

Abstract
Myotonic dystrophy type-1 (DM1), the most prevalent form of adult muscular dystrophy, is caused by expansion of a CTG repeat in the 3′ untranslated region of the DM protein kinase (DMPK) gene. The pathogenic effects of the CTG expansion arise from the deleterious effects of the mutant transcript. RNA with expanded CUG tracts alters the activities of several RNA binding proteins, including muscleblind-like 1 (MBNL1). MBNL1 becomes sequestered in nuclear foci in complex with the expanded CUG repeat RNA. The resulting loss of MBNL1 activity causes mis-regulated alternative splicing of multiple genes, leading to symptoms of DM1. The binding interaction between MBNL1 and mutant RNA could be a key step in the pathogenesis of DM1 and serves as a potential target for therapeutic intervention. We have developed two high throughput screen (HTS) suitable assays using both homogenous time-resolved fluorescence energy transfer (HTRF) and AlphaScreen technologies to detect the binding of a C-terminally His-tagged MBNL1 and a biotinylated (CUG)12 RNA. These assays are homogenous and successfully miniaturized to 1536-well plate format. Both assays were validated and show robust signal-to-basal ratios and Z’ factors.
Keywords: Myotonic dystrophy type 1, DM1, Muscleblind-like 1, MBNL1
Introduction
Myotonic dystrophy type 1 (DM1), an autosomal dominant neuromuscular disorder affecting 1/8000 individuals, is the most common form of muscular dystrophy in adults. The disease has a wide range of multi-systemic clinical presentations, which includes myotonia, cardiac conduction defects, progressive muscle wasting, endocrine imbalance and cataracts, causing progressive disability and premature death [1]. The genetic lesion that causes DM1 is an expansion of CTG repeats in the 3′ untranslated region (UTR) of the DM protein kinase (DMPK) gene. The size of the triplet repeat expansion ranges from hundreds to thousands of repeats and correlates with disease severity [2,3]. DM represents a novel disease paradigm where the mutant RNA exerts a dominant deleterious effect that is independent of the protein it encodes. The pathogenic expanded CUG repeat (CUGexp) RNA accumulates in nuclear foci and sequesters proteins of the muscleblind-like (MBNL) family, of which MBNL1 is the most common family member in skeletal muscle [4,5]. MBNL proteins regulate alternative splicing, and their sequestration alters the splicing patterns of a myriad of transcripts. Of the affected transcripts, cardiac troponin T (cTNT), sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA1), insulin receptor (IR) and muscle-specific chloride channel (ClC-1) are the most well studied [6-9].
The binding interaction between MBNL proteins and CUG repeat RNA is a key molecular step in the pathogenesis of DM1. Murine knockouts of MBNL1 recapitulate certain symptoms and splicing patterns of the disease [10], while over-expression of MBNL1 in a DM1 mouse model that expresses a (CUG)250 repeat transcript reverses myotonia and RNA mis-splicing [11]. In addition, proof of principle that blocking the CUGexp/MBNL1 interaction in vivo could be beneficial for DM was demonstrated by the use of morpholino oligo to disrupt MBNL1 CUGexp binding, resulting in the correction of misregulated splicing [12].
Currently, there is no treatment to halt disease progression and therapy for DM1 is limited to supportive care. Recent drug development efforts have aimed at finding ligands that bind the expanded CUG RNA using combinatorial chemistry and peptoid synthesis, as well as a targeted small molecule screen. Several compounds that are able to inhibit the interaction between expanded CUG RNA and MBNL1 protein have been found [13-17]. Of these, only pentamidine has shown some in vivo activity but the therapeutic window is narrow due to its cytotoxicity [18]. Continued development of small molecule inhibitors to disrupt the binding interaction between MBNL1 and CUG expansion remains an attractive approach for the treatment of DM1.
Protein-RNA interactions control many aspects of RNA splicing, translation and decay. Compounds that modulate these interactions could become useful research tools as well as potential therapeutics. Traditional methods for detection of protein-RNA binding include gel mobility shift, filter-binding and yeast-three-hybrid (Y3H) assays [19,20,15,21-24]. While these assays are useful in low throughput applications, they lack adaptability to high throughput screening (HTS) since they require either an electrophoresis apparatus or multiple wash steps. In addition, yeast-three-hybrid assay has added disadvantages due to the indirect nature of the assay. Two types of homogeneous assays, scintillation proximity and AlphaScreen technology, have been used to assay the bindings of HIV RNAs and host proteins in a high throughput manner [24,25]. A down side of scintillation proximity assays is the requirement of radio-labeled reagents that generate large amounts of radioactive waste. In addition, the advantages of a homogenous assay format are the simplification of assay workflow and reduction of assay noise due to plate wash steps. However, elimination of wash steps may lead to increased number of false positives. With sizes of many compound libraries currently expanding to over a million, there is increasing need of additional HTS-ready assays for orthogonal screening to eliminate detection related artifacts and allow fast hit triaging. Thus, the strategy of using one homogenous protein-RNA binding assay for HTS and the other as an orthogonal screen would facilitate hit prioritization efforts by rapidly eliminating detection-based false positives.
We have developed and compared two homogeneous assays to measure the binding of MBNL1 to CUG repeat RNA. One uses the AlphaScreen technology and the other uses homogeneous time-resolved fluorescence energy transfer (HTRF). Since these two assays apply different detection methods, they can be used as an orthogonal pair to eliminate false positive compounds. Both assays were optimized, miniaturized to 1536-well format and validated using a DMSO control plate. Our results indicate that while both assays are suitable for HTS, the HTRF assay is preferred for increased throughput and easier implementation, as no light shielding is needed. The HTRF assay was further validated in a pilot screen of 1280 pharmacologically active compounds (LOPAC1280, Sigma-Aldrich) and showed performance and hit rate that are acceptable for HTS. The hits were confirmed with the HTRF assay and assay artifacts were eliminated with the AlphaScreen assay as an orthogonal screen. The results indicate that the two assays can be paired for HTS and orthogonal screening use.
