

Abstract
AIM: To investigate the expression and methylation status of the secreted frizzled-related protein 2 (SFRP2) in esophageal squamous cell carcinoma (ESCC) and explore its role in ESCC carcinogenesis.
METHODS: Seven ESCC cell lines (KYSE 30, KYSE150, KYSE410, KYSE510, EC109, EC9706 and TE-1) and one immortalized human esophageal epithelial cell line (Het-1A), 20 ESCC tissue samples and 20 paired adjacent non-tumor esophageal epithelial tissues were analyzed in this study. Reverse-transcription polymerase chain reaction (RT-PCR) was employed to investigate the expression of SFRP2 in cell lines, primary ESCC tumor tissue, and paired adjacent normal tissue. Methylation status was evaluated by methylation-specific PCR and bisulfite sequencing. The correlation between expression and promoter methylation of the SFRP2 gene was confirmed with treatment of 5-aza-2’-deoxycytidine. To assess the potential role of SFRP2 in ESCC, we established stable SFRP2-transfected cells and examined them with regard to cell proliferation, colony formation, apoptosis and cell cycle in vivo and in vitro.
RESULTS: SFRP2 mRNA was expressed in the immortalized normal esophageal epithelial cell line but not in seven ESCC cell lines. By methylation-specific PCR, complete methylation was detected in three cell lines with silenced SFRP2 expression, and extensive methylation was observed in the other four ESCC cell lines. 5-aza-2’-deoxycytidine could restore the expression of SFRP2 mRNA in the three ESCC cell lines lacking SFRP2 expression. SFRP2 mRNA expression was obviously lower in primary ESCC tissue than in adjacent normal tissue (0.939 ± 0.398 vs 1.51 ± 0.399, P < 0.01). SFRP2 methylation was higher in tumor tissue than in paired normal tissue (95% vs 65%, P < 0.05). The DNA methylation status of the SFRP2 correlated inversely with the SFRP2 expression. To assess the potential role of SFRP2 in ESCC, we established stable SFRP2 transfectants and control counterparts by introducing pcDNA3.1/v5 hisA -SFRP2 or pcDNA3.1/v5 hisA -empty vector into KYSE30 cells lacking SFRP2 expression. After transfection, the forced-expression of SFRP2 was confirmed by the RT-PCR. In comparison with the control groups, stably-expressed SFRP2 in KYSE 30 cells significantly reduced colony formation in vitro (47.17% ± 15.61% vs 17% ± 3.6%, P = 0.031) and tumor growth in nude mice (917.86 ± 249.35 mm3 vs 337.23 ± 124.43 mm3, P < 0.05). Using flow cytometry analysis, we found a significantly higher number of early apoptotic cells in SFRP2-transfected cells than in the control cells (P = 0.025). The mean cell number in the S and G2-M phases of the cell cycle was also significantly lower in SFRP2-transfected KYSE30 cells compared with mock transfected counterparts.
CONCLUSION: Silencing of SFRP2 expression through promoter hypermethylation may be a factor in ESCC carcinogenesis through loss of its tumor-suppressive activity.
Keywords: Esophageal squamous cell carcinoma, Secreted frizzled-related protein 2, Methylation, Tumor suppressor gene, Wnt signaling pathway
INTRODUCTION
Esophageal cancer is the sixth leading cancer-related cause of death worldwide. Although esophageal adenocarcinoma is the most rapidly increasing cancer in Western countries, esophageal squamous cell carcinoma (ESCC) remains the predominant histological subtype in China[1]. Despite recent improvements in its diagnosis and treatment, the prognosis for patients with ESCC is still unsatisfactory[2]. Among the reasons cited for this situation are the lack of understanding of the carcinogenic mechanism of ESCC, and the lack of sensitive and specific molecular markers to detect these cancers at an early stage.
In addition to genetic changes, epigenetic modifications and in particular DNA methylation, are recognized as common molecular alteration in human tumors. Functional inactivation of tumor suppressor genes (TSG) through promoter methylation has been shown to be involved in the pathogenesis of various cancers, including ESCC[3-7]. Importantly, DNA methylation changes have been reported to occur not only in advanced cancers, but also in premalignant lesions[8,9]. DNA methylation changes are, therefore, potentially good early indicators for the clinical application of sensitive cancer detection.
Aberrant promoter methylation of secreted frizzled-related protein (SFRP) genes, a new group of tumor suppressor genes, has been documented in several human malignancies such as colorectal cancer[10], gastric cancer[11], and hepatocellular carcinoma[12]. SFRPs are a family of secreted glycoproteins that act as negative regulators of the Wnt signaling pathway. Every member in this family of genes contains a frizzled-like cysteine-rich domain through which they either interact with Wnt proteins to prevent them from binding to Fz proteins, or form non-functional complexes with Fz and then block the Wnt signaling pathway[13,14]. As the Wnt pathway plays a crucial role in various human carcinogenesis[15,16], its aberrant activation by epigenetic inactivation of SFRPs may induce tumorigenesis.
