

Abstract
A chemical investigation of an ethyl acetate extract of the Red Sea soft coral Sarcophyton glaucum has led to the isolation of two peroxide diterpenes, 11(S) hydroperoxylsarcoph-12(20)-ene (1), and 12(S)-hydroperoxylsarcoph-10-ene (2), as well as 8-epi-sarcophinone (3). In addition to these three new compounds, two known structures were identified including: ent-sarcophine (4) and sarcophine (5). Structures were elucidated by spectroscopic analysis, with the relative configuration of 1 and 2 confirmed by X-ray diffraction. Isolated compounds were found to be inhibitors of cytochrome P450 1A activity as well as inducers of glutathione S-transferases (GST), quinone reductase (QR), and epoxide hydrolase (mEH) establishing chemo-preventive and tumor anti-initiating activity for these characterized metabolites.
Keywords: Sarcophyton glaucum, soft coral, diterpenes, cancer chemo-preventive activity
1. Introduction
Marine natural products are diverse in terms of chemical structures as well as biological activities. The Red Sea serves as an epicenter for marine bio-diversity with a high endemic biota. Indeed of the 180 soft corals species identified world-wide, approximately 40% are native to the Red Sea [1]. Soft corals are marine invertebrates possessing a vast range of terpenoid metabolites. These terpenes, mostly cembranoids, represent the main chemical defense for coral against natural predators [2]. Soft corals of the genus Sarcophyton (family Alcyoniidae) are particularly rich in cembrane terpenes [3]. Cembranoids contain a 14-membered macro cyclic skeleton and exhibit a wide range of biological activities including anti-tumor, neuro-protective, antimicrobial, calcium-antagonistic, and anti-inflammatory activity [4,5,6,7]. The cembranoid diterpene sarcophine has been investigated since 1998 for its potential as a chemo-preventive agent [8], cytotoxic agent, anti-microbial agent [9], competitive cholinesterase inhibitor [10], noncompetitive phosphofructokinase inhibitor [11], and a Na+, K+-ATPase inhibitor [12]. Recent studies focusing on the treatment of human diseases have shown that sarcophine and sarcophine derivatives (e.g., hydroxylated sarcophine) are potent cancer chemo-preventive agents [8,9,13,14,15].
Cancer chemoprevention is based on chemical constituents that block, inhibit, or reverse the development of cancer in normal or pre-neoplastic tissue [16]. During the past 20 years, thousands of novel marine metabolites have been identified and assayed for anticancer activity [17]. Most of these drug leads are identified by high-throughput in vitro screening via a cost-effective testing of cancer cell lines derived from human and rodent sources. Indeed several marine-derived drug leads have reached phase II human clinical trials based on promising anticancer results, although toxicity testing has mostly screened out such candidate drugs. Sarcophine anti-tumor potency appears to at least in part involve inhibition of cell transformation that can be induced in vitro by 12-O-tetradecanoyl phorbol-13-acetate (TPA) with irreversible acquisition of tumorigenicity [7,13]. In many cases, carcinogenesis is initiated by pro-carcinogens in combination with phase I enzymes such as cytochrome P450 1A and oxidative stress leading to DNA damage. This process can be mitigated at least in part by phase II detoxification enzymes such as glutathione S-transferases (GSTs), quinone reductase (QR), and epoxide hydrolase (mEH).
Herein, we report the isolation of three new and two known cembranolides (Chart 1) from an ethyl acetate extraction of the Red Sea soft coral Sarcophyton glaucum. Structures of these isolated metabolites were elucidated by 1D and 2D spectroscopic techniques, while the absolute configuration of 1 and 2 were confirmed by X-ray diffraction and circular dichroism (CD) analyses. Compounds 2 and 3 were found to be promising inhibitors of cytochrome P450 1A activity as well as inducers of GST and QR activity in in vitro assays.
Chart 1.

Structures of metabolites 1–5.
2. Results and Discussion
Freshly collected specimens of S. glaucum were immediately frozen in dry ice and kept at −20 °C until ready for organic-solvent extraction. The EtOAc-soluble fraction was subjected to normal and reverse phase chromatography to afford new hydroperoxyl cembranolides (1 and 2), a cembrene derivative 8-epi-sarcophinone (3) along with two known cembranolides, ent-sarcophine (4) [18,19], and sarcophine (5) [18,19,20,21].
Preliminary 1H NMR analysis established that all fractions shared a common carbon skeleton, differing either in the degree of oxidation or the configuration of one or more chiral centers. Precedent from soft coral literature led to the assumption of a cembranoid-skeleton backbone [19]. Compound 1 was obtained as colorless crystals, 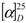 +12.6 (c 0.09, CHCl3). The HR-FAB-MS exhibited a [M + Na]+ ion at m/z 371.18281, indicating a molecular formula of C20H28O5Na and seven degrees of unsaturation that was supported by NMR data. An IR spectrum indicated the presence of an α,β-unsaturated-γ-lactone (1750 and 1686 cm−1), a carbonyl (1707 cm−1), an olefin (1669 cm−1), an epoxide (1256 cm−1) and a broad absorption band for OH stretching (3000–3353 cm−1). The 13C NMR and DEPT spectrum (Table 1) exhibited 20 carbon signals establishing: three methyls, seven methylenes, four methines, and six quaternary carbons. The spectrum also revealed the presence of an exomethylene functionality at δC 113.4/144.5, two oxymethine carbons at δC 60.8 and 86.5, one oxygenated quaternary carbon at δC 61.1, and two olefinic carbons at δC 119.6 and 146.0.