Materials and methods
Reagents
5′-Biotinylated RNA oligo Biot-(CUG)12 with the sequence 5′-GCGCUGCUGCUGCUGCUGCUGCUGCUGCUGCUGCUGCUGCGC was purchased from IDT (Skokie, IL). (CUG)12 was designed to maintain a uniform hairpin structure after denaturation/renaturation, by adding a three nucleotides at both ends which form the GC clamp at the base of hairpin [26]. The unlabeled mixed RNA/DNA oligo ssCAG-21 with the sequence 5′-rUrArGrCrArGrCrArGrCrArGrCrArGrCrArGrCTT (DNA sequence is underlined) was custom synthesized by IDT (Skokie, IL). The HTRF reagents, anti-His6-Terbium cryptate (anti-His-Tb) and streptavidin-conjugated XL665 (SA-XL665), were purchased from CisBio (Bedford, MA). The AlphaScreen reagents, nickel-coated acceptor beads and streptavidin-conjugated donor beads, were purchased from PerkinElmer (Waltham, MA). The library of pharmacologically active compounds (LOPAC1280) consists of a collection of small molecules with characterized biological activities and was purchased from Sigma-Aldrich (St. Louis, MO). More information on LOPAC1280 can be found at http://www.sigmaaldrich.com/chemistry/drug-discovery/validation-libraries/lopac1280-navigator.html).
Recombinant protein purification
BL21(DE3)pLysS E. coli (Invitrogen) were transformed with the pGEX-6P-MBNL1-δ105-His plasmid, which expresses recombinant MBNL1-His6 protein with a 105 amino acid C-terminal deletion, an N-terminal GST tag and a C-terminal 6x His tag. 500 ml LB broth cultures with 200 g/ml carbenicillin were grown at 37 °C to OD600 = 1.7-2.0, then induced with 250 M IPTG overnight at 25°C. Cells were pelleted (all centrifugation was performed at 4 °C), resuspended in 25 ml 1x His binding buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM imidazole and 0.1% IGEPAL CA-630) and incubated on ice for 30 min. Cell lysis was achieved with alternating rounds of sonication and icing. After clearing of cell debris via centrifugation, the supernatant, along with 1 mM PMSF, was added to 5 ml of Ni-agarose resin, prepared as 10 ml Ni-NTA Agarose slurry (Invitrogen) equilibrated with 1x His binding buffer. The lysate/bead mixture was incubated for 2 hrs at room temperature with gentle agitation. The supernatant was then discarded, and the resin was washed 3 times in 25 ml 1x His binding buffer with increasing imidazole concentrations of 5 mM, 50 mM and 100 mM. The bound MBNL1-His6 protein was eluted in two rounds by vigorous shaking of the Ni-agarose resin in 3 ml 1x His binding buffer with 250 mM imidazole. The eluates were added to PD-10 Desalting Columns (GE Healthcare) that were equilibrated with 1x FBB buffer (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 50 mM KCl and 1 mM MgCl2). MBNL1 protein was eluted from the columns in 2 rounds with 2 ml 1x FBB buffer each. Glycerol was added to 50% in each sample, and total protein concentrations were determined with the Bio-Rad Protein Assay. Each sample was resolved on a 10% SDS-PAGE Tris-glycine gel, stained by SYPRO Ruby (Invitrogen) and scanned with a GE Healthcare Typhoon 9400 Variable Mode Imager. MBNL1-His6 and contaminating protein bands were quantitated with ImageQuant software, and MBNL1-His6 purity was calculated as the ratio of the MBNL1 band versus the sum of all protein bands (typically ~0.60-0.75). The MBNL1-His6 concentration for each sample was then calculated by multiplying this ratio by the total protein concentration.
Binding assay in 384-well plate format
Assay development and optimization was performed in white solid-bottom 384-well plates (Greiner Bio-One, Monroe, NC). For HTRF binding assays, reagents were sequentially added manually as 5 μl Biot-(CUG)12, 5μl MBNL1-His6, 5μl anti-His-Tb and 5μl SA-XL665. For AlphaScreen binding assays, reagents were sequentially added manually as 5μl Biot-(CUG)12, 5μl MBNL1-His6, 5μl acceptor bead and 5μl donor bead. All reagents were diluted in binding buffer composed of 25 mM HEPES pH 7.4, 110 mM KCL, 10 mM NaCl, 1 mM MgCl2, 15μM ZnCl2, 0.02% Tween-20, 0.1% BSA and 5 mM freshly added DTT. After 60 minute incubation at room temperature, the fluorescence (HTRF assay) or luminescence (AlphaScreen assay) was measured using an EnVision plate reader (PerkinElmer, Boston, MA). All results were generated from duplicate data points.
Binding assays in 1536-well plate format
The binding assay was miniaturized to the 1536-well plate format for HTS. Briefly, 2μl/well of a premixed protein-RNA solution (20 nM Biot-(CUG)12 and 20 nM MBNL1-His6) was dispensed to a white solid-bottom 1536-well plate (Greiner Bio-One, Monroe, NC), followed by the addition of 23 nl/well compound in DMSO solution. After a 15 minute incubation period at room temperature, the detection reagents were simultaneously added as 1λl/well each (0.22 ng/μl anti-His-Tb and 20 nM SA-XL665 for the HTRF assay or 25 ng/μl acceptor bead and 25 ng/μl donor beads for the AlphaScreen assay). The binding reactions were allowed to equilibrate for 60 minutes. The assay signal was then measured on an EnVision plate reader (PerkinElmer, Boston, MA). Dose-response of ssCAG-21 and confirmation of LOPAC1280 screen hits were tested in duplicates.
Instruments used
A BioRAPTR FRDTM Microfluidic Workstation (Beckman Coulter, Inc. Fullerton, CA) was used to dispense reagents into 1536-well plates at volumes of 1-2μl. For experiments using untagged oligos as binding inhibitors, the oligos were serially diluted in 384-well plates and then reformatted into 1536-well plates as 7μl/well using a CyBi-Well dispensing station with a 384-well head (Cybio Inc., Woburn, MA). For the DMSO plate tests and LOPAC1280 screen, 23 nl of liquids were transferred to 1536-well assay plates using an automated pin-tool station (Kalypsys, San Diego, CA). An EnVision filter-based imaging plate reader (PerkinElmer, Boston, MA) was used for both the HTRF and AlphaScreen assays. For the two channel HTRF detection, an excitation of 340 nm, emission of 665 nm and 545 nm, count delay of 70μs and count window of 400μs was used. AlphaScreen assays were read using the AlphaScreen setting on the EnVision plate reader with either the 1536-well or 384-well aperture.