The aberrant hypermethylation of the SFRP2 promoter has been reported to be a good molecular marker in gastric and colorectal cancer[17,18], suggesting that SFRP2 is a tumor suppressor. However, other reports indicate that SFRP2 promotes tumor progression in glioma[19], renal cancer cells[20], and decreases apoptosis in breast cancer cells[21]. In spite of these studies, to our knowledge there have not yet been any reports describing the significance of epigenetic inactivation of the SFRP2 gene in ESCC progression and its potential as a diagnostic and therapeutic target. We therefore analyzed the methylation and expression status, as well as the function, of this gene in ESCC.
Here, we first determine SFRP2 methylation and its expression level in 7 ESCC cell lines and 20 paired primary ESCC tissues. We then explore the functional significance of methylation-induced silencing of SFRP2 expression in ESCC cell lines both in vitro and in vivo.
MATERIALS AND METHODS
Cell lines and cell culture
A total of seven ESCC cell lines (KYSE 30, KYSE150, KYSE410, KYSE510, EC109, EC9706 and TE-1) were maintained in our laboratory. One human immortalized normal esophageal epithelial cell line (Het-1A), which was used as a “normal” control for ESCC cell lines, was purchased from the American Type Culture Collection (Manassas, VA, United States). ESCC cell lines were cultured in RPMI 1640 (Invitrogen, Carlsbad, CA, United States) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, United States) and antibiotics (100 U/mL penicillin G and 100 μg/mL streptomycin) at 5% CO2, 37 °C, and 95% humidity. Het-1A cells were cultured in bronchial epithelial basal medium with growth supplements (Clonetics, San Diego, CA, United States).
Human esophageal tissue samples
Paired specimens of human ESCC and adjacent non-cancerous esophageal squamous epithelium (n = 20) were obtained from patients who underwent resection for ESCC without chemotherapy or radiation therapy at the Beijing Friendship Hospital. Samples were stored in liquid nitrogen. All subjects gave informed consent for obtaining the study materials. The study was approved by the Ethics Committee of Beijing Friendship Hospital.
RNA extraction and reverse-transcription polymerase chain reaction
Total RNA was extracted from the 20 pairs of human tissue and eight cell lines by Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. For semi-quantitative reverse-transcription polymerase chain reaction (RT-PCR), 2 μg of RNA was reversely transcribed using Superscript II reverse transcriptase according to the manufacturer’s protocol (Invitrogen). The mRNA expression levels of the SFRP2 were determined by conventional RT-PCR with Taq polymerase (Takara, Dalian, China). Glyceraldehyde-3-phosohate dehydrogenase was used as an internal control of RNA integrity. The RT-PCR procedure consisted of 35 cycles with an annealing temperature of 56 °C. The primers used are listed in Table 1.
Table 1.
List of primer sequences
| Primer | Forward primer (5'-3') | Reverse primer(5'-3') |
| RT-PCR | ||
| SFRP2 | 5’-GATGATGACAACGACATAATGGAAACG -3’ | 5’-GAGTGTGCTTGGGGAACGGGAGCT-3’ |
| GAPDH | 5’-CCCTTCATTGACCTCAACTACATGG-3’ | 5’-CATGGTGGTGAAGACGCCAG-3’ |
| SFRP2(CDS) | 5’-CCAAGCTTATGCTGCAGGGCCCTGGCTCGC-3’ | 5’-CGGAATTCCTAGCACTGCAGCTTGCGGATGCTG-3’ |
| MSP | ||
| SFRP2-M | 5’-GGGTCGGAGTTTTTCGGAGTTGCGC-3’ | 5’-CCGCTCTCTTCGCTAAATACGACTCG-3’ |
| SFRP2-U | 5’-TTTTGGGTTGGAGTTTTTTGGAGTTGTGT-3’ | 5’-AACCCACTCTCTTCACTAAATACAACTCA-3’ |
| BSP | ||
| SFRP2 | 5’-AAAAAGGTTAAGAAAATTTTGGT-3’ | 5’-AACCAAAACCCTACAACATC-3’ |
SFRP2: Secreted frizzled-related protein 2; GAPDH: Glyceraldehyde-3-phosohate dehydrogenase; MSP: Methylation-specific.
Isolation and bisulfite modification of genomic DNA
Genomic DNA was obtained from esophageal tissues and cell lines by standard phenol-chloroform extraction. Genomic DNA was treated with sodium bisulfite using a Zymo DNA Modification kit (Zymo Research, Orange, CA). Bisulfite induces deamination of unmethylated cytosines, converting unmethylated CpG sites to UpG without modifying methylated sites. This allows their differentiation by methylation-specific PCR, or sequencing.
Demethylation with the DNA demethylating agent 5-aza-2-deoxycytidine
Three human ESCC cell lines (TE-1, KYSE-30 and KYSE-510) were treated with DNA demethylating agent 5-Aza-dC by addition of fresh medium containing 5-Aza-dC (10 μmol/L, Sigma) every day for 3 consecutive days. Cells were harvested for DNA and RNA extractions. Control cells received no drug treatment.
Methylation-specific polymerase chain reaction
The methylation status of the SFRP2 in esophageal tissues and cell lines was determined by MSP. Briefly, bisulfite-treated DNA was used as a template to amplify MSP using primers specific for methylated and unmethylated sequences of the gene. The PCR reaction was set up using 0.5 U hot-start Taq-polymerase (Takara) per reaction in a total volume of 20 μL and run for 40 cycles with an annealing temperature of 60 °C. MSP primers are listed in Table 1.
Bisulfite genomic sequencing
For bisulfite genomic sequencing, a fragment of the SFRP2 promoter CpG islands that contained 34 CpG sites was amplified from the bisulfite-treated DNA using specific primers (Table 1). The PCR procedure consisted of 35 cycles with an annealing temperature of 54 °C. The PCR products were then cloned into the pEasy-T1 vector (Transgene, Beijing, China). Eight to ten colonies were randomly chosen and sequenced.
Cloning of SFRP2 and construction of expression vector
Expression vectors containing the human full-length cDNA fragment of SFRP2 were generated by PCR cloning. Primers are listed in Table 1. Amplified PCR products were cloned into pEasy-T1 vector (Transgene), and the fragments verified by sequencing, then cut using EcoRIHF and HindIII, and ligated into the EcoRIHF/HindIII sites of pcDNA3.1/ V5-HisA vector (Invitrogen).
Cell cycle analysis
Cell cycle analysis was carried out by flow cytometry. KYSE30 cells were transfected with 4 μg of pcDNA3.1/V5-HisA-SFRP2-expression vector or pcDNA3.1 V5-HisA empty vector using Lipofectamine2000 (Invitrogen). After 48 h, cells were harvested and fixed in 70% ethanol for 30 min. The samples were concentrated by removing the ethanol and treating with 10 μg/mL RNase (Roche). They were then stained with propidium iodine (Sigma) and analyzed for cell cycle distribution by DNA content analysis using flow cytometry.
Stable clone establishment
To prepare stable cell lines re-expressing SFRP2, we transfected KYSE30 cells with the pcDNA3.1/V5-HisA-SFRP2 expression vector encoding SFRP2 cDNA using Lipofectamine2000 (Invitrogen), according to the manufacturer’s instructions. Cells transfected with pcDNA3.1 V5-HisA empty vector were used as controls. Transfected cells were selected by culturing with G418 (Merck, Darmstadt, Germany) at 300 μg/mL for 1 mo. Single colonies of stable transfectants were isolated and expanded for further analysis.
Detection of apoptosis by annexin V/FITC
KYSE30 cells stably transfected with pcDNA3.1/V5-HisA-SFRP2-expression vector or pcDNA3.1 V5-HisA empty vector were harvested, washed twice in phosphate buffered saline (PBS) (4 °C), and re-suspended in 250 μL binding buffer. One hundred microliters of cells from each sample were aliquoted into FACS tubes and stained with 5 μL annexin V/FITC and 10 μL of 20 μg/mL propidium iodine (Sigma). After gentle mixing, cells were incubated for 15 min at room temperature in the dark. Four hundred microliters of binding buffer was added immediately prior to analysis by flow cytometry.
Cell proliferation assay
Stable transfected KYSE30 cells with or without SFRP2 expression vector and parental cells were selected for measurement of cell proliferation. A quantity of 5 × 103 cells were reseeded in 96-well plates. After incubation, the medium was removed and 20 μL 3-(4,5-dimethyl-thiazol-2yl)-2,5-diphenyltetrazolium bromide (5 mg/mL, Gibco) was added to each well. DMSO was added and the optical density of each well was read at 490 nm. Each experiment was performed in triplicate.
Colony formation assay
A quantity of 2 × 103 KYSE30 cells transfected with either SFRP2 expression vector or empty vector were cultured in 60-mm plates with G418 (Merck, Darmstadt, Germany) at 0.3 mg/mL for 4 wk in regular culture medium. Colonies with cell numbers of more than 50 cells per colony were counted. Colonies were fixed with methanol for 15 min and stained with Giemsa. All the experiments were performed in triplicate wells in three independent experiments.
In vivo tumorigenicity
Stable KYSE30 cells transfected with either the SFRP2 expression vector or empty vector cells and parental cells (1.5 × 106 cells in 0.2 mL PBS) were inoculated subcutaneously into the dorsal flank of 5-wk-old male Balb/c nude mice. Each group was comprised of 5 mice, and all were maintained under sterile conditions. Tumor formation was observed 2 wk later. Tumor diameter was measured every 3 d for 5 wk, and tumor volume (mm3) estimated by measuring the longest and shortest diameter of the tumor and calculating the volume as follows: volume = (shortest diameter)2 × (longest diameter) × 0.5. After 5 wk of observation, the mice were sacrificed. Tumors were excised, measured, and weighed. Care of animals and all experimental procedures were approved by the Animal Ethics Committee of Beijing Friendship Hospital, and were carried out in accordance the “Guideline for the Welfare of Animals in Experimental Neoplasia”[22].
Statistical analysis
All statistical analyses were conducted using SPSS 11.5 software (SPSS Inc, Chicago, IL, United States). Data were expressed as the mean ± SD. P values < 0.05 were considered statistically significant. Statistical comparison between two groups was performed using the non-parametric Mann-Whitney U-test or Student’s t-test. For comparison of more than three groups, we used one-way analysis of variance, followed by Tukey’s multiple comparison.
RESULTS
Frequent transcriptional silencing of SFRP2 in ESCC cell lines
We first examined mRNA expression of SFRP2 by means of reverse-transcription-PCR in seven ESCC cell lines (TE-1, KYSE-30, KYSE-150, KYSE-410, KYSE-510, EC-109 and EC-9706), and one immortalized normal esophageal epithelial cell line (Het-1A). RT-PCR showed that the SFRP2 transcript was silenced in all seven ESCC cell lines, but not in the immortalized normal esophageal epithelial cell line (Het-1A) (Figure 1A). We next examined the role of promoter methylation in the silencing of SFRP2. By methylation-specific PCR, complete methylation was detected in three cell lines with silenced SFRP2 expression, and extensive methylation was observed in the other four silenced cell lines (Figure 1B). We analyzed SFRP2 methylation in more detail using high-resolution bisulfite genomic sequencing analysis. Extensive methylation was detected in three ESCC cell lines (KYSE-30, KYSE-510 and TE-1) with silenced SFRP2 expression, whereas no methylated CpG site was detected in the immortalized normal esophageal epithelial cell lines (Het-1A) (Figure 2).
Figure 1.
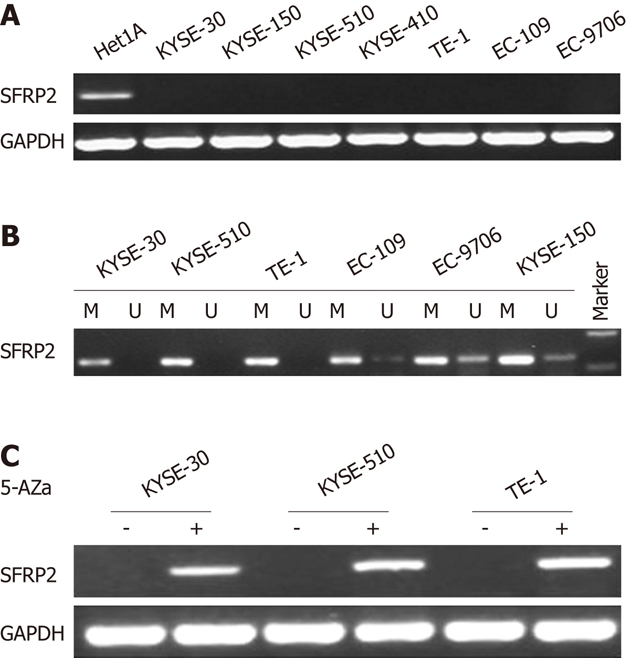
Expression and promoter methylation of secreted frizzled-related protein 2 in esophageal squamous cell carcinoma cell lines. A: Secreted frizzled-related protein 2 (SFRP2) mRNA expression as determined by reverse-transcription polymerase chain reaction (RT-PCR); B: The methylation status of the SFRP2 promoter as determined by methylation specific PCR; C: mRNA expression of SFRP2 (determined by RT-PCR) restored after treatment with the demethylation agent 5-Aza-dC in three esophageal squamous cell carcinoma cell lines. M: Methylated; U: Unmethylated.
Figure 2.

A representative picture of bisulfite genomic sequencing in the secreted frizzled-related protein 2 gene promoter. A-293bp region with 34 CpG site was analyzed. B
SFRP2 reactivation in ESCC cell lines treated with 5-Aaza-dC
Since we had established that the SFRP2 mRNA expression was related to its methylation status, we next investigated whether DNA demethylation could restore the expression of SFRP2 mRNA in the three ESCC cell lines lacking SFRP2 expression using 5-Aza-dC. 5-Aza-dC restored the expression of SFRP2 in all of the cell lines (Figure 1C), further supporting the role of promoter methylation as a primary mechanism of SFRP2 inactivation.
Expression and methylation status of the SFRP2 in primary ESCC tumors
We determined the mRNA expression level of SFRP2 in 20 pairs of primary human ESCC tumors and adjacent non-cancerous tissues using RT-PCR. When compared with adjacent non-cancerous tissues, SFRP2 was found to be clearly down-regulated in primary ESCC tumor specimens (1.51 ± 0.399 vs 0.939 ± 0.398, P < 0.001) (Figure 3). SFRP2 promoter methylation was observed in 95% (19/20) of the ESCC tissues and 65% (13/20) of the paired normal esophageal epithelial tissues, as measured by methylation-specific PCR. The positive rate of methylation in ESCC tissue was significantly higher compared with that in the normal control (P = 0.044). Notably, using high-resolution bisulfite genomic sequencing analysis, no SFRP2 methylation was seen in normal esophageal epithelium specimens (Figure 2).
Figure 3.

Expression of secreted frizzled-related protein 2 in esophageal squamous cell carcinoma and paired non-cancerous tissues. A: Reverse-transcription polymerase chain reaction (RT-PCR) analysis of secreted frizzled-related protein 2 (SFRP2) mRNA expression in 10 primary esophageal squamous cell carcinoma (ESCC) tissues and their adjacent non-cancerous tissues; B: The SFRP2 mRNA expression level normalized according to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA level. Data are expressed as mean ± SD. ESCC tissues vs adjacent non-tumor tissues (aP < 0.01).
SFRP2 inhibits tumor cell proliferation and induces apoptosis
A cell proliferation assay was performed on stably transfected KYSE30 cells with or without the SFRP2 expression, and on parental cells. Re-expression of SFRP2 in the transfected KYSE30/SFRP2 cells was confirmed by reverse-transcription PCR (Figure 4A). Cells stably expressing SFRP2 grew significantly slower than the empty vector-transfected cells and parental cells (P < 0.05, Figure 4B). We carried out apoptosis and cell cycle analysis to investigate whether SFRP2 over-expression affects these parameters in ESCC cells. We found a significantly higher number of early apoptotic cells in SFRP2-transfected cells than in the control cells (P = 0.025, Figure 5). The mean cell number in the S and G2-M phases of the cell cycle was also significantly lower in SFRP2-transfected cells, suggesting SFRP2-induced G1 arrest in KYSE30 cells (Table 2).
Figure 4.

Effect of secreted frizzled-related protein 2 ectopic expression on cell proliferation. A: Strong secreted frizzled-related protein 2 (SFRP2) mRNA expression in KYSE30 cells transfected with pcDNA3.1/SFRP2, but not in KYSE30 and cells transfected with the empty vector; B: SFRP2-transfected KYSE30 cells growing significantly slower than control and parental cells (aP < 0.05).
Figure 5.
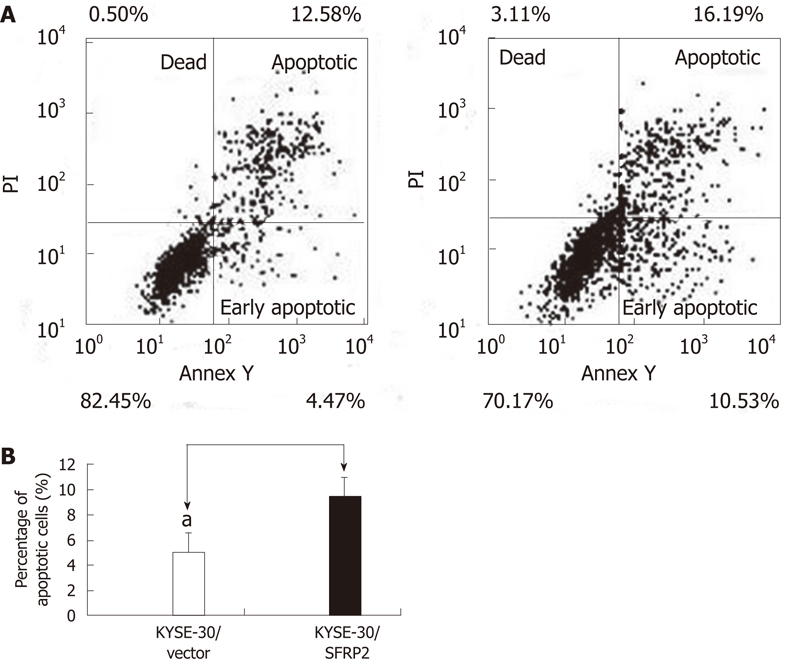
Ectopic expression of SFRP2 induces apoptosis in KYSE30 cells. A: The representative images of flow cytometry analysis; B: The data are the percentage of early apoptotic out of the total cell population of KYSE30 cells transfected with SFRP2 and cells with the empty vector.The early apoptotic cells in SFRP2-transfected cells were significantly higher than that in the control cells (aP < 0.05).
Table 2.
Effect of secreted frizzled-related protein 2 re-expression on the cell cycle distribution of KYSE30 cells (%)
| Groups | G0/G1 | S | G2/M |
| KYSE-30/vector | 45.76 ± 3.74 | 48.64 ± 4.90 | 5.59 ± 5.18 |
| KYSE-30/SFRP2 | 64.49 ± 6.11 | 30.52 ± 5.07 | 4.97 ± 6.60 |
SFRP2 suppresses colony formation of ESCC cell lines
Frequent silencing of SFRP2 by promoter methylation in ESCC cell lines, but not in the immortalized normal esophageal epithelial cell line, suggests a potential tumor-suppressor role of this gene. To test this speculation, we examined the effect of SFRP2 re-expression on growth characteristics of ESCC cells using colony formation assays. The colonies formed by SFRP2-transfected KYSE30 cells were significantly fewer in number (47.17% ± 15.61% vs 17% ± 3.6%, P = 0.031) and smaller in size than those in control cells (Figure 6), indicating that SFRP2 has a definite growth inhibitory effect.
Figure 6.

Colony formation assays. A: The representative dishes of transfection with pcDNA3.1V5HisA/secreted frizzled-related protein 2 (SFRP2) or empty vector (pcDNA3.1 V5HisA). Left: colonies formed by control cells. Right: colonies formed by cell strains with stably transfected SFRP2; B: Quantitative analyses of colony formation efficiency. Values are the mean ± SD of at least 3 independent experiments. The relative colony formation efficiency of SFRP2-transfected esophageal squamous cell carcinoma cells were lower than those of mock-transfected cells (aP < 0.05).
SFRP2 inhibits tumor growth in nude mice
After studying the effect of SFRP2 on cell proliferation in vitro, the involvement of SFRP2 in carcinogenesis in vivo was investigated. SFRP2-transfected KYSE30 cells, control cells, and parental cells were injected into nude mice. The tumor growth curve of stably transfected KYSE30/SFRP2 expression vector and KYSE30/vector control in nude mice is shown in Figure 7. The tumor size was significantly lower in SFRP2 transfected nude mice than in the vector control mice (917.86 ± 249.35 mm3 vs 337.23 ± 124.43 mm3, P < 0.05); however, no difference was found between the weight and volume of tumors from parental cells and those from the control, suggesting that SFRP2 functions as a tumor suppressor in esophageal carcinogenesis.
Figure 7.

Secreted frizzled-related protein 2 inhibits growth of tumors derived from KYSE-30 in vivo. A: A representative picture of nude mice: at week 5 nude mice injected with KYSE-30, KYSE-30/vector, KYSE30/SFRP2 and phosphate buffered saline; B: The tumor growth curves of nude mice. The growth of tumors derived from the SFRP2-transfected esophageal squamous cell carcinoma cells cells were significantly slower than those from the mock-transfected cells (aP < 0.05).
DISCUSSION
The SFRPs are a group of negative modulators of the Wnt signaling pathway, of which five members (SFRP1-5) have been identified to date. Using microarray analysis, we previously confirmed that the expression of SFRP2 was 3.6-fold lower in ESCC tissues than in paired noncancerous tissue samples, implying that down-regulation of SFRP2 may be involved in the carcinogenesis of ESCC. In this study, we extended our previous work on SFRP2 to ESCC and found that SFRP2 was silenced in seven ESCC cell lines, but not in an immortalized esophageal epithelial cell line. The silencing of expression was linked closely to promoter methylation, as confirmed by demethylation treatment and methylation analyses, suggesting that promoter methylation is the principal regulatory mechanism of SFRP2 inactivation in ESCC. There are unmethylated alleles in 4 ESCC cell lines with no expression of SFRP2 detected by reverse-transcription PCR, indicating the existence of other transcription regulatory mechanisms that repress SFRP2 expression, such as histone remodeling or transcriptional repressors.
Similar results were found in the experiments on primary ESCC tissues and adjacent non-tumor tissues. SFRP2 was clearly down-regulated in primary ESCC tumor tissues when compared to the paired non-cancerous tissues. MSP analysis of the SFRP2 promoter revealed a higher prevalence of CpG methylation in ESCC tissues than in adjacent non-tumorous tissues, suggesting that aberrant methylation of the SFRP2 promoter region is not a cell line-specific event and is frequent in ESCC carcinogenesis.
Functional inactivation of SFRP2 by promoter methylation, which resulted in tumor cell proliferation and invasion, has been confirmed in many human cancers[23-27], suggesting that SFRP2 functions as a tumor suppressor. In contrast to these results, others have found that SFRP2 reduced apoptosis and promotes tumor progression[19,21]. Yamamura et al[20] found that ectopic expression of SFRP2 in renal cancer cells reduced UV-induced apoptosis and increased the G2 phase of the cell cycle, significantly promoting the growth of xenografts in nude mice. However, there has been no detailed report thus far describing the functional significance of SFRP2 in ESCC. We therefore analyzed the functional role of SFRP2 in esophageal carcinogenesis by both in vitro and in vivo assays. Ectopic expression of SFRP2 in KYSE30 cells with no SFRP2 expression induced low cloning efficacy in colony formation assay, along with reduced tumor size in nude mice. FACScan analysis of SFRP2-re-expressed KYSE30 cells revealed a significant decrease in cell proliferation, and an increase in apoptotic cells. Taken together, these results confirmed the role of SFRP2 as a functional TSG through suppression of cell proliferation and induction of cell apoptosis in ESCC. Previous studies of other tumors, such as colorectal and gastric cancer, showed that SFRP2 functioned as tumor suppressor through attenuating the Wnt signaling pathway[10-12]. When it came to ESCC in the present study, although we confirmed that ectopic expression of SFRP2 significantly reduced the expression lever of c-myc and cyclinD1, which were downstream genes of the Wnt pathway (data not shown), further experiments such as immunofluorescence and luciferase reporter assay are necessary to reach a similar conclusion.
SFRP2 methylation was proven to be a highly promising predictive biomarker in many human cancers. Cheng et al[17] reported that methylated SFRP2 was detected in 66.7% of serum samples from cancer patients, but not in normal controls and the frequency of SFRP2 hypermethylation decreased from 73.3% in gastric cancer to 37.5% in intestinal metaplasia and 20% in adjacent non-cancer tissues, indicating that SFRP2methylation may have the potential for identifying individuals at risk of further histological progression. Muller et al[28] proposed SFRP2 hypermethylation as a sensitive marker able to detect 77%-90% of colorectal cancers (CRC); Huang et al[18] reported that methylated SFRP2 occurred in 94.2%, 52.4%, 37.5% and 16.7% of patients with CRC, adenoma, hyperplastic polyp, and ulcerative colitis, respectively. However, the clinical impact of SFRP2 hypermethylation in ESCC is unknown. Further studies of clinical correlation of SFRP2 methylation are required to answer this question.
In summary, we have presented the first convincing evidence that the epigenetic silencing of SFRP2 is a frequent event in human ESCC, and that SFRP2 is a potent TSG with key roles in suppressing cell proliferation and inducing apoptosis in the development of esophageal cancer. Further studies should be conducted to assess whether SFRP2 methylation might serve as a useful bio-marker for early detection and a potential target for treatment of esophageal carcinoma through reversal of SFRP2 inactivation by demethylating agents.
COMMENTS
Background
Esophageal squamous cell carcinoma (ESCC) is one of the most frequent malignant neoplasms in China. Although the exact mechanisms remain unclear, several studies have indicated that functional inactivation of tumor suppressor genes (TSG) through promoter methylation were involved in the pathogenesis of ESCC. Secreted frizzled-related protein 2 (SFRP2) promoter methylation has been documented in several human malignancies. We therefore analyzed the methylation and expression status, as well as the function of this gene in ESCC.
Research frontiers
Aberrant promoter methylation of SFRP2 gene has been documented in several human malignancies, such as colorectal cancer, gastric cancer, and breast cancer. This study investigated the role of SFRP2 in ESCC and showed for the first time that the hypermethylation of SFRP2 gene promoter was the predominant mechanism for SFRP2 gene silence in esophageal cancer.
Innovations and breakthroughs
The authors present the first convincing evidence that the epigenetic inactivation of SFRP2 is a frequent event in human ESCC, and that SFRP2 is a potent TSG with key roles in suppressing cell proliferation and inducing apoptosis in the development of ESCC.
Applications
SFRP2 methylation might serve as a useful bio-marker for early detection and a potential target for treatment of esophageal carcinoma.
Terminology
DNA methylation is an epigenetic modification that is important for normal development in higher organisms. Aberrant changes in methylation patterns play an important role in carcinogenesis, cancer progression and chemosensitivity.
Peer review
This is a study looking for the significance of epigenetic inactivation of SFRP2 in the development of ESCC. The data suggest that SFRP2 methylation might serve as a potential target for treatment of ESCC in the future. Overall ,this study is well designed and the data are convicing.
Footnotes
Supported by National Natural Science Foundation of China, No. 81050016; Research Fund for the Doctoral Program of Higher Education of China, No. 200800250003
Peer reviewers: Piero Marco Fisichella, MD, Assistant Professor of Surgery, Medical Director, Swallowing Center, Loyola University Medical Center, Department of Surgery, Stritch School of Medicine, 2160 South First Avenue, Room 3226, Maywood, IL 60153, United States; Liang-Shun Wang, MD, Professor, Vice-superintendent, Shuang-Ho Hospital, Taipei Medical University, No. 291, Jhongjheng Rd., Jhonghe City, New Taipei City 237, Taiwan, China
S- Editor Lv S L- Editor Rutherford A E- Editor Xiong L
References
- 1.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 2.Tamoto E, Tada M, Murakawa K, Takada M, Shindo G, Teramoto K, Matsunaga A, Komuro K, Kanai M, Kawakami A, et al. Gene-expression profile changes correlated with tumor progression and lymph node metastasis in esophageal cancer. Clin Cancer Res. 2004;10:3629–3638. doi: 10.1158/1078-0432.CCR-04-0048. [DOI] [PubMed] [Google Scholar]
- 3.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 4.Feng Q, Balasubramanian A, Hawes SE, Toure P, Sow PS, Dem A, Dembele B, Critchlow CW, Xi L, Lu H, et al. Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst. 2005;97:273–282. doi: 10.1093/jnci/dji041. [DOI] [PubMed] [Google Scholar]
- 5.Yu J, Cheng YY, Tao Q, Cheung KF, Lam CN, Geng H, Tian LW, Wong YP, Tong JH, Ying JM, et al. Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology. 2009;136:640–51.e1. doi: 10.1053/j.gastro.2008.10.050. [DOI] [PubMed] [Google Scholar]
- 6.Ramírez N, Bandrés E, Navarro A, Pons A, Jansa S, Moreno I, Martínez-Rodenas F, Zárate R, Bitarte N, Monzó M, et al. Epigenetic events in normal colonic mucosa surrounding colorectal cancer lesions. Eur J Cancer. 2008;44:2689–2695. doi: 10.1016/j.ejca.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Palomaki GE, Knight GJ, Holman MS, Haddow JE. Maternal serum alpha-fetoprotein screening for fetal Down syndrome in the United States: results of a survey. Am J Obstet Gynecol. 1990;162:317–321. doi: 10.1016/0002-9378(90)90377-j. [DOI] [PubMed] [Google Scholar]
- 8.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 9.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, Pretlow TP, Yang B, Akiyama Y, Van Engeland M, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 11.Nojima M, Suzuki H, Toyota M, Watanabe Y, Maruyama R, Sasaki S, Sasaki Y, Mita H, Nishikawa N, Yamaguchi K, et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene. 2007;26:4699–4713. doi: 10.1038/sj.onc.1210259. [DOI] [PubMed] [Google Scholar]
- 12.Takagi H, Sasaki S, Suzuki H, Toyota M, Maruyama R, Nojima M, Yamamoto H, Omata M, Tokino T, Imai K, et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J Gastroenterol. 2008;43:378–389. doi: 10.1007/s00535-008-2170-0. [DOI] [PubMed] [Google Scholar]
- 13.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 14.Rattner A, Hsieh JC, Smallwood PM, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc Natl Acad Sci USA. 1997;94:2859–2863. doi: 10.1073/pnas.94.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolligs FT, Bommer G, Göke B. Wnt/beta-catenin/tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion. 2002;66:131–144. doi: 10.1159/000066755. [DOI] [PubMed] [Google Scholar]
- 16.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 17.Cheng YY, Yu J, Wong YP, Man EP, To KF, Jin VX, Li J, Tao Q, Sung JJ, Chan FK, et al. Frequent epigenetic inactivation of secreted frizzled-related protein 2 (SFRP2) by promoter methylation in human gastric cancer. Br J Cancer. 2007;97:895–901. doi: 10.1038/sj.bjc.6603968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Z, Li L, Wang J. Hypermethylation of SFRP2 as a potential marker for stool-based detection of colorectal cancer and precancerous lesions. Dig Dis Sci. 2007;52:2287–2291. doi: 10.1007/s10620-007-9755-y. [DOI] [PubMed] [Google Scholar]
- 19.Roth W, Wild-Bode C, Platten M, Grimmel C, Melkonyan HS, Dichgans J, Weller M. Secreted Frizzled-related proteins inhibit motility and promote growth of human malignant glioma cells. Oncogene. 2000;19:4210–4220. doi: 10.1038/sj.onc.1203783. [DOI] [PubMed] [Google Scholar]
- 20.Yamamura S, Kawakami K, Hirata H, Ueno K, Saini S, Majid S, Dahiya R. Oncogenic functions of secreted Frizzled-related protein 2 in human renal cancer. Mol Cancer Ther. 2010;9:1680–1687. doi: 10.1158/1535-7163.MCT-10-0012. [DOI] [PubMed] [Google Scholar]
- 21.Lee JL, Chang CJ, Chueh LL, Lin CT. Secreted frizzled related protein 2 (sFRP2) decreases susceptibility to UV-induced apoptosis in primary culture of canine mammary gland tumors by NF-kappaB activation or JNK suppression. Breast Cancer Res Treat. 2006;100:49–58. doi: 10.1007/s10549-006-9233-9. [DOI] [PubMed] [Google Scholar]
- 22.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang DR, Tang D. Hypermethylated SFRP2 gene in fecal DNA is a high potential biomarker for colorectal cancer noninvasive screening. World J Gastroenterol. 2008;14:524–531. doi: 10.3748/wjg.14.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sogabe Y, Suzuki H, Toyota M, Ogi K, Imai T, Nojima M, Sasaki Y, Hiratsuka H, Tokino T. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int J Oncol. 2008;32:1253–1261. doi: 10.3892/ijo_32_6_1253. [DOI] [PubMed] [Google Scholar]
- 25.Chung MT, Lai HC, Sytwu HK, Yan MD, Shih YL, Chang CC, Yu MH, Liu HS, Chu DW, Lin YW. SFRP1 and SFRP2 suppress the transformation and invasion abilities of cervical cancer cells through Wnt signal pathway. Gynecol Oncol. 2009;112:646–653. doi: 10.1016/j.ygyno.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 26.Veeck J, Noetzel E, Bektas N, Jost E, Hartmann A, Knüchel R, Dahl E. Promoter hypermethylation of the SFRP2 gene is a high-frequent alteration and tumor-specific epigenetic marker in human breast cancer. Mol Cancer. 2008;7:83. doi: 10.1186/1476-4598-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki H, Toyota M, Carraway H, Gabrielson E, Ohmura T, Fujikane T, Nishikawa N, Sogabe Y, Nojima M, Sonoda T, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008;98:1147–1156. doi: 10.1038/sj.bjc.6604259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller HM, Oberwalder M, Fiegl H, Morandell M, Goebel G, Zitt M, Mühlthaler M, Ofner D, Margreiter R, Widschwendter M. Methylation changes in faecal DNA: a marker for colorectal cancer screening? Lancet. 2004;363:1283–1285. doi: 10.1016/S0140-6736(04)16002-9. [DOI] [PubMed] [Google Scholar]