+12.6 (c 0.09, CHCl3). The HR-FAB-MS exhibited a [M + Na]+ ion at m/z 371.18281, indicating a molecular formula of C20H28O5Na and seven degrees of unsaturation that was supported by NMR data. An IR spectrum indicated the presence of an α,β-unsaturated-γ-lactone (1750 and 1686 cm−1), a carbonyl (1707 cm−1), an olefin (1669 cm−1), an epoxide (1256 cm−1) and a broad absorption band for OH stretching (3000–3353 cm−1). The 13C NMR and DEPT spectrum (Table 1) exhibited 20 carbon signals establishing: three methyls, seven methylenes, four methines, and six quaternary carbons. The spectrum also revealed the presence of an exomethylene functionality at δC 113.4/144.5, two oxymethine carbons at δC 60.8 and 86.5, one oxygenated quaternary carbon at δC 61.1, and two olefinic carbons at δC 119.6 and 146.0.
The low field oxymethine carbon at δC 86.5 (C-11) suggested the presence of a peroxide functionality that is consistent with the presence of a broad singlet at δH 8.25 in the 1H NMR spectrum [18]. 13C NMR analysis indicated that two oxygens contribute to an α,β-unsaturated-γ-lactone with appropriate signals at δC 174.5 and 78.9 for the carbonyl and oxymethine carbons, respectively. The olefinic methyl group at δH 1.85 (H3-17) exhibited an HMBC correlation with a low-filed 13C NMR resonance for a keto group in association with the α,β-unsaturated-γ-lactone ring at δC 174.5 (C-16). Carbon signals at δC 124.2 (C-15) and 162.2 (C-1) were consistent with α and β olefinic carbons of the α,β-unsaturated-γ-lactone system. The carbon signal at δC 78.9 (C-2) is consistent with an oxymethine carbon while the oxymethine proton at δH 5.50 (d, J = 15.0 Hz; H-2) exhibited a strong correlation with a one-proton doublet at δH 5.09 (J = 15.0 Hz; H-3) in the 1H-1H COSY spectrum (Figure 1). The olefinic methyl group at δH 1.94 (H-18) also shows an HMBC correlation with an olefinic methine at δC 119.6 (C-3). The methyl signal at δH 1.27 (H-19) indicates a proximal oxygen functionality identified from 13C NMR to be an epoxide. The location of the epoxide ring at C7/C-8 was detected from HMBC correlations (Figure 1), as there are clear correlations between C-7 (δC 60.8) and H-6 (2.59, td, J = 5.0, 13.5 Hz; 2.39, m), H-5 (2.20, m; 2.39, m), H3-19, and H-9 (1.30, m; 1.79, m); and between C-8 (δC 61.1) and H-7 (2.50, dd, J = 4.5, 8.5 Hz), H-9, H-10, and H-6. A triplet-like signal at δH 4.35 (J = 5.0 Hz; H-11) revealed the presence of a peroxide at δC 86.5 that showed a strong correlation with methylene signals at δH 1.50 (m) and 1.70 (m) (H-10) in the 1H-1H COSY spectrum (Figure 1). The position of the peroxide was established through HMBC correlation between H-11 and C-9 (32.1, t), C-10 (26.7, t), C-12 (144.5, s), and C-13 (30.1, t). Exomethylene protons at δH 5.12 (s) and 5.16 (s) (H2-20) showed strong correlations with carbon signals at δC 113.4 (C-20) and δC 144.5 (C-12) in HMQC and HMBC analyses, respectively.
Table 1.
1H and 13C NMR spectral data of 1–3.
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | -- | 162.2 | -- | 162.5 | -- | 163.3 |
| 2 | 5.50 (d, 15.0) | 78.9 | 5.44 (d, 16.0) | 79.2 | 5.47 (dd, 1.5, 10.0) | 78.9 |
| 3 | 5.09 (d, 15.0) | 119.6 | 4.98 (d, 16.0) | 118.9 | 5.12 (brd, 10.5) | 122.1 |
| 4 | -- | 146.0 | -- | 146.7 | -- | 141.9 |
| 5 | 2.20 (m) | 35.9 | 2.02 (m) | 37.1 | 2.66 (m) | 37.9 |
| 2.39 (m) | 2.38 (dt, 4.5, 13.5) | 2.76 (m) * | ||||
| 6 | 2.59 (td, 5, 13.5) | 25.4 | 1.77 (m) | 25.0 | 2.06 (m) | 32.8 |
| 2.39 (m) | 2.73 (m) * | |||||
| 7 | 2.50 (d, 4.5, 8.5) | 60.8 | 2.53 (dd, 5.0, 6.0) | 59.0 | -- | 212.1 |
| 8 | -- | 61.1 | -- | 59.2 | 2.44 (m) | 46.6 |
| 9 | 1.30 (m) | 32.1 | 2.25 (m) | 39.0 | 1.56 (m) | 32.4 |
| 1.79 (m) | 2.46 (m) | 1.95 (m) | ||||
| 10 | 1.50 (m) | 26.7 | 5.42 (ddd, 16.0, 10.5, 7.5) | 124.6 | 1.88 (brd, 11.0) | 26.5 |
| 1.70 (m) | 2.26 (m) | |||||
| 11 | 4.35 (t like, 5) | 86.5 | 5.56 (d, 16.0) | 136.1 | 4.78 (td, 7.5, 1) | 124.1 |
| 12 | -- | 144.5 | -- | 84.0 | -- | 134.9 |