Filter binding assay
The RNAs used [(CUG)20 and (CAG)20] were chemically synthesized at Metabion International AG (Jena, Germany). Structures formed by these transcripts have been described earlier [27]. All transcripts were 5′-end-labeled with T4 polynucleotide kinase and [γ32P]ATP (4500 Ci/mmol; ICN). The labeled RNAs were purified by electrophoresis in a denaturing 10% polyacrylamide gel, excised, eluted from the gel (0.3 M sodium acetate pH 5.2, 0.5 mM EDTA and 0.1% SDS) and precipitated. Prior to use, the 32P-labeled RNAs were subjected to a denaturation and renaturation procedure in 2 x assay buffer containing 200 mM Tris-HCl (pH 8.0), 100 mM NaCl, 100 mM KCl, 2 mM MgCl2, by heating the sample at 85°C for 1 min. and then slowly cooling to 37°C. RNA degradation assay was initiated by mixing 5 l of the RNA sample (200 pM) with 5 l of a compound solution. Incubation was performed at 37°C for 30 min. and stopped by adding an equal volume of stop solution (7.5 M urea and 20 mM EDTA with dyes). Samples were analyzed on a denaturing 12% polyacrylamide gel electrophoresis.
Filter binding assay was performed as described earlier [28]. Briefly, 15μl of 5′-labeled RNA (200 pM) after denaturation-renaturation procedure (described above) was mixed with 15μl of aqueous solution of different concentrations of compound and then 30 μl of 40 nM MBNL1 solution in 1 x assay buffer was added. After 30 minutes of incubation at 37°C samples were loaded on filter sandwich (nitrocellulose [BioRad], Hybond N+ [Amerscham], three sheets of 3 mm Whatman paper) mounted on slot-blot apparatus. To determine a kinetic of RNA/MBNL1 interaction similar procedure was used with exception of compound adding.
Data analysis
The quantitative data were calculated as mean ± standard error. The results for the assay optimization and inhibitor dose response experiments were analyzed with Prism (Graphpad, San Diego, CA). For assay validation, signal-to-basal (S/B) ratios, Z’ factors and coefficient of variation (CV) values were calculated from 64 wells of DMSO only treatments (total signal) and 32 wells of binding reaction lacking MBNL-His6 (basal signal). For LOPAC1280 hit confirmation, percent binding is calculated from 48 wells of DMSO only treatments (normalized as 100% binding) and 24 wells of binding reaction without MBNL-His6 (normalized as 0% binding). The Kd and IC50 values were calculated based on three independent experiments using the one-site binding model or dose-response inhibition curve, respectively, with use of GraphPad Prism 5.00 (GraphPad Software Inc.).
Results and Discussion
Assay designs and principles
We intended to design a strategy for high throughput screening to identify small compounds which prevent or disrupt the pathogenic interaction of MBNL1 splicing factor with RNA hairpin structure formed by CUG repeats. To this end, we have developed the two proximity-based assays to measure the binding of MBNL1-His6 protein to (CUG)12 RNA. The MBNL1 protein preparation used here retains all four zinc finger domains responsible for interaction with target RNAs, though it was C-terminally truncated by 105 amino acids (MBNL1δ105) in order to increase protein stability in both bacteria and solution. Comparison of MBNL1δ105 and MBNL1-FL (full length protein) showed no differences in affinity to a (CUG)109 transcript (Fig. 1). A hexa-histidine tag was attached to the C-terminus of MBNL1δ105 (MBNL1-His6) and was used for protein purification and as the capture point in the HTS binding assays. A 12-repeat (CUG)12 RNA bearing “clamp” sequences of GCG and CGC at the 5′ and 3′ ends, respectively, was modified at its 5′ end with biotin using a 6 carbon linker. Previous work has shown high-affinity MBNL1 binding to CUG-repeat sequences as short as four units provided that the hairpin structure of the RNA is stabilized by a tetraloop cap [19]. The “clamp” sequences stabilized the RNA hairpin structure and the biotin tag served to capture the streptavidin conjugated detection reagents in both assays. In preliminary experiments, RNA with the clamp showed higher optimal signal than the RNA without GC clamp (data not shown). This could either be due to increased affinity of MBNL1 for hairpin RNA structures or the stabilization of distance between the detection reagents to promote more efficient energy transfer reactions, because un-clamped triplet repeats is known to form several alternative hairpin structures [24].
Figure 1. RNA binding activity of MBNL1 constructs.
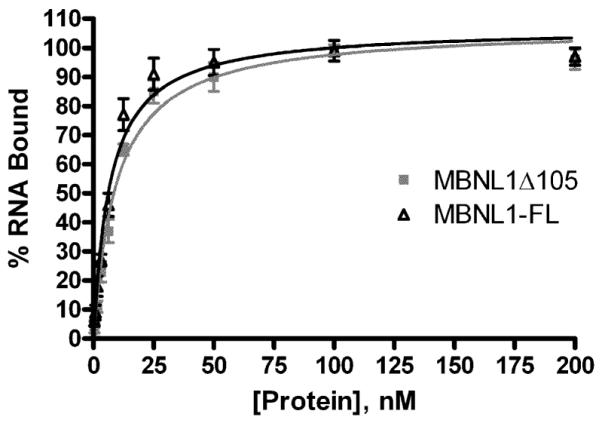
Filter binding assay was used to compare (CUG)109 binding affinity of full-length (MBNL1-FL) and truncated recombinant MBNL1 (MBNL1 105). Experimentally calculated KD was 9.63 ± 1.02 for MBNL1 105 and 6.92 ± 0.78 for MBNL1-FL.
An HTRF assay was used to quantify binding of MBNL1 protein with CUGexp RNA. This method utilizes terbium, a lanthanide cryptate, as the FRET donor, and XL665 as the FRET acceptor. MBNL1-His6 is captured by a terbium-conjugated anti-His6 antibody (Anti-His-Tb), while Biot-(CUG)12 is captured by a streptavidin-conjugated XL-665 (SA-XL665). Binding of (CUG)12 RNA to MBNL1 brings terbium and XL-665 within close proximity to enable time resolved FRET detection in a homogenous assay format (Fig. 2a).
Figure 2. Schematic of assay principles.

(a) In the HTRF assay, the MBNL1-His6 and Biot-(CUG)12 complex is captured by the HTRF reagents anti-His-Terbium and streptavidin-XL665. The formation of protein-RNA complex brings terbium cryptate within close proximity to XL665 to allow time-resolved fluorescence energy transfer. (b) In the AlphaScreen assay, streptavidin-conjugated donor beads are brought into close proximity to nickel-coated acceptor beads upon capture by the protein-RNA complex. Excitation with 680nm light induces release from donor bead of singlet oxygen that is capable of energy transfer with thioxene derivatives in the acceptor bead, culminating in 520-620nm photoemission by acceptor bead.
