

Abstract
Features of end-stage renal disease such as oxidative stress, inflammation, hypertension, and dyslipidemia are associated with accelerated atherosclerosis and increased risk of death from cardiovascular disease. By inhibiting the formation and increasing the disposal of oxidized lipids, HDL exerts potent antioxidant and anti-inflammatory actions. Given that apolipoproteinA-1 can limit atherosclerosis, we hypothesized that an apolipoproteinA-1 mimetic peptide, 4F, may reduce the proinflammatory properties of LDL and enhance the anti-inflammatory properties of HDL in uremic plasma. To test this, plasma from each of 12 stable hemodialysis patients and age-matched control subjects was incubated with 4F or vehicle. The isolated HDL and LDL fractions were added to cultured human aortic endothelial cells to quantify monocyte chemotactic activity, thus measuring their pro- or anti-inflammatory index. The LDL from the hemodialysis patients was more pro-inflammatory and their HDL was less anti-inflammatory than those of the control subjects. Pre-incubation of the plasma from the hemodialysis patients with 4F decreased LDL pro-inflammatory activity and enhanced HDL anti-inflammatory activity. Whether 4F or other apolipoproteinA-1 mimetic peptides will have any therapeutic benefit in end-stage renal disease will have to be examined directly in clinical studies.
Keywords: anti-inflammatory agents, atherosclerosis, cardiovascular disease, inflammation, lipid disorders, oxidative stress
Chronic kidney disease (CKD) is associated with accelerated atherosclerosis and increased risk of death from cardiovascular disease.1, 2, 3, 4 This is primarily due to oxidative stress, inflammation, hypertension, and dyslipidemia, which are the common features of advanced CKD.5, 6, 7, 8, 9, 10, 11 Oxidative stress and inflammation drive plaque formation by promoting low-density lipoprotein (LDL) oxidation, monocyte adhesion, infiltration, differentiation and foam cell transformation in the artery wall, and by limiting high-density lipoprotein (HDL)-mediated reverse cholesterol transport.12, 13, 14, 15, 16, 17 HDL protects against plaque formation and progression by mediating reverse cholesterol transport and by exerting potent antioxidant, anti-inflammatory and antithrombotic actions.18, 19, 20, 21, 22, 23 However, in the presence of systemic inflammation, HDL is transformed to a pro-oxidant and proinflammatory agent.24, 25, 26 Plasma apolipoprotein A-1 (apoA-1), the principal apolipoprotein constituent of HDL, and HDL-cholesterol content are significantly reduced27, 28, 29, 30, 31, 32 and the antioxidant activity of HDL is markedly impaired in patients with end-stage renal disease (ESRD).33, 34 The reduction in plasma HDL concentration and HDL antioxidant activity can contribute to atherogenic diathesis by limiting reverse cholesterol transport in this population. Overexpression of human apoA-1 (a protein comprising 243 amino acids) or intravenous administration of apoA-I Milano has been shown to ameliorate atherosclerosis in experimental animals and humans.35, 36, 37, 38 However, widespread clinical application of apoA-1 has been hampered by its limited supplies, formidable cost and lack of mass-production capability. In an attempt to overcome these limitations, a series of synthetic short apoA-1 mimetic peptides have been developed, which have proven to be highly effective in attenuating atherosclerosis in experimental animals. Comprehensive assessment of the physical–chemical characteristics of these peptides has revealed the critical role of the hydrophobic region in their biological activity.
Administration of the apoA-1 mimetic peptide, 4F, has been shown to significantly improve HDL function in mice and monkeys,39, 40 reduce the size and macrophage content of atherosclerotic plaques in aged mice,41, 42 improve vascular function and reduce endothelial damage in other rodents.43, 44, 45 In addition, use of apoA-1 I mimetic peptides has been effective in a wide range of inflammatory conditions in experimental animals.46, 47, 48 The beneficial actions of apoA-1 I mimetic peptides are largely related to their ability to remove oxidation products from lipoproteins and cell membranes and restore structure and function of LDL and HDL.45
In view of the documented LDL and HDL abnormalities in CKD and demonstrated efficacy of apoA-1 I mimetic peptides in experimental animals, we hypothesized that ApoA-1 I mimetic peptide may reduce the proinflammatory properties of LDL and enhance anti-inflammatory properties of HDL in uremic plasma. To test this hypothesis, plasma from 12 ESRD patients and 12 control individuals were treated with 4F or vehicle, HDL and LDL fractions were then isolated and added to cultures of human aortic endothelial cells and the resulting monocyte chemotactic activity was quantified.