| 13 | 2.07 (m) | 30.1 | 1.41 (dd, 4) * | 37.6 | 1.92 (m) | 36.1 |
| 2.20 (m) | 2.07 (td, 13.0, 4.5) | 2.00 (m) | ||||
| 14 | 2.07 (m) | 24.8 | 2.42 (m) * | 21.2 | 2.16 (brt, 12.0) | 26.1 |
| 1.50 (m) | 2.50 (m) * | 2.60 (m) | ||||
| 15 | -- | 124.2 | -- | 123.8 | -- | 122.3 |
| 16 | -- | 174.5 | -- | 174.9 | -- | 175.0 |
| 17 | 1.85 (s) | 8.9 | 1.87 (brs) | 9.1 | 1.82 (t, 1.5) | 8.9 |
| 18 | 1.94 (s) | 16.0 | 1.89 (s) | 16.2 | 1.84 (s) | 16.2 |
| 19 | 1.27 (s) | 16.7 | 1.30 (s) | 18.2 | 1.06 (d, 7.5) | 18.8 |
| 20 | 5.12 (s) | 113.4 | 1.43 (s) | 22.8 | 1.60 (s) | 15.7 |
| 5.16 (s) | ||||||
| -OOH | 8.25 (brs) | -- | 7.70 (brs) | -- | -- | -- |
a Recorded in CDCl3 and obtained at 500 and 125 MHz for 1H and 13C NMR, respectively. * Overlapping signals.
Figure 1.

Selected 1H-1H COSY ( ) and HMBC (
) and HMBC ( ) correlations of 1–3.
) correlations of 1–3.
Comparison of the above data with those structural relatives isolated from the same species [22,23], strongly indicated a cembranoid molecular framework containing the rare 11-peroxid-12(20)-exomethylene as confirmed by X-ray analysis (Figure 2). The relative configuration of 1 was determined on the basis of coupling constants and NOESY experiments. The vicinal coupling constant of 15.0 Hz between H-2 and H-3 as well as a NOESY correlation of H-2 with H3-18 established a trans configuration between the γ-lactone (H-2) and the olefinic proton (H-3). In order to confirm the position of the peroxyl group, as well as the relative stereochemistry, X-ray structure analysis was performed. The absolute stereochemistry of 1 at C-2 was determined via circular dichroism (CD) analysis (Figure 3). The observed positive Cotton effect {[θ]248 +0.7} followed by a negative value {[θ]225 −3.23} observed in the CD spectrum for the electronic transitions of the 2(5H)-furanone moiety, indicated a left hand (M) helix configuration for the five-membered α,β-unsaturated-γ-lactone ring [24]. Supporting CD data for 1, CD spectral comparison between 1 and ent-sarcophine (4) indicated the same R absolute configuration for the two compounds at C-2 [18,19,21,22]. Therefore, 1 was assigned as 11(S)-hydroperoxylsarcoph-12(20)-ene.
Figure 2.

ORTEP depiction for X-ray crystal structures of 1–2.
Compound 2 was obtained as color-less crystals, −20.1 (c 0.1, CHCl3) with much of the spectral data identical to 1 (Table 1). The HR-FAB-MS showed an [M + Na]+ ion at m/z 371.18293 indicating a molecular formula C20H8O5Na and seven degrees of unsaturation that was supported by NMR data. The analysis of 1H, 13C NMR and DEPT spectra revealed the presence of four methyls, five methylenes, five methines (two of them oxygenated, δC 59.0, and 79.2) , and six quaternary carbons (two of them oxygenated, δC 59.2, and 84.0). NMR spectra also revealed the presence of four olefinic functionalities at δC 118.9, 124.6, 136.1 and 146.7. The presence of an α,β-unsaturated-γ-lactone functionality was assigned based on NMR parallels with 1. From HMBC (Figure 1), a methyl unit (1.43, s; H3-20) was observed proximal to C-12 determined from correlations between C-12 (δC 84.0) and H3-20 (1.43, s), H-11 (5.56, d, J = 16.0 Hz), H-10 (5.42, ddd, J = 16.0, 10.5, 7.5 Hz), H-13 (2.07, td, J = 13.0, 4.5 Hz; 1.41, dd, J = 4 Hz, overlapped with H3-18). HMBC correlations (Figure 1) were also observed between C-7 (δC 59.0) and H-6 (1.77, m, 2H), H-5 (2.02, m; 2.38, dt, J = 4.5, 13.5 Hz), H3-19 (1.30, s), and H2-9 (2.25, m; 2.46, m), and C-8 (δC 59.2) and H-7 (2.53, dd, J = 5.0, 6.0 Hz), H2-9 (2.25, m; 2.46, m), H-10 (5.42), and H2-6 (1.77, m, 2H) indicating the same epoxide location as in 1 bridging C-7 and C-8. The olefinic proton signal at δH 5.56 (H-11, d, J = 16.0 Hz) showed an HMBC correlation with an oxygenated carbon at δC 84.0 (C-12), a methyl signal at δC 22.8 (C-20), and an olefinic carbon at δC 124.6 (C-10) establishing that the peroxyl and double bond functionalities are located at C-12 and C-10/C-11, respectively.
The combined spectral data indicated a cembranoid molecular framework containing a rare 12-peroxid-10-ene. This chemical configuration was confirmed by X-ray analysis (Figure 2) and HMBC correlations (Figure 1). The relative configuration of 2 was determined on the basis of coupling constants and NOESY experiments. The germinal coupling between H-2 and H-3 (16.0 Hz) and a NOESY correlation between H-2 and H3-18 indicated a trans configuration between the γ-lactone (H-2) and olefinic protons (H-3). The absolute stereochemistry of 2 was determined via CD analysis with the CD spectra (Figure 3) of 2 nearly equivalent with 1 and 4 establishing the same (R) configuration at C-2 [18,19,21,22]. Therefore, compound 2 was assigned to be 12-hydroperoxylsarcoph-10-ene (2).
Figure 3.

Circular dichroism (CD) spectra of 1–4.
Compound 3 was obtained as a color-less oil, +19.2 (c 0.1, CHCl3). The HR-FAB-MS showed an [M + Na]+ ion at m/z 339.19313 suggesting a molecular formula of C20H28O3Na that was supported by NMR data. Spectral data suggested that 3 was similar to the sarcophinone previously isolated from the soft coral Sarchophyton molle Tix [23], except for an up-field shift for H3-19 (δH 1.06) and an increase in its coupling constant (7.5 Hz) in comparison with sarcophinone H3-19 (δH 1.13, J = 6.4 Hz). This up-field shift for such a methyl attached to a methine carbon can be explained by an alternative stereochemistry since the β-configuration methyl group is down-field relative to the α-stereochemistry [23,24].
The location of the ketone carbonyl group at C-8 was determined from HMBC data that established clear correlations with H-8 (δH 2.44, m)/H3-19 (δH 1.06, d, J = 7.5), H-9 (δH 1.56 and 1.95, m, 2H), H-6 (δH 2.06 and 2.73, m, 2H), and H-5 (δH 2.66 and 2.76, m, 2H) (Figure 1). The absolute stereochemistry at C-2 was determined by CD analysis in which the spectrum was dominated by negative and positive Cotton effects {[θ]245 −1.9, [θ]220 +4.8} (Figure 3) due to the electronic transitions of the 2(5H)-furanone moiety [15]. These Cotton effects indicated a right-handed (P) helix for the five-membered α,β-unsaturated-γ-lactone ring. Similar CD spectra for 3 and sarcophine (5) show a common S configuration at C-2 [11,12,14,15]. Compound 3 was therefore identified as 8-epi-sarcophinone. There are two reports that have the same structure as 3 and are referred to as iso-sarcophinone [25,26]; however with an absence of spectral data, direct comparisons cannot be made. In a more comprehensive study of iso-sarcophinone by Su et al. [23] full 1H and 13C NMR data is provided and the reported compound is an epimer of 3 with the opposite stereochemistry at C-8; this epimer of 3 has also been named as iso-sarcophinone. Since only spectral data comparisons are possible for the Su et al. study [23] and the NMR data for compound 3 reported here are not consistent with iso-sarcophinone, we propose that iso-sarcophinone has not been isolated in the present study but instead a new 8-epi-sarcophinone as shown in 3. Whether 8-epi-sarcophinone was isolated and not appropriately named or iso-sarcophinone was isolated but incorrectly identified by Czarkie et al. [25] is uncertain with an absence of key spectral data. With this study, spectral data is now available for both iso-sarcophinone [23] and 8-epi-sarcophinone.
To examine the anti-cancer activity of characterized S. glaucum metabolites, individual components were assayed for inhibition of the phase I enzyme cytochrome P450 1A since the enzyme in combination with pro-carcinogens and/or oxidative stress can lead to DNA damage. Compounds 2, 3, and 4 were identified as inhibitors of Cyp1A activity (p < 0.01) with IC50 values of 2.7, 3.7 and 3.4 nM respectively (Figure 4), compared with the initial activity of β-naphthoflavone-stimulated cells. Assayed compounds 1 and 5 exhibited insignificant inhibition of Cyp1A activity (p > 0.05).
To examine induction of protective enzymes of oxidative stress by characterized S. glaucum metabolites, individual components were assayed for induction of glutathione-S-transferase activity, quinone reductase (QR) and epoxide hydrolase (mEH). GSTs are responsible for the detoxification of a wide range of substrates including xenobiotics as well as occupational and environmental carcinogens such as pesticides and polycyclic aromatic hydrocarbons [27]. Total GST activity was investigated in cultured Hepa1c1c7 cells. Forty eight hours after murine hepatoma cell culture incubation with 10 µg/mL of each metabolite, total GSTs activity was significantly induced by 2–3 (p < 0.01 and p < 0.05, respectively) (Figure 5A). While free thiols that serve as non-enzymatic antioxidants assisting in counteracting the deleterious effect of ROS were significantly elevated in cell cultures only when treated with 2 (10 µg/mL) (p < 0.05) (Figure 5A).
QR that is induced coordinately with other Phase II enzymes such as GSTs and contributes to quinone detoxification was investigated in murine hepatoma cell culture. After 48 h incubation, 2–3 resulted in a significant induction of QR activity (p < 0.01 and p < 0.05, respectively) (Figure 5B). In contrast, epoxide hydrolase mEH, an important metabolic enzyme that catalyzes the addition of water to alkene epoxides and arene oxides [28] was significantly elevated in cell cultures only when treated with 4 (10 µg/mL) (p < 0.05) (Figure 5C).