The second assay was developed with the same binding pair, MBNL1-His6 and Biot-(CUG)12, using the AlphaScreen assay format. This assay is a bead based assay using phthalocyanine (a photosensitizer) coated donor beads that convert ambient oxygen to an excited singlet oxygen upon excitation. The singlet oxygen has a 4 msec half-life, which allows a diffusion radius of approximately 200 nm in aqueous environment. Energy in the singlet oxygen form is transferred to an acceptor bead containing thioxene derivatives culminating in light production if the acceptor bead is within close proximity to the donor bead. Capture of the MBNL1-His6 and Biot-(CUG)12 complex by the nickel-charged acceptor beads and streptavidin-conjugated donor beads brings the AlphaScreen detection beads to close proximity that allows energy transfer in a homogeneous detection format (Fig. 2b). Both assays have the advantage of being homogenous and can be easily implemented in a “mix and read” format. The detection mechanisms in these two assays are different as the HTRF assay is based on the time resolved fluorescence while AlphaScreen uses chemiluminance for detection. Thus, these two assays can be used as orthogonal screens to eliminate detection related false positive compounds.
Assay optimization
The concentrations of both MBNL1 protein and (CUG)12 RNA were titrated to optimize the signal-to-basal ratio (S/B) for 384-well format assays. The basal fluorescence was defined by the same assay mixture except the MBNL1 protein. At first, the concentration of Biot-(CUG)12 was kept constant at 20 nM while MBNL1-His6 was titrated from 5 nM to 80 nM in both HTRF and AlphaScreen assay formats. The concentrations of detection reagents were kept constant at 0.11 ng/μl anti-His-Tb and 10 nM SA-XL665 for the HTRF assay, and 12.5 ng/μl acceptor bead and 12.5 ng/μl donor beads for the AlphaScreen assay. The results from both assays for the MBNL1 protein titration were almost identical, and 20 nM MBNL1-His6 showed the best S/B ratio of 6-fold in the HTRF assay and 29-fold in the AlphaScreen assay (Fig. 3a). For subsequent experiments, the amount of MBNL1-His6 to Biot-(CUG)12 was kept at the optimal ratio of 1:1 that uses 20 nM concentration for each of the binding partners.
Figure 3. Assay optimization.

(a) MBNL1-His6 was titrated against 20 nM final concentration of Biot-(CUG)12 in both HTRF and AlphaScreen assays performed in 384-well format. The optimal ratio of MBNL1 protein and (CUG)12 ligand was 1:1 for both assays. (b) Detection reagents were titrated against 20 nM each of MBNL1-His6 and Biot-(CUG)12 for the HTRF and AlphaScreen assays. (c) The dose response curves for ssCAG-21, a competitive inhibitor of MBNL1-His6/Biot-(CUG)12 binding, was determined using the HTRF and AlphaScreen assays. RLU = relative luminescence unit; S/B = signal to background ratio.
The amount of detection reagents were titrated against a premixed solution containing 20 nM each of MBNL1-His6 and Biot-(CUG)12. For the HTRF assay, a titration of detection reagents carried out using the mixtures of anti-His-Tb (0.055 to 0.44 ng/μl) and the SA-XL665 (2.5 to 20 nM). The results showed that the optimal signal-to-basal ratio for the binding of 20 nM MBNL1-His6 to 20 nM Biot-(CUG)12 complex was achieved with 10 nM SA-XL665 to 0.11 or 0.22 ng/μl anti-His-Tb (Fig. 3b). Since 0.11 ng/μl and 0.22 ng/μl anti-His-Tb yielded almost identical S/B ratio, 0.11 ng/μl anti-His-Tb and 10 nM SA-XL665 was used for further experiments.
For the AlphaScreen assay, the manufacturers recommended using a 1:1 ratio of donor to acceptor beads. Therefore, the two detection reagents were titrated against premixed 20 nM MBNL1-His/Biot-(CUG)12 at a 1:1 ratio. Optimal S/B ratio was achieved at 12.5 ng/μl of each type of bead (Fig. 3b). This concentration was selected for further experiments.
Unlabeled oligonucleotide ssCAG-21 was used as a positive control to test for its ability to competitively inhibit binding of Biot-(CUG)12 to MBNL1 in both the HTRF and AlphaScreen assays (Fig. 3c). This oligonucleotide was shown previously as highly potent competitor of CUGexp/MBNL1 interaction using two different methodological platforms: the agarose gel electrophoretic mobility shift assay and β-galactosidase enzyme complementation assay [15,17]. The dose dependent inhibitory effects of ssCAG-21 on MBNL1-(CUG)12 binding in both HTS assays showed comparable and low IC50 values, indicating that both assays possessed similar detection sensitivity.
Assay miniaturization and validation
The optimized 384-well plate assay was miniaturized to the 1536-well plate format by proportionally reducing the assay volume from 20μl/well to 4 l/well without significantly affecting the assay window. The concentration-response of ssCAG-21 oligo was measured in both 1536-well assay formats. The IC50 values of the oligo were found to be comparable to those obtained from the 384-well assays (Fig. 4a). Thus, both assays were successfully miniaturized to the 1536-well plate format for HTS.
Figure 4. Assay miniaturization and validation.

(a) The assays were miniaturized to 4 ul final volume in 1536-well format. The dose response for inhibition by ssCAG-21 was similar to that of 384-well format. (b) Scatter plot representations of the DMSO plate test results for the HTRF and AlphaScreen assays. The treatments were used: column 1 was treated with titrations of ssCAG-21 (from 125 to 0.004 nM), column 2-3 and 5-48 with DMSO (total signal) and column 4 with DMSO and lacked MBNL1-His6 (basal signal).
To further validate these two binding assays, we carried out a test with a plate containing DMSO, a solvent for the compound libraries. For the HTRF assay, the S/B ratio was 5.35 fold, CV was 2.4% and Z’ factor was 0.89 as determined from the DMSO plate test (Fig. 4b). Similarly, for the AlphaScreen assay, the S/B ratio was 55.5, CV was 6.7% and Z’ factor was 0.75 (Fig. 4b). These results demonstrate that both assays are robust and suitable for compound screens in the 1536-well plate format.
Although both the AlphaScreen and HTRF assay gave statistical calculations that are suitable for HTS, the HTRF assay produced cleaner results (CV 2.4%) versus the AlphaScreen assay (CV 6.7%) in the DMSO plate test. The ratiometric calculation of the results from HTRF assay may contribute to the lower well-to-well variations. Additional advantages of the HTRF assay are that: (1) the HTRF assay lacks light sensitivity, which facilitates reagent additions and time course studies during assay development. (2) The detection speed of HTRF assay is much faster as it can be read on charge-coupled device (CCD) based microplate imager while AlphaScreen signal requires a laser-based excitation that is read by a photomultiplier tube (PMT). The ability of the CCD imager to collect signal from the entire plate at once allows for shorter read times and higher assay throughput for screens using the HTRF assay. For these reasons, the HTRF assay was chosen for validation screening against a small set of compounds.
LOPAC1280 library screening and hit confirmation
The HTRF assay was used for screening a library of 1280 pharmacologically active compounds (LOPAC1280, Sigma) using quantitative HTS (qHTS) format [29]. Each compound was screened at 5 concentration points between 92 nM - 57.5 μM to allow dose response curve fitting from primary screen data. Results from the screen showed 7 compounds with inhibitory activity in the 665 nm channel while having no effect on donor signal at 545 nm. Of these 7 actives, one was a biotin analog based on structure and was not selected for follow-up. The six remaining compounds were re-plated as 12-point 1:3 intra-plate titrations (covering a concentration range of 0.3 nM - 57.5 μ M) for confirmation with the HTRF assay and the orthogonal AlphaScreen assay to eliminate hits that interfere with the HTRF labels. Upon retesting, 5 out of 6 compounds were confirmed in the HTRF assay, and of these, 2 out of 5 were confirmed in the AlphaScreen assay, with <2-fold shift in IC50 values (Table 1). The 3 compounds that were inactive in the AlphaScreen assay may affect the detection system in the HTRF assay rather than block the interaction between MBNL1 protein and (CUG)12 RNA. Another possibility for the low confirmation rate is the low potency of the hits. Because all of the hits had IC50 >10 μM, the calculation of dose-response curves was only based on one asymptote, which made calculations of IC50 less accurate.
Table 1. Summary of LOPAC1280 library screening hits.
| Sample ID | Sample Name | IC50 (μM) | ||
|---|---|---|---|---|
| 1′ Screen1 | HTRF2 | AlphaScreen | ||
| NCGC00094139-01 | nor-Binaltorphimine dihydrochloride | 31.6 | 21.9 | 12.5 |
| NCGC00094284-01 | Pergolide methanesulfonate | 15.8 | 24.8 | >100 |
| NCGC00094191-01 | NF449 octasodium salt | 28.2 | 31.0 | 31.5 |
| NCGC00094069-01 | LY-53,857 maleate | 25.1 | 38.0 | 94.7 |
| NCGC00022834-04 | Metergoline | 25.1 | 52.0 | 81.8 |
| NCGC00093637-01 | SB 200646 HCl | 20.0 | 55.7 | 99.2 |
IC50 calculated from HTRF assay in 5-point 1:5 titration covering a concentration range of 92 nM – 57.5 μM.
IC50 calculated from from HTRF assay in 12-point 1:3 titration covering a concentration range of 0.3 nM – 57.5 μM.
The two confirmed hits, NF449 and nor-Binaltorphimine dihydrochloride (nor-BNI), were reordered as powder samples and studied further in secondary assays. One potential mechanism for decreased signal in the binding assays would be compound-induced degradation of RNA. To test this, (CUG)20 and (CAG)20 RNA were incubated for 30 minutes with different concentrations of each compound in assay buffer at 37°C. For nor-BNI significant RNA degradation was observed for both the (CUG)20 and (CAG)20 transcripts (Fig. 5b), indicating a non-selective RNA degradation effect. No RNA degradation was observed for NF449 up to 800 M compound concentration for either transcript tested (Fig. 5b). To further confirm the activity of NF449, the compound was tested for its ability to inhibit binding of MBNL1 to (CUG)20 and (CAG)20 RNA in a filter binding assay. The IC50 for binding inhibition to (CUG)20 RNA was 29.7 μM (Fig. 5c), which was similar to the IC50 calculated from the homogenous assays in spite of the significant differences in the nature of these assays. The potency of NF449 was also 3-fold lower for (CAG)20 RNA than the (CUG)20 sequence, indicating selectivity between the two similar sequences (Fig. 5c). However, it is unclear whether NF449 functions through binding to the protein or RNA targets. More in-depth studies are needed to evaluate its mode of action. Because both assays use His6 and biotin tags for detection reagent capture, compounds inhibiting the binding of these tags would appear as false positives. However, these compounds are likely to be histidine and biotin analogs and can either be eliminated based on their structure or testing in tertiary assays using protein and RNA without such tags.
Figure 5. Confirmation of LOPAC1280 hits.

(a) Dose response of nor-Binaltorphimine dihydrochloride (nor-BNI) and NF449. The IC50 values are 21.9 M in HTRF and 12.5 μM in AlphaScreen assays for nor-BNI, and 31.0 μM in HTRF and 31.5 M in AlphaScreen assays for NF449. (b) Nor-BNI has nonspecific RNA degradation activity. 100 pM of 5′-end labeled (CUG)20 and (CAG)20 was incubated for 30 min with varying concentrations of compounds in assay buffer at 37°C. Significant RNA degradation was observed for nor-BNI for both RNA sequences, while no degradation was observed for NF449 up to 800 μM compound concentration against either RNA species. (c) NF449 shows specificity to inhibit MBNL1 binding to CUG repeat hairpin. Filter binding assay results for two 5′-end labeled transcripts (CUG)20 and (CAG)20 incubated with different compound concentration and 200 nM MBNL1 protein. The IC50 for (CUG)20 is 29.7±4.2 M and for (CAG)20, 98.7±9.6 μM.
Conclusions
We have developed two robust assays for detection of protein-RNA binding and validated their use for HTS and orthogonal screen. These assays apply AlphaScreen and HTRF technologies in a homogenous, easy to use format that is suitable for HTS. While the AlphaScreen system has previously been applied to a host protein-HIV RNA binding assay [25], the HTRF assay format has only been applied to protein-peptide, protein-protein and protein-DNA binding assays [30-32], but not protein-RNA binding. In addition to utilizing these assays for compound library screens to identify small molecule disruptors of the preformed RNA-protein complexes, these assays could also be utilized to screen for compounds that prevent complex formation by altering the order of addition of the target reagents and the compounds. Both the AlphaScreen and the HTRF assays were optimized in 1536-well plate format for HTS. The HTRF assay was better suited for HTS based on assay throughput, while the AlphaScreen assay was useful as a homogenous orthogonal assay for hit confirmation and elimination of false positives that interfere with the HTRF assay. Therefore, pairing the HTRF and AlphaScreen assays in a primary and orthogonal secondary screen is an effective strategy for HTS to identify protein-RNA binding inhibitors.
Acknowledgements
This research was supported by the Molecular Libraries Initiative of the NIH Roadmap for Medical Research, National Institutes of Health, the US grants MH087421, AR049077 and NS48843, and the Polish Ministry of Science and Higher Education grant N302 260938.
REFERENCES
- Aït-Aïssa S, Porcher J-M, Arrigo AP, Lambrè C. Activation of the hsp70 promoter by environmental inorganic and organic chemicals: relationships with cytotoxicity and lipophilicity. Toxicology. 2000;145(2-3):147–157. doi: 10.1016/s0300-483x(00)00145-1. [DOI] [PubMed] [Google Scholar]
- ASTM Standard guide for acute toxicity test with the rotifer. Brachionus. 2004:1440–91. [Google Scholar]
- Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, Staels B, Auwerx J, Laville M, Vidal H. Tissue distribution and quantification of the expression of mRNAs of peroxisome proloferator-activated receptors and liver X receptor-alpha in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46(8):1319–1327. doi: 10.2337/diab.46.8.1319. [DOI] [PubMed] [Google Scholar]
- Baker ME, Ruggeri B, Sprague J, Eckhardt C, Lapira J, Wick I, Soverchia L, Ubaldi M, Polzonetti-Magni AM, Vidal-Dorsch D, Bay S, Gully JR, Reyes JA, Kelley KM, Schlenk D, Breen EC, Sásik R, Hardiman G. Analysis of endocrine disruption in southern California coastal fish using an aquatic multi-species microarray. Environ. Health Perspect. 2009;117(2):223–230. doi: 10.1289/ehp.11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay S, Berry W, Chapman P, Fairey R, Gries T, Long ER, McDonald D, Weisberg SB. Evaluating consistency of best professional judgment in the application of a multiple lines of evidence sediment quality triad. Integrated Environmental Assessment and Management. 2007;3:491–497. doi: 10.1897/ieam_2007-002.1. [DOI] [PubMed] [Google Scholar]
- Blair RM, Fang H, Branham WS, Hass BS, Dial SL, Moland CL, Tong W, Shi L, Perkins R, Sheehan DM. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands. Toxicol. Sci. 1999;54(1):138–153. doi: 10.1093/toxsci/54.1.138. [DOI] [PubMed] [Google Scholar]
- Brown SB, Adams BA, Cyr DG, Eales JG. Contaminant effects on the teleost fish thyroid. Environ. Toxicol. Chem. 2004;23(7):1680–1701. doi: 10.1897/03-242. [DOI] [PubMed] [Google Scholar]
- Burton GA, Jr., Baudo R, Beltrami M, Rowland C. Assessing sediment contamination using six toxicity assays. Limnology. 2001;60(2):263–267. [Google Scholar]
- Burton GA., Jr. Sediment quality criteria in use around the world. Limnology. 2002;3(2):65–67. [Google Scholar]
- Carnevali O, Maradonna F. Exposure to xenobiotic compounds: looking for new biomarkers. Gen. Comp. Endocrinol. 2003;131(3):203–209. doi: 10.1016/s0016-6480(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Carnevali O, Tosti L, Speciale C, Peng C, Zhu Y, Maradonna F. DEHP impairs zebrafish reproduction by affecting critical factors in oogenesis. PLoS One. 2010;5(4):e10201. doi: 10.1371/journal.pone.0010201. doi:10.1371/journal.pone.0010201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celiz MD, Tso J, Aga DS. Pharmaceutical metabolites in the environment: analytical challenges and ecological risks. Environ. Toxicol. Chem. 2009;28(12):2473–2484. doi: 10.1897/09-173.1. [DOI] [PubMed] [Google Scholar]
- Chapman PM. The sediment quality triad approach to determining pollution induced degradation. Science of the Total Environment. 1990;97:815–825. [Google Scholar]
- Chapman PM, Mann GS. Sediment quality values (SQVs) and ecological risk assessment (ERA) Mar. Pollut. Bull. 1999;38(5):339–344. [Google Scholar]
- Chapman PM, Wang F, Jassen C, Goulet FF, Kamunde CN. Conducting of ecological risk assessment of inorganic metals and metalloides-current status. Hum. Ecol. Risk. Assess. 2003;9(4):641–697. [Google Scholar]
- Chapman PM, Anderson J. A decision making framework for sediment contamination. Integrated Environmental Assessment and Management. 2005;1:163–173. doi: 10.1897/2005-013r.1. [DOI] [PubMed] [Google Scholar]
- Coumoul X, Diry M, Barouki R. PXR dependent induction of human CYP3A4 gene expression by organochlorine pesticides. Biochem. Pharmacol. 2002;64(10):1513–1519. doi: 10.1016/s0006-2952(02)01298-4. [DOI] [PubMed] [Google Scholar]
- de March BGE. Mixture toxicity indices in acute lethal toxicity tests. Arch. Environ. Contam. Toxicol. 1987;16(1):33–37. [Google Scholar]
- Den Besten PJ, de Deckere E, Babut MP, Power B, DelValls DA, Zago C, Oen AMP, Heise S. Biological effects-based sediment quality in ecological risk assessment for european waters. J. Soils Sediments. 2003;3(3):144–162. [Google Scholar]
- Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an endocrine society scientific statement. Endocr. Rev. 2009;30(4):293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIN 38414-20: 1996-01. German standard methods for the examination of water, waste water and sludge - Sludge and sediments (group S) - Determination of six selected polychlorinated biphenyls by gas chromatography (S 20).
- D.Lgs. 152/2006. Norme in materia ambientale. Gazz. Uff. n. 88 - Suppl. Ord. n. 96.
- D.M. 471/1999. Regolamento recante criteri, procedure e modalità per la messa in sicurezza, la bonifica e il ripristino ambientale dei siti inquinati, ai sensi dell’articolo 17 del decreto legislativo 5 febbraio 1997, n. 22, e successive modificazioni e integrazioni. Gazz. Uff. Suppl. Ord. n. 293.
- D.M. 367/2003. Regolamento concernente la fissazione di standard di qualità nell’ambiente acquatico per le sostanze pericolose, ai sensi dell’articolo 3, comma 4, del D.Lgs. 152/1999. Gazz. Uff. n. 5.
- Fallone F, Villard PH, Decome L, Sèrèe E, De Mèo M, Chacon C, Durand A, Barra Y, Lacarelle B. PPARα activation potentiates AhR-induced CYP1A1 expression. Toxicology. 2005;216(2-3):122–128. doi: 10.1016/j.tox.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Fisher MA, Mehne C, Means JC, Ide CF. Induction of CYP1A mRNA in Carp (Cyprinus carpio) from the Kalamazoo River. Arch. Environ. Contam. Toxicol. 2006;50(1):14–22. doi: 10.1007/s00244-004-0171-4. [DOI] [PubMed] [Google Scholar]
- Gambrell RP, Reddy CN, Khalid RA. Characterization of trace and toxic materials in sediments of a lake being restored. J. Wat. Pollut. Cont. Fed. 1983;55(9):1201–1210. [Google Scholar]
- George S, Gubbins M, MacIntosh A, Reynolds W, Sabine V, Scott A, Thain J. A comparison of pollutant biomarker response in European flounders (Platicthys flesus) subjected to estuarine pollution. Mar. Environ. Res. 2004;58(2):571–575. doi: 10.1016/j.marenvres.2004.03.047. [DOI] [PubMed] [Google Scholar]
- Giguère V. Orphan nuclear receptors: from gene to function. Endocr. Rev. 1999;20(5):689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- Goksôyr A, Förlin L. The cytochrome P450 system in fish, aquatic toxicology and environmental monitoring. Aquat. Toxicol. 1992;22(4):287–312. [Google Scholar]
- Great Lakes Water Quality Board . Guidelines and register for the evaluation of Great Lakes dredging projects. International Joint Commission; Windsor, Ontario, Canada: 1982. [Google Scholar]
- Grôsvik BE, Goksôyr A. Biomarker protein expression in primary cultures of salmon (Salmo salar L.) hepatocytes exposed to environmental pollutants. Biomarkers. 1996;1(1):45–53. doi: 10.3109/13547509609079346. [DOI] [PubMed] [Google Scholar]
- Grün F, Blumberg B. Endocrine disrupters as obesogens. Mol. Cell. Endocrinol. 2009;304(1-2):19–29. doi: 10.1016/j.mce.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu YZ, Hogenesc JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- Hamelink JL, Landrum PF, Bergman HL, Benson WH. Bioavailability: physical, chemical and biological interactions. 1 ed CRC-Press; Boca Raton, FL: 1994. [Google Scholar]
- Hardiman G. Microarray technology--advances, applications, future prospects. Pharmacogenomics. 2007;8(12):1639–1642. doi: 10.2217/14622416.8.12.1639. [DOI] [PubMed] [Google Scholar]
- Honkakoski P, Negishi M. Regulation of cytochrome P450 (CYP) genes by nuclear receptors. Biochem. J. 2000;347(Pt2):321–337. doi: 10.1042/0264-6021:3470321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama GK, Vijayan MM, Forsyth RB, Ackerman PA. Heat shock proteins and physiological stress in fish. Am. Zool. 1999;39(6):901–909. [Google Scholar]
- Janosek J, Hilscherova K, Blaha L, Holoubek I. Environmental xenobiotics and nuclear receptors--interactions, effects and in vitro assessment. Toxicol. in Vitro. 2006;20(1):18–37. doi: 10.1016/j.tiv.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29(14):2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraaij RH. Sequestration and bioavailability of hydrophobic chemicals in sediment. Thesis institute for risk assessment sciences Utrecht university; Utrecht, The Netherlands: 2001. [Google Scholar]
- Letcher RJ, Bustnes JO, Dietz R, Jenssen BM, Jørgensen EH, Sonne C, Verreault J, Vijayan MM, Gabrielsen GW. Exposure and effects assessment of persistent organohalogen contaminants in arctic wildlife and fish. Sci. Total Environ. 2010;408(15):2995–3043. doi: 10.1016/j.scitotenv.2009.10.038. [DOI] [PubMed] [Google Scholar]
- Li W, Zha J, Spear PA, Li Z, Yang L, Wang Z. Changes of thyroid hormone levels and related gene expression in chinese rare minnow (Gobiocypris rarus) during 3-amino-1,2,4-triazole exposure and recovery. Aquat. Toxicol. 2009;92(1):50–57. doi: 10.1016/j.aquatox.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Long ER, Chapman PM. A sediment quality triad-measures of sediment contamination toxicity and infaunal community composition in Puget Sound. Marine Pollution Bulletin. 1985;16:405–415. [Google Scholar]
- Luoma SN. Bioavailability of trace metals to aquatic organisms--a review. Sci. Total Environ. 1983;28:1–22. doi: 10.1016/s0048-9697(83)80004-7. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Maradonna F, Carnevali O. Vitellogenin, zona radiata protein, cathepsin D and heat shock protein 70 as biomarkers of exposure to xenobiotics. Biomarkers. 2007;12(3):240–255. doi: 10.1080/13547500601070859. [DOI] [PubMed] [Google Scholar]
- Marlatt VL, Lakoff J, Crump K, Martyniuk CJ, Watt J, Jewell L, Atkinson S, Blais JM, Sherry J, Moon TW, Trudeau VL. Sex- and tissue-specific effects of waterborne estrogen on estrogen receptor subtypes and E2-mediated gene expression in the reproductive axis of goldfish. Comp Biochem Physiol A Mol Integr Physiol. 2010;156(1):92–101. doi: 10.1016/j.cbpa.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Miller KA, Addison RF, Bandiera SM. Hepatic CYP1A levels and EROD activity in English sole: biomonitoring of marine contaminants in Vancouver Harbour. Mar. Environ. Res. 2004;57(1-2):37–54. doi: 10.1016/S0141-1136(03)00059-X. [DOI] [PubMed] [Google Scholar]
- Mortensen AS, Arukwe A. The persistent DDT metabolite, 1-1-dichloro-2,2-bis(p-chlorophenyl)ethylene, alters thyroid hormone-dependent genes, hepatic cytochrome P4503A, and pregnane X receptor gene expressions in atlantic salmon (Salmo salar) Parr. Environ. Toxicol. Chem. 2006;25(6):1607–1615. doi: 10.1897/05-376r1.1. [DOI] [PubMed] [Google Scholar]
- Nagler JJ, Cyr DG. Exposure of male american plaice (Hippoglossoides platessoides) to contaminated marine sediments decreases the hatching success of their progeny. Environ. Toxicol. Chem. 1997;16(8):1733–1738. [Google Scholar]
- Nakajima T, Ichihara G, Kamijima M, Itohara S, Aoyama T. Functional activation of peroxisome proliferator-activated receptor alpha (PPARalpha) by environmental chemicals in relation to their toxicities. Nagoya J. Med. Sci. 2002;65(3-4):85–94. [PubMed] [Google Scholar]
- Nilsson CB, Hoegberg P, Trossvik C, Azaïs-Bræsco V, Blaner WS, Fex G, Harrison EH, Nau H, Schmidt CK, van Bennekum AM, Håkansson H. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases serum and kidney retinoic acid levels and kidney retinol esterification in the rat. Toxicol. Appl. Pharmacol. 2000;169(2):121–131. doi: 10.1006/taap.2000.9059. [DOI] [PubMed] [Google Scholar]
- Power DM, Llewellyn L, Faustino M, Nowell MA, Björnsson BT, Einarsdottirc IE, Canarina AVM, Sweeney GE. Thyroid hormones in growth and development of fish. Comp. Biochem. Phisiol. C: Toxicol. Pharmacol. 2001;130:447–459. doi: 10.1016/s1532-0456(01)00271-x. [DOI] [PubMed] [Google Scholar]
- Rouse RJ, Field K, Lapira J, Lee A, Wick I, Eckhardt C, Bhasker CR, Soverchia L, Hardiman G. Development and application of a microarray meter tool to optimize microarray experiments. BMC Res. Notes. 2008;11:1–45. doi: 10.1186/1756-0500-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S. Endocrine disruptors and human health: is there a problem? Toxicology. 2004;205(1-2):3–10. doi: 10.1016/j.tox.2004.06.032. [DOI] [PubMed] [Google Scholar]
- Sásik R, Woelk CH, Corbeil J. Microarray truths and consequences. J. Mol. Endocrinol. 2004;33(1):1–9. doi: 10.1677/jme.0.0330001. [DOI] [PubMed] [Google Scholar]
- Sérée E, Villard PH, Pascussi JM, Pineau T, Maurel P, Nguyen QB, Fallone F, Martin PM, Champion S, Lacarelle B, Savouret JF, Barra Y. Evidence for a new human CYP1A regulation pathway involving PPAR-alpha and 2 PPRE sites. Gastroenterology. 2004;127(5):1436–1445. doi: 10.1053/j.gastro.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Sonnenschein C, Soto AM. An updated review of environmental estrogen and androgen mimics and antagonists. J. Steroid Biochem. Mol. Biol. 1998;65(1-6):143–150. doi: 10.1016/s0960-0760(98)00027-2. [DOI] [PubMed] [Google Scholar]
- Soto AM, Vandenberg LN, Maffini MV, Sonnenschein C. Does breast cancer start in the womb? Basic Clin. Pharmacol. Toxicol. 2008;102(2):125–133. doi: 10.1111/j.1742-7843.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegeman JJ, Livingstone DR. Forms and functions of cytochrome P450. Comp. Biochem. Physiol. C: Toxicol. Pharmacol. Endocrinol. 1998;121(1-3):1–3. [PubMed] [Google Scholar]
- Thomas RL. A protocol for the selection of process oriented remedial options to control in situ sediment contaminants. Hydrobiologia. 1987;149(1):247–258. [Google Scholar]
- U.S.EPA . EPA 905/9-88-002. Office of water regulations and standards; Washington, DC, and EPA region 5, Chicago: 1987. An overview of sediment quality in the United States. [Google Scholar]
- U.S.EPA Tetra-through octa-chlorinated dioxins and furans byiIsotope dilution HRGC/HRMS. Method. 1994;1613 [Google Scholar]
- U.S.EPA Semivolatile organic compounds by gas chromatography/mass spectrometry (GC/MS) Method. 1996;8270C [Google Scholar]
- U.S.EPA Nonhalogenated organics using Gc/Fid. Method. 1996 8015B/96. [Google Scholar]
- U.S.EPA Volatile organic compounds b gas chromatography/mass spectrometry (GC/MS) Method. 1996;8260B [Google Scholar]
- U.S.EPA . EPA 823-R-97-006. 1-3. Science and technology office; Washington, DC: 1997. The incidence and severity of sediment contamination in surface waters of the united states. [Google Scholar]
- U.S.EPA Inductively coupled plasma-mass spectrometry. Method. 1998;6020A Revision 2007. [Google Scholar]
- U.S.EPA . EPA-823-B-01-002. Office of water; Washington, DC: 2001. Methods for collection, storage and manipulation of sediments for chemical and toxicological analyses: technical manual. [Google Scholar]
- Van der Oost R, Beyer J, Vermeulen NPE. Fish bioaccumulation and biomarkers in environmental risk assessment: a review. Environ. Toxicol. Pharmacol. 2003;13(2):57–149. doi: 10.1016/s1382-6689(02)00126-6. [DOI] [PubMed] [Google Scholar]
- Vanhaecke P, Persoone G, Claus C, Sorgeloos P. Proposal for a short-term toxicity test with Artemia nauplii. Ecotoxicol. Environ. Saf. 1981;5(3):382–387. doi: 10.1016/0147-6513(81)90012-9. [DOI] [PubMed] [Google Scholar]
- Van Veld PA, Vogelbein WK, Cochran MK, Goksøyr A, Stegeman JJ. Route-specific cellular expression of cytochrome P4501A (CYP1A) in fish (Fundulus heteroclitus) following exposure to aqueous and dietary benzo[a]pyrene. Toxicol. Appl. Pharmacol. 1997;142(2):348–359. doi: 10.1006/taap.1996.8037. [DOI] [PubMed] [Google Scholar]
- Vijayan MM, Pereira C, Kruzynski G, Iwama GK. Sublethal concentrations of contaminant induce the expression of hepatic heat shock protein 70 in two salmonids. Aquat. Toxicol. 1998;40(1-3):101–108. [Google Scholar]
- Von Danwitz B. Thesis. University of Bremen; Bremen, Germany: 1992. Zur Abschätzung der Schadwirkung von Stoffkombinationen autf aquatische organismen (On the estimation of mixture toxicity for aquatic organisms, in German) [Google Scholar]
- Wang J, Wei Y, Wang D, Chan LL, Dai J. Proteomic study of the effects of complex environmental stresses in the livers of goldfish (Carassius auratus) that inhabit Gaobeidian Lake in Beijing, China. Ecotoxicology. 2008;17(3):213–220. doi: 10.1007/s10646-007-0187-4. [DOI] [PubMed] [Google Scholar]
- Wong CKC, Yeung HY, Cheung KKL, Yung KKL, Wong MH. Ecotoxicological assessment of persistent organic and heavy metal contamination in Hong Kong coastal sediment. Arch. Environ. Contam. Toxicol. 2000;38(4):486–493. doi: 10.1007/s002449910064. [DOI] [PubMed] [Google Scholar]
- Woods CG, Vanden Heuvel JP, Rusyn I. Genomic profiling in nuclear receptor-mediated toxicity. Toxicol. Pathol. 2007;35:474–494. doi: 10.1080/01926230701311351. [DOI] [PubMed] [Google Scholar]
- Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor α. Biochem. Biophys. Res. Commun. 2001;287(2):391–396. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]