google Scholar]
- Gillespie DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976;22:403–434. [Google Scholar]
- Harvey PH, May RM, Nee S. Phylogenies without fossils. Evolution. 1994;48:523–529. doi: 10.1111/j.1558-5646.1994.tb01341.x. [DOI] [PubMed] [Google Scholar]
- Hubbell SP. Princeton (NJ): Princeton University Press; 2001. The Unified Neutral Theory of Biodiversity and Biogeography. [Google Scholar]
- Jablonski D, Roy K, Valentine JW, Price RM, Anderson PS. The impact of the pull of the recent on the history of marine diversity. Science. 2003;300:1133–1135. doi: 10.1126/science.1083246. [DOI] [PubMed] [Google Scholar]
- Kendall DG. On some modes of population growth giving rise to R.A. Fisher's logarithmic series distribution. Biometrika. 1948;35:6–15. [PubMed] [Google Scholar]
- Losos J, Adler FR. Stumped by trees? A generalized null model for patterns of organismal diversity. Am. Nat. 1995;145:329–342. [Google Scholar]
- McPeek MA. The ecological dynamics of clade diversification and community assembly. Am. Nat. 2008;172:E270–E284. doi: 10.1086/593137. [DOI] [PubMed] [Google Scholar]
- Nee S. Inferring speciation rates from phylogenies. Evolution. 2001;55:661–668. doi: 10.1554/0014-3820(2001)055[0661:isrfp]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Nee S. Birth-death models in macroevolution. Annu. Rev. Ecol. Syst. 2006;37:1–17. [Google Scholar]
- Nee S, Holmes EC, May RM, Harvey PH. Extinction rates can be estimated from molecular phylogenies. Philos. Trans. Biol. Sci. 1994;344:77–82. doi: 10.1098/rstb.1994.0054. [DOI] [PubMed] [Google Scholar]
- Nee S, May RM, Harvey PH. The reconstructed evolutionary process. Philos. Trans. R. Soc. Lond. B. 1994;344:305–311. doi: 10.1098/rstb.1994.0068. [DOI] [PubMed] [Google Scholar]
- Nee S, Ø Mooers A, Harvey PH. Tempo and mode of evolution revealed from molecular phylogenies. Proc. Natl. Acad. Sci. U. S. A. 1992;89:8322–8326. doi: 10.1073/pnas.89.17.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odling-Smee F, Laland KN, Feldman MW. Princeton (NJ): Princeton University Press; 2003. Niche construction: the neglected process in evolution. [Google Scholar]
- Paradis E. Analysis of diversification: combining phylogenetic and taxonomic data. Proc. R. Soc. Lond. B. 2003;270:2499–2505. doi: 10.1098/rspb.2003.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E. Can extinction rates be estimated without fossils? J. Theor. Biol. 2004;229:19–30. doi: 10.1016/j.jtbi.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Phillimore AB, Price TD. Density-dependent cladogenesis in birds. PLoS Biol. 2008;6:0483–0489. doi: 10.1371/journal.pbio.0060071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigot A, Phillimore AB, Owens IPF, Orme CDL. The shape and temporal dynamics of phylogenetic trees arising from geographic speciation. Syst. Biol. 2010;59:660–673. doi: 10.1093/sysbio/syq058. [DOI] [PubMed] [Google Scholar]
- Pinelis I. Evolutionary models of phylogenetic trees. Proc. R. Soc. Lond. B. 2003;279:1425–1431. doi: 10.1098/rspb.2003.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price T, Phillimore AB, Awodey M, Hudson R. Ecological and geographical influences on the allopatric phase of island speciation. In press. In: Grant PR, Grant BR, editors. From field observations to mechanisms. A program in evolutionary biology. Princeton (NJ): Princeton University Press; 2010. [Google Scholar]
- Purvis A, Orme CDL, Toomey NH, Pearson PN. Temporal patterns in diversification rates. Chapter 15, p. 278–300. In: R.Butlin DSchluter, J.Bridle, editors. Speciation and patterns of diversity. Cambridge (UK): Cambridge University Press; 2009. [Google Scholar]
- Pybus OG, Harvey PH. Testing macro-evolutionary models using incomplete molecular phylogenies. Proc. R. Soc. Lond. B. 2000;267:2267–2272. doi: 10.1098/rspb.2000.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky DL. Extinction rates should not be estimated from molecular phylogenies. Evolution. 2010;64:1816–1824. doi: 10.1111/j.1558-5646.2009.00926.x. [DOI] [PubMed] [Google Scholar]
- Rabosky DL, Lovette IJ. Density-dependent diversification in North American wood warblers. Proc. R. Soc. Lond. B. 2008;275:2363–2371. doi: 10.1098/rspb.2008.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raup DM. Biases in the fossil record of species and genera. Bull. Carnegie Mus. Nat. Hist. 1979;13:85–91. [Google Scholar]
- Raup DM, Gould SJ, Schopf TJM, Simberloff DS. Stochastic models of phylogeny and the evolution of diversity. J. Geol. 1973;81:525–542. [Google Scholar]
- Ricklefs RE. Estimating diversification rates from phylogenetic information. Trends Ecol. Evol. 2007;22:601–610. doi: 10.1016/j.tree.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Ricklefs RE. Evolutionary diversification, coevolution between populations and their antagonists, and the filling of niche space. Proc. Natl. Acad. Sci. U. S. A. 2010;107:1265–1272. doi: 10.1073/pnas.0913626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JS. Central moments and probability distributions of three measures of phylogenetic tree imbalance. Syst. Biol. 1996;45:99–110. [Google Scholar]
- Rosindell J, Cornell SJ, Hubbell SP, Etienne RS. Protracted speciation revitalizes the neutral theory of biodiversity. Ecol. Lett. 2010;13:716–727. doi: 10.1111/j.1461-0248.2010.01463.x. [DOI] [PubMed] [Google Scholar]
- Schluter D. The ecology of adaptive radiation. New York: Oxford University Press; 2000. [Google Scholar]
- Schluter D. Evidence for ecological speciation and its alternative. Science. 2009;323:737–741. doi: 10.1126/science.1160006. [DOI] [PubMed] [Google Scholar]
- Weir JT, Schluter D. The latitudinal gradient in recent speciation and extinction rates of birds and mammals. Science. 2007;315:1574–1576. doi: 10.1126/science.1135590. [DOI] [PubMed] [Google Scholar]
- Yule G. A mathematical theory of evolution based on the conclusions of Dr. J. C. Willis, FRS. Philos. Trans. R. Soc. Lond. 1924 B 213:21–87. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.