Figure 4.

Enzyme regulation of cancer metabolism by extracted soft coral components. Cytochrome P450 1A inhibition (assayed concentration 1 µg/mL, mean ± SD, n = 4). Asterisk (*) indicates significant different p < 0.05.
Figure 5.

Anti-initiating activity through the modulation of carcinogen metabolism. Effect of treatment with 10 µg/mL of each sample for 48 h on glutathione-S-transferase activity (bars) and non-enzymatic antioxidant activity GSH (circles) (A), quinone reductase (QR) (B) and epoxide hydrolase mEH (C) activities in Hepa1c1c7 cells. Data expressed as mean ± SD (n = 4).
3. Experimental Section
3.1. General Experimental Procedures
1H and 13C NMR spectra were recorded in CDCl3 on a Varian MercuryPlus 300 MHz and Varian Unity INOVA 500 spectrometer (300/500 MHz for 1H and 75/125 MHz for 13C, respectively). All chemical shifts (δ) are given in ppm units with reference to TMS as an internal standard and the coupling constants (J) are given in Hz. FAB-MS was performed on a Finnigan LCQ ion trap mass spectrometer and HR-FAB-MS experiments were performed on Fourier transform ion cyclotron mass spectrometer (Ion Spec, Varian). The spectra were recorded by infusion into the ESI (electrospray ionization) source using chloroform as the solvent. High performance liquid chromatography (HPLC) was performed on an Agilent pump equipped with an Agilent-G1314 variable wavelength UV detector at 254 nm and a semi-preparative reverse-phase column (Econosphere™, RP-C18, 5 μm, 250 × 4.6 mm). Optical rotation was determined at 589 nm (sodium D line) using a Perkin–Elmer-341 MC digital polarimeter; [α]D-values are given in the unit of 10 deg−1·cm2·g−1. CD was measured with an OLIS, DSM-10 UV/Vis CD.
Silica gel 60 (Merck, 230–400 mesh) and Sephadex LH-20 (Sigma) were used for column chromatography. Pre-coated silica gel plates (Merck, Kieselgel 60 F254, 0.25 mm) were used for TLC analyses. Spots were visualized by heating after spraying with 10% H2SO4.
3.2. Animal Material
Soft coral S. glaucum was collected from the Egyptian Red Sea coast of Hurghada in June, 2009. A voucher specimen (03RS24) was deposited in the National Institute of Oceanography and Fisheries, Marine Biological Station, Hurghada, Egypt.
3.3. Extraction and Separation
The frozen soft coral was chopped into small pieces (4 kg, wet weight) and extracted with ethyl acetate at room temperature (4 L × 5). The combined ethyl acetate extracts were concentrated to a brown gum. The dried EtOAc-soluble material (20.0 g) was subjected to gravity chromatography on silica gel column (6 × 120 cm) using n-hexane–EtOAc (gradient separation) into 8 fractions. Fraction 3 (2.2 g) eluted with n-hexane–EtOAc (8:1) was subjected to silica gel column separation to afford 5 (50 mg). The remaining samples of this fraction were collected and purified by Sephadex LH-20 using hexane–CHCl3–MeOH (7:4:0.5) followed by reverse phase HPLC using acetonitrile H2O (1:1) to afford 1 (35 mg), 2 (23 mg) and 3 (14 mg). Fraction 4 eluted with n-hexane–EtOAc (6:1) was re-purified on reverse phase HPLC using acetonitrile/H2O (50–100% H2O) 4 (9 mg).
11-hydroperoxylsarcoph-11(20)-ene (1): colorless crystal; = +12.6 (c 0.09, CHCl3); IR (KBr) νmax 3353, 3000, 1750, 1707, 1686, 1669, 1256 cm−1; 1H NMR and 13C NMR data, see Table 1; HR-FAB-MS [M + Na]+
m/z 371.18281 (41%) (calc. 371.18284, C20H28O5Na).
3.3.1. Single-Crystal X-ray Crystallography of 1
X-ray intensity data were measured on a Bruker Smart Apex II automated X-ray diffractometer equipped with a CCD detector. The frames were integrated with the Bruker SAINT Software package (Version 6) using a narrow-frame algorithm. Integration of the data using a monoclinic unit cell yielded a total of 4644 reflections to a maximum θ angle of 18.45° (1.12 Å resolution), of which 1364 were independent (average redundancy 3.405, completeness = 99.9%, Rint = 3.57%, Rsig = 3.55%) and 1257 (92.16%) had intensities greater than 2σ(F2). The final cell constants are based upon the refinement of the XYZ-centroids of 56 reflections with intensities greater than 20 σ(I) and 2θ values in the range 6.72° < 2θ < 23.48°. The calculated minimum and maximum transmission coefficients (based on crystal size) are 0.9274 and 0.9982, respectively.
The structure was solved and refined using the Bruker SHELXTL Software Package in the space group P21 (No. 4 in the International Tables for X-ray Crystallography [29]), with Z = 2 for the formula C20H28O5. The final anisotropic full-matrix least-squares refinement on F2 with 339 variables converged at R1 = 2.54% for the observed data (intensities greater than 4σ(F2)) and wR2 = 5.13% for all data, with a goodness-of-fit value of 1.075. The largest peak in the final difference electron density synthesis was 0.071 e−/Å3, and the largest hole was −0.079 e−/Å3, with an RMS deviation of 0.020 e−/Å3.