RESULTS
General data
General data are shown in Table 1. The ESRD group showed marked elevations of plasma creatinine and urea nitrogen and a significant reduction of blood hemoglobin and plasma HDL-cholesterol level and a moderate reduction of total cholesterol concentration.
Table 1.
Age, gender, serum creatinine, urea nitrogen, total cholesterol, HDL-cholesterol, and Hgb concentrations in the ESRD and control groups, as well as Kt/V in the ESRD group
| Control | ESRD | |
|---|---|---|
| Age | 47.7±3.2 | 52.7±4.3 |
| Male/female | 6/6 | 5/7 |
| Creatinine (mg per 100 ml) | 0.8±0.1 | 8.81±0.65*** |
| BUN (mg per 100 ml) | 13±0.8 | 68.3±4.1*** |
| Hgb (g per 100 ml) | 14.4±0.41 | 11.1±0.33*** |
| Total cholesterol (mg per 100 ml) | 160±1.6 | 137±6.6 |
| HDL-cholesterol (mg per 100 ml) | 46.6±2.4 | 38.0±1.7* |
| Kt/V | NA | 1.54±0.06 |
BUN, blood urea nitrogen; ESRD, end-stage renal disease; HDL, high-density lipoprotein; Hgb, blood hemoglobin; NA, not applicable.
P<0.05,
P<0.005.
Markers of oxidative stress and inflammation
The ESRD group showed marked elevations of plasma, tumor necrosis factor-α, interleukin (IL)-6, and IL-8 (Table 2) and significant increases in plasma malondialdehyde (MDA) and carbonylated proteins (Table 2), pointing to the presence of systemic oxidative stress and inflammation.
Table 2.
Plasma concentrations of MDA, protein carbonyl, IL6, IL-8, and TNF-α in the ESRD and control groups
| Control | ESRD | |
|---|---|---|
| MDA (μmol/l) | 1.09±0.09 | 1.87±0.07*** |
| Protein carbonyl content (nmol/mg) | 0.49±0.82 | 10.8±3.3* |
| IL-6 (pg/ml) | 1.43±0.36 | 5.80±1.4* |
| TNF-α (pg/ml) | 2.01±0.33 | 10.64±1.4*** |
| IL-8 (pg/ml) | 1.87±0.38 | 5.70±0.34*** |
ESRD, end-stage renal disease; IL, interleukin; MDA, malondialdehyde; TNF-α, tumor necrosis factor.
P<0.05,
P<0.005.
ApoA-I, LCAT, and antioxidant enzymes
The reduction of HDL-cholesterol level was accompanied by a marked reduction of serum apoA-1 and lecithin:cholesterol acyltransferase (LCAT) concentrations (Table 3). This was also accompanied by significant reductions of plasma paraoxonase and glutathione peroxidase (GPX) activities in the ESRD group (Table 3).
Table 3.
Plasma concentrations of apo-A1 and LCAT and activities of paraoxonase and GPX in the ESRD and control groups
| Control | ESRD | |
|---|---|---|
| ApoA-I (nmol/mg) | 158.2±30.3 | 72.2±7.6* |
| LCAT (pg/ml) | 7.6±0.47 | 4.7±0.54*** |
| Paraoxonase activity (pg/ml) | 143.1±17.3 | 91.2±7.4* |
| GPX activity (pg/ml) | 531.0±63.7 | 227.5±13.4*** |
ESRD, end-stage renal disease; ApoA-1, apolipoprotein-A1; GPX, glutathione peroxidase; LCAT, lecithin cholesterol acyltransferase.
P<0.05,
P<0.005.
HDL antioxidant activity
As expected, addition of normal HDL lowered the pro-oxidant activity of the standard oxidized LDL substrate used in this assay. In contrast, HDL from ESRD patients failed to lower the pro-oxidant activity of the standard oxidized LDL, and on average increased it (Figure 1).
