

Abstract
RNAs Interference plays a very important role in gene silencing. In vitro identification of miRNAs is a slow process as it is difficult to isolate them. Nucleotide sequences of miRNAs are highly conserved among the plants and, this form the key feature behind the identification of miRNAs in plant species by homology alignment. In silico identification of miRNAs from EST database is emerging as a novel, faster and reliable approach. Here EST sequences of Senecio vulgaris (Groundsel) were searched against known miRNA sequences by using BLASTN tool. A total of 10 miRNAs were identified from 1956 EST sequences and 115 GSS sequences. The most stable miRNA identified is svu-mir-1. This approach will accelerate advance research in regulation of gene expression in Groundsel by interfering RNAs.
Background:
Senecio vulgaris also known as Groundsel, is an herb belonging to the family Asteraceae. Groundsel has a long history of herbal use and although not an official plant. It is still often used by herbalists. The whole herb is anthelmintic, diaphoretic and diuretic. It has been also reported to act as a remedy for scurvy. It is often used as a poultice and is said to be useful in treating sickness of the stomach, whilst a weak infusion is used as a simple and easy purgative. The plant can be harvested in May and dried for later use, or the fresh juice can be extracted and used as required. A homeopathic remedy is made from the plant. It is used in the treatment of menstrual disorders and nose bleeds [1].
To date RNA interference (RNAi) has mainly been used as a readily available, rapid, reverse genetic tool to create medicinal plants with novel chemical phenotypes and to determine the phenotypes of genes responsible for the synthesis of many pharmaceutically important secondary metabolites [2]. MicroRNAs (miRNAs) are a type of interfering RNAs. miRNAs are produced from non-coding DNA region [3]. They represent a newly identified class of non-protein-coding small (˜20 nt) RNAs, which negatively regulate the gene expression at the post-transcriptional level by repressing gene translation or degrading targeted mRNAs. They can regulate gene expression by controlling the protein translation mechanism during variety of cell phenomena. miRNAs originate from endogenous transcripts that can form local hairpin structures and are processed such that a single miRNA molecule accumulates from one arm of a hairpin precursor molecule [4]. In silico identification of miRNAs has gained momentum because miRNAs are evolutionary conserved. There are four approaches for identifying miRNAs: (1) genetic screening [5, 6]. (2) Direct cloning after isolation of small RNAs [7], (3) computational strategy [8], and (4) expressed sequence tags (ESTs) analysis [9]. The basics of computational miRNA prediction are based on few parameters like calculation of optimum free energies (dG), Structural continuity, and number of G: C base pairing etc. [3]. Identification of miRNAs by homology search analysis with ESTs is also an emerging approach. Several miRNAs are evolutionarily conserved from species to species [10, 11] and suggests a powerful base to predict homologues or orthologues of previously known miRNAs. More importantly this approach is very useful for predicting miRNAs in multiple species. Recently a set of miRNA and their targets were reported using the largest data set of Triticeae ESTs [12]. In rice, six new miRNAs were identified which are conserved in other monocots [13].
Methodology:
The present work used EST analysis method for the identification of miRNAs. miRNAs from known plants Arabidopsis, Brassica, Glycine, Saccharum, Sorghum, Vitis, Solanum, Oryza, Triticum, Chlamydomonas, and other plant species were downloaded from the miRNA database miRBase Release 17. 16772 miRNAs are available on the site (http://www.mirbase.org/). 1956 ESTs and 115 GSS sequences of S vulgaris are available on (http://www.ncbi.nlm.nih.gov). Local databases of EST and GSS sequences were created by Bioedit sequence alignment editor tool (version 7.0.9.0). BLASTN tool was used to reveal homology between ESTs and miRNA sequences and GSS and miRNA sequences. An E-value cut off 0.01 and word-match length 7 between query miRNA and ESTs sequences was used as criteria to assign identity to any sequence. On the basis of the parameters like expectation value (E-value) and word match size between queries and databases, precursor-miRNAs (pre-miRNAs) were identified. The pre-miRNAs are aproximately 70-nucleotides in length and are folded into imperfect stem-loop structures [14]. So, we fetched 100 sequences upstream and downstream from the matching region. Resultant sequences were predicted as premiRNAs. For the functional annotation of EST and GSS hits, they were searched against Swissprot database by using BLASTX tool. The RNA structure and free energy of the hits that did not show significant homology with proteins in Swissprot database were calculated by RNA structure software version 5.2. The flow chart of miRNA prediction is given in Figure 1. The following criteria were considered necessary for miRNA homologs:
Figure 1.

Flowchart of miRNA prediction in Senecio vulgaris
(1) The RNA sequence folding into an appropriate stem-loop hairpin secondary structure, (2) a mature miRNA sequence located in one arm of the hairpin structure, (3) predicted mature miRNAs with no more than 3 nt substitutions as compared with the known miRNAs, (4) miRNAs having less than 6 mismatches with the opposite miRNA sequence in the other arm, (miRNA*) (5) no loop or break in miRNA* sequences, and (6) predicted secondary structures with higher minimal folding free energy (MFE) and minimal folding free energy index (MFEI), the MEFI usually being over 0.85 [15]. Also the AU contents of premiRNA within 30% to 70% were considered significant [16].
Nomenclature of miRNAs:
Names were assigned to the predicted miRNAs in similar pattern to miRBase [17]. The mature sequences are designated ‘miR’, and the precursor hairpins are labeled as ‘mir’ with the prefix ‘svu’ for S. vulgaris. In the cases where distinct precursor sequences have identical miRNAs with different resources and mismatch pattern, they were named as svu-mir-1-a and svumir- 1-b. In this research work both suv-mir-3a and suv-mir-3b have identical miRNA sequences, however resources were different.
Discussion:
The main principle behind the computational miRNA prediction is sequence and structure homologies. As mentioned above in materials and methods, after BLASTX searches, all ESTs and GSS hits with noncoding sequences were maintained for secondary structure analysis, only those in line with the screening criteria of RNA structure prediction were selected as candidates. In this analysis we identified 10 potential miRNAs of S. vulgaris. Nomenclatures of these miRNA were based on the scheme proposed by Ambros [18]. The basic information of predicted miRNA as gene Id, source of the data,percentage of AU in pre-miRNA and free energy of the structure are given in Table 1, (see supplementary material) Structures with energy of -90kcal/mol or higher were rejected (higher means less negative).
As biological database grow exponentially in size of complexity, on web (WWW), the identification of miRNAs using computational approaches is more feasible and faster than experimental approaches. The value of computational methods increases due to advantages includes low cost and high efficiency associated with it. According to Zhang [19], 10,000 ESTs contain 1 miRNA, so about 1 miRNA should have been predicted theoretically from the total of 1956 ESTs and 115 GSSs in this work. However, we obtained 10 potential miRNA candidates from the present work. Generally, plant miRNA clusters have not been frequently observed, so only one plant miRNA can come from the same transcript. In this work, different length of mature miRNAs from the same precursor were regarded as different ones, considering they corresponded to different target genes.
Conclusions:
In this paper, with a computational approach 10 miRNAs were identified from the EST and GSS databases of S vulgaris. Six out of ten most stable secondary structure of miRNA are shown in Figure 3. The free energies of different miRNAs are displayed in Figure 2. There lies great potential in mining the huge information from databases and transfer that to divergent plant species. Comparative genomics is useful for understanding the regulation and function of genes in the plant genomes for which genome sequence and related genomic resources are not available. S vulgaris is a less studied plant member of Asteraceae family, having medicinal value. This effort will accelerate research in S vulgaris structural and functional genomics. Data mining was done from biological database resource, dbEST and dbGSS of NCBI. This led the identification of miRNA in S vulgaris, the effort will accelerate the development of genomic resources for this lesser studied plant member of Asteraceae family, with great economic value.
Figure 3.

Secondary structure of predicted miRNA of Senecio vulgaris(Groundsel), red color shown miRNA sequence.
Figure 2.
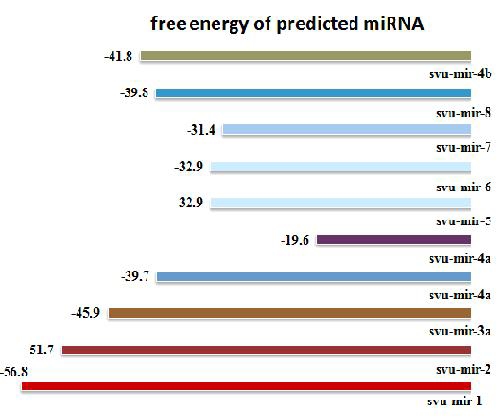
Energy plot among the predicted miRNA of Senecio vulgaris (Groundsel).
Supplementary material
Footnotes
Citation:Sahu et al, Bioinformation 7(8): 375-378 (2011)
References
- 1. http://www.maltawildplants.com/ASTR/Senecio_vulgaris.php.
- 2.JF Borgio. Journal of Medicinal Plants Research. 2009;3:1176. [Google Scholar]
- 3.K Shallu, S Sahu. Indian Journal of Scientific Research. 2011;2:44. [Google Scholar]
- 4.Ambros, et al. Curr biol. 2003;13:807. doi: 10.1016/s0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- 5.RC Lee, et al. Cell. 1993;75:843. [Google Scholar]
- 6.B Wightman, et al. Cell. 1993;75:855. [Google Scholar]
- 7.EA Mead, Z Tu. BMC Genomics. 2008;9:244. doi: 10.1186/1471-2164-9-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DH Mathews, et al. J Mol Biol. 1999;288:911. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 9.B Zhang, et al. Dev Biol. 2006;289:3. [Google Scholar]
- 10.BJ Reinhart, et al. Genes Dev. 2002;16:1616. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MJ Axtell, DP Bartel. Plant Cell. 2005;17:1658. [Google Scholar]
- 12.A Dryanova, et al. Genome. 2008;51:433. [Google Scholar]
- 13.R Sunkar, et al. Plant Cell. 2005;17:1397. [Google Scholar]
- 14.Y Lu, X Yang. Comp Funct Genomics. 2010;pii:128297. [Google Scholar]
- 15.BH Zhang, et al. Cell mol life sci. 2006;63:246. doi: 10.1007/s00018-005-5467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.FL Xie, et al. FEBS Lett. 2007;581:1464. doi: 10.1016/j.febslet.2007.02.074. [DOI] [PubMed] [Google Scholar]
- 17.S Griffiths-Jones, et al. Nucleic Acids Res. 2006;34:D140. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.V Ambros, et al. RNA. 2003;9:277. [Google Scholar]
- 19.B Zhang, et al. Plant J. 2006;46:243. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.