12-Hydroperoxylsarcoph-10-ene (2): colorless crystal; = −20.1 (c 0.1, CHCl3); IR (KBr) νmax 3353, 3000, 1750, 1707, 1686, 1669, 1256 cm−1; 1H NMR and 13C NMR data, see Table 1; HR-FAB-MS [M + Na]+
m/z 371.18293 (33%) (calc. 371.18290, C20H28O5Na).
3.3.2. Single-Crystal X-ray Crystallography of 2
X-ray intensity data were measured on a Bruker Smart Apex II automated X-ray diffractometer equipped with a CCD detector. The frames were integrated with the Bruker SAINT Software package (Version 6) using a narrow-frame algorithm. Integration of the data using an orthorhombic unit cell yielded a total of 9176 reflections to a maximum θ angle of 17.97° (1.15 Å resolution), of which 1332 were independent (average redundancy 6.889, completeness = 100%, Rint = 4.16%, Rsig = 2.43%) and 1255 (94.22%) had intensities greater than 2σ(F2). The final cell constants are based upon the refinement of the XYZ-centroids of 105 reflections with intensities greater than 20 σ(I) and 2θ values in the range 4.15° < 2θ < 36.10°. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.899. The structure was solved and refined using the Bruker SHELXTL Software Package in the space group P212121 (No. 19 in the International Tables for X-ray Crystallography [29]), with Z = 4 for the formula C20H28O5. The final anisotropic full-matrix least-squares refinement on F2 with 235 variables converged at R1 = 2.15% for the observed data (intensities greater than 4σ(F2)) and wR2 = 4.66% for all data, with a goodness-of-fit value of 1.042 and a data-to-parameter ratio of 5.7. The largest peak in the final difference electron density synthesis was 0.069 e−/Å3, and the largest hole was −0.079 e−/Å3, with an RMS deviation of 0.017 e−/Å3.
8-epi-Sarcophinone (3): colorless crystal; = +19.2 (c 0.1, CHCl3); IR (KBr) νmax 1730, 1700, 1410, 1230, 960 cm−1; 1H NMR and 13C NMR data, see Table 1; HR-FAB-MS [M + Na]+
m/z 339.19313 (100%) (calc. 339.19317, C20H28O5Na).
ent-Sarcophine (4): = −20.0 (c 0.03, CHCl3); lit. = −80.0 (c 0.3, CHCl3) [19].
(+)-Sarcophine (5): = +95.0 (c 0.5, CHCl3); lit. = +92 (c 1.0, CHCl3) [20].
3.4. Cell Culture
Murine hepatoma cells (Hepa1c1c7) was purchased from the American Type Culture Collection. Cells were cultured on Dulbeco’s Modified Eagle’s medium (DMEM). Media were supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, containing 100 U/mL penicillin G sodium, 100 U/mL streptomycin sulfate, and 250 ng/mL amphotericin B. Cells were maintained in humidified air containing 5% CO2 at 37 °C. The monolayer cells were harvested using trypsin/EDTA. All experiments were repeated four times, unless mentioned, and the data were represented as mean ± SD. The extract, fractions and compounds were dissolved in DMSO (99.9%) and diluted 1000 fold for each assay. In all cellular experiments, results were compared with DMSO-treated cells. All cell culture material was obtained from Cambrex, BioScience (Copenhagen, Denmark).
3.5. Evaluation of Carcinogen Metabolizing Enzymes
Cytochrome P450 1A (Cyp1A) activity was determined by the rate of dealkylation of 3-cyano-7-ethoxycoumarin (CEC) to the fluorescent 3-cyano-7-hydroxycoumarin based on Crespi et al. [30], and modified by Gerhäuser et al. [31]. Homogenates from cultured Hepa1c1c7 cells, induced with β-naphthoflavone (1 µg/mL final concentration) were used as a source of Cyp1A. The rate of CEC conversion was measured kinetically at excitation 408/20 nm and emission 460/40 nm by a microplate fluorescence reader (FluoStarOptima, BMG lab technologies, Durham, NC, USA). Inhibition of Cyp1A activity was calculated in comparison with the initial fluorescence of a complete reaction mixture with cell homogenate and buffer instead of the assay compound.
Hepa1c1c7 cells (1 × 106) were incubated with the compounds (10 µg/mL) for 48 h. Glutathione-S-transferase (GST) activity was measured in the cell lysate according to Habig et al. [32] and based on GST-catalyzed reaction between GSH and 1-chloro-2,4-dinitrobenzene that acts as an electrophilic substrate for GST. In the kinetic analysis, the absorbance was assessed at 340 nm. GSTs were normalized to the protein content as measured by bicinchoninic acid assay [33]. Quinone reductase (QR) activity was determined by measuring the reduction of 2,6-dichloroindophenol [34]. The specific QR activity was expressed as nmol of 2,6-dichloroindophenol reduced by 1 mg of protein within 1 min. Enzyme activity for mEH was assessed by the production rate of 7-(29,39-dihydroxy) propoxycoumarin (DHC) from 7-glycidoxycoumarin (GOC), as described by Inoue et al. [35]. The fluorescence intensity was measured at excitation 325 nm and emission 391 nm. DHC in methanol was used as a standard. The enzyme activity was expressed as µM DHC/min/mg protein.
3.6. Statistical Analysis
Data were analyzed by a one-way ANOVA followed by a post hoc Turkey test; p < 0.05 indicated statistical significance.
4. Conclusions
Three new (1–3) and two known cembranolides (4 and 5) were isolated and chemically characterized from the Red Sea soft coral Sarcophyton glaucum. The absolute configuration of 1 and 2 were confirmed by X-ray diffraction and circular dichroism (CD) analyses. Compounds 2 and 3 were found to be promising inhibitors of cytochrome P450 1A activity as well as inducers of GST and QR activity in in vitro assays.
Acknowledgements
Financial assistance was provided in part by a grant from the Robert Welch Foundation (D-1478).
Supplementary Files
PDF-Document (PDF, 13611 KB)
Footnotes
Samples Availability: Available from the authors.
References
- 1.Edwards A.J., Head S.M. Key Environments-Red Sea. Pergamon Press; Oxford, UK: 1987. p. 440. [Google Scholar]
- 2.Roethle P.A., Trauner D. The chemistry of marine furanocembranoids, pseudopteranes, gersolanes, and related natural products. Nat. Prod. Rep. 2008;25:298–317. doi: 10.1039/b705660p. [DOI] [PubMed] [Google Scholar]
- 3.Blunt J.W., Copp B.R., Hu W.P., Munro M.H.G., Northcote P.T., Prinsep M.R. Marine natural products. Nat. Prod. Rep. 2008;25:35–94. doi: 10.1039/b701534h. [DOI] [PubMed] [Google Scholar]
- 4.Gross H., Wright A.D., Beil W., Koenig G.M. Two new bicyclic cembranolides from a new Sarcophyton species and determination of the absolute configuration of sarcoglaucol-16-one. Org. Biomol. Chem. 2004;2:1133–1138. doi: 10.1039/b314332e. [DOI] [PubMed] [Google Scholar]
- 5.Sawant S., Youssef D., Mayer A., Sylvester P., Wali V., Arant M., El Sayed K. Anticancer and anti-inflammatory sulfur-containing semisynthetic derivatives of sarcophine. Chem. Pharm. Bull. 2006;54:1119–1123. doi: 10.1248/cpb.54.1119. [DOI] [PubMed] [Google Scholar]
- 6.Sawant S.S., Youssef D.T.A., Reiland J., Ferniz M., Marchetti D., El Sayed K.A. Biocatalytic and antimetastatic studies of the marine cembranoids sarcophine and 2-epi-16-deoxysarcophine. J. Nat. Prod. 2006;69:1010–1013. doi: 10.1021/np050527v. [DOI] [PubMed] [Google Scholar]
- 7.Wahlberg I., Eklund A.M. Cembranoids, pseudopteranoids, and cubitanoids of natural occurrence. Prog. Chem. Org. Nat. 1992;59:141–294. [Google Scholar]
- 8.El Sayed K.A., Hamann M.T., Waddling C.A., Jensen C., Lee S.K., Dunstan C.A., Pezzuto J.M. Structurally novel bioconversion products of the marine natural product sarcophine effectively inhibit JB6 cell transformation. J. Org. Chem. 1998;63:7449–7455. doi: 10.1021/jo9813134. [DOI] [PubMed] [Google Scholar]
- 9.Grote D., Soliman H.S.M., Shaker K.H., Hamza M., Seifert K. Cembranoid diterpenes and a briarane diterpene from corals. Nat. Prod. Res. 2005;20:285–291. doi: 10.1080/14786410500087657. [DOI] [PubMed] [Google Scholar]
- 10.Neeman I., Fishelson I., Kashman Y. Sarcophine—a new toxin from the soft coral Sarcophyton glaucum (alcyonaria). Toxicon. 1974;12:593–598. doi: 10.1016/0041-0101(74)90192-5. [DOI] [PubMed] [Google Scholar]
- 11.Erman A., Neeman I. Inhibition of phosphofructokinase by the toxic cembranolide sarcophine isolated from the soft-bodied coral Sarcophyton glaucum. Toxicon. 1976;15:207–215. doi: 10.1016/0041-0101(77)90046-0. [DOI] [PubMed] [Google Scholar]
- 12.El Sayed K.A., Orabi K.Y., Dunbar D.C., Hammann M.T., Avery M.A., Sabnis Y.A., Mossa J.S., El feraly F.S. Transformation of lactone to lactam in sarcophine and antimalarial activity of resulting N-substituted azasarcophines. Tetrahedron. 2002;58:3699–3708. [Google Scholar]
- 13.Fahmy H., Khalifa S.I., Konoshima T., Zjawiony J.K. An improved synthesis of 7,8-epoxy-1,3,11-cembratriene-15R(α),16-diol, a cembranoid of marine origin with a potent cancer chemopreventive activity. Mar. Drugs. 2004;2:1–7. [Google Scholar]
- 14.Katsuyama I., Fahmy H., Zjawiony J.K., Khalifa S.I., Kilada R.W., Konoshima T., Takasaki M., Harakuni T. Semisynthesis of new sarcophine derivatives with chemopreventive activity. J. Nat. Prod. 2002;65:1809–1814. doi: 10.1021/np020221d. [DOI] [PubMed] [Google Scholar]
- 15.Abouzied A.M., Sawant S.S., Sylvester P.W., Avery M.A., Desai P., Youssef D.T.A., El Sayed K.A. Bioactive rearranged and halogenated semisynthetic derivatives of the marine natural product sarcophine. J. Nat. Prod. 2004;67:2017–2023. doi: 10.1021/np0497393. [DOI] [PubMed] [Google Scholar]
- 16.Hong W.K., Sporn M.B. Recent advances in chemoprevention of cancer. Science. 1997;278:1073–1077. doi: 10.1126/science.278.5340.1073. [DOI] [PubMed] [Google Scholar]
- 17.Arif J.M., Al-Hazzani A.A, Kunhi M., Al-Khodairy F. Novel marine compounds: Anticancer or genotoxic? J. Biomed. Biotechnol. 2004;2:93–98. doi: 10.1155/S1110724304307060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin S.W., Shi Y.P., Li X.M., Wang B.G. A novel hydroperoxyl-substituted cembranolide diterpene from marine soft coral Lobophytum crassum. Chin. Chem. Lett. 2005;1:1489–1491. [Google Scholar]
- 19.Yao L.-G., Liu H.L., Guo Y.W., Mollo E. New cembranoids from the hainan soft coral Sarcophyton glaucum. Helv. Chim. Acta. 2009;92:1085–1091. [Google Scholar]
- 20.Bowden B.F., Coll J.C., Mitchell S.J. Studies of Australian soft corals. XVIII. Further cembranoid diterpenes from soft corals of the genus Sarcophyton. Aust. J. Chem. 1980;33:879–884. doi: 10.1071/CH9800879. [DOI] [Google Scholar]
- 21.Kashman Y., Zadock E., Neeman I. New cembrane derivatives of marine origin. Tetrahedron. 1974;30:3615–3620. [Google Scholar]
- 22.Kamel H.N., Ferreira D., Garcia-Fernandez L.F., Slattery M. Cytotoxic diterpenoids from the hybrid soft coral Sinularia maxima × Sinularia polydactyla. J. Nat. Prod. 2007;70:1223–1227. doi: 10.1021/np070074p. [DOI] [PubMed] [Google Scholar]
- 23.Su J.Y., Yan S.J., Zeng L.M. New diterpene lactone from the soft coral Sarcophyton molle Tix. Dur. Chem. J. Chin. Univ. 2001;22:1515–1517. [Google Scholar]
- 24.Nii K., Tagami K., Kijima M., Munakata T., Ooi T., Kusumi T. Acid-catalyzed reactions of sarcophytoxide, a marine cembranoid: An apparently enantio-directive reaction, unusual products and stereochemical reconsideration of epoxide-ketone rearrangement. Bull. Chem. Soc. Jpn. 2008;81:562–573. [Google Scholar]
- 25.Czarkie D., Carmely S., Groweiss A., Kashman Y. Attempted acid-catalyzed transannular reactions in the cembranoids. Tetrahedron. 1985;41:1049–1056. [Google Scholar]
- 26.Swapnali S.S., Sylvester P.W., Avery M.A., Desai P., Youssef D.T.A., El Sayed K.A. Bioactive rearranged and halogenated semisynthetic derivatives of the marine natural product sarcophine. J. Nat. Prod. 2004;67:2017–2023. doi: 10.1021/np0497393. [DOI] [PubMed] [Google Scholar]
- 27.Rooseboom M., Commandeur J.N.M., Vermeulen N.P.E. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol. Rev. 2004;56:53–102. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 28.Cannady E.A., Dyer C.A., Christian P.J., Sipes G., Hoyer P.B. Expression and activity of microsomal epoxide hydrolase in follicles isolated from mouse ovaries. Toxicol. Sci. 2002;68:24–31. doi: 10.1093/toxsci/68.1.24. [DOI] [PubMed] [Google Scholar]
- 29.Henry N.F.M., Lonsdale K. International Tables for X-ray Crystallography. Vol. 1. Kynoch Press; Birmingham, UK: 1952. p. 78. [Google Scholar]
- 30.Crespi C.L., Miller V.P., Penman B.W. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal. Biochem. 1997;248:188–190. doi: 10.1006/abio.1997.2145. [DOI] [PubMed] [Google Scholar]
- 31.Gerhäuser C., Klimo K., Heiss E., Neumann I., Gamal-Eldeen A., Knauft J., Liu J.-U., Sitthimonchai S., Frank N. Mechanism-based in vitro screening of potential cancer chemopreventive agents. Mutat. Res. 2003;523–524:163–172. doi: 10.1016/s0027-5107(02)00332-9. [DOI] [PubMed] [Google Scholar]
- 32.Habig W.H., Pabst M.J., Jakoby W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 33.Smith P.K., Krohn R.I., Hermanson G.T., Mallia A.K., Gartner F.H., Provenzano M.D., Fujimoto E.K., Goeke N.M., Olson B.J., Klenk D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 34.Yu R., Mandlekar S., Lei W., Fahl W.E., Tan T.-H., Kong A.T. p38 Mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J. Biol. Chem. 2000;275:2322–2327. doi: 10.1074/jbc.275.4.2322. [DOI] [PubMed] [Google Scholar]
- 35.Inoue N., Yamada K., Imai K., Aimoto T. Sex hormone-related control of hepatic EH activities in mice. Biol. Pharm. Bull. 1993;16:1004–1007. doi: 10.1248/bpb.16.1004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDF-Document (PDF, 13611 KB)