Figure 1.
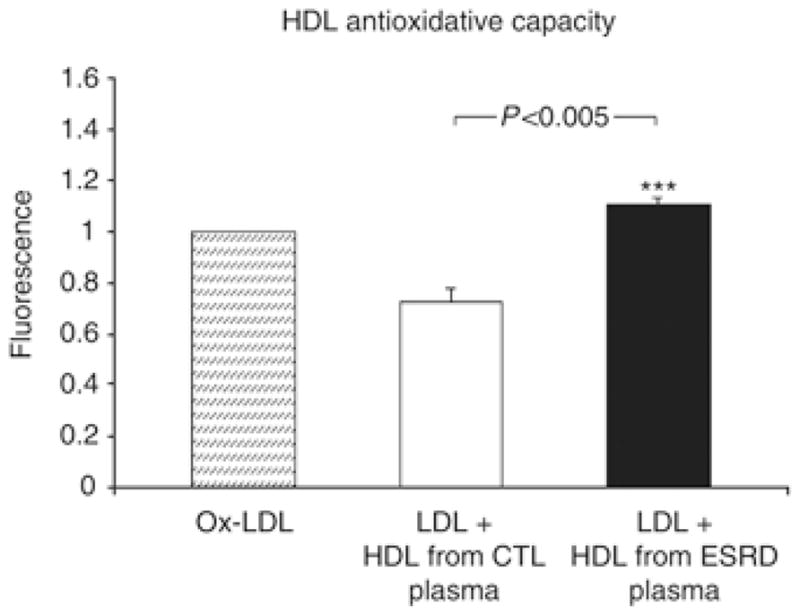
High-density lipoprotein antioxidative capacity in the normal control and ESRD groups. Oxidized LDL (25 μl, 250 μg/ml) was incubated at room temperature for 2 h alone (Ox-LDL) or together with HDL from control individuals and ESRD patients. Fluorescence intensity was determined after 2 h of incubation at room temperature as described in the Materials and Methods section. *P<0.005 ESRD versus control HDL. CTL, control; ESRD, end-stage renal disease.
LDL and HDL inflammatory activity
Low-density lipoprotein isolated from the ERSD patients was markedly proinflammatory compared with LDL isolated from the control group (Figure 2). Treatment of ERSD plasma in vitro with the apoA-1 mimetic peptide 4F significantly reduced the inflammatory properties of the ERSD LDL (Figure 2).
Figure 2.
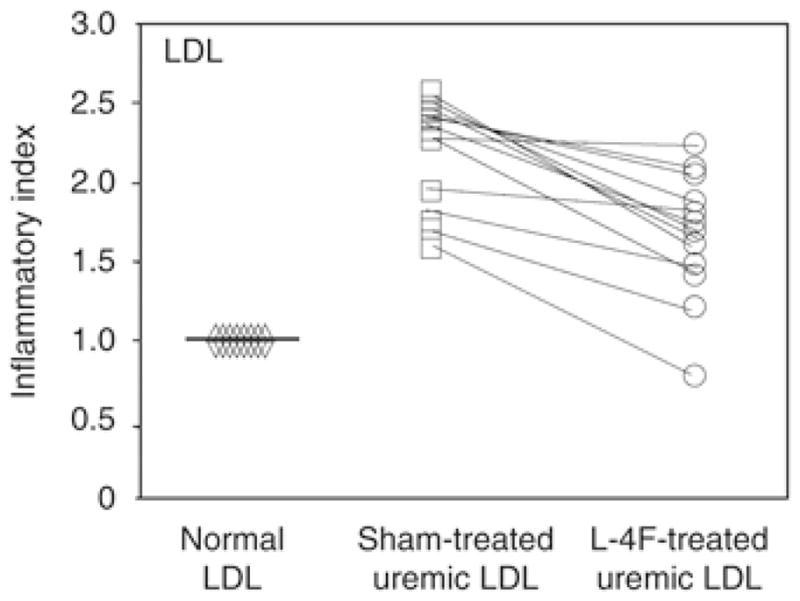
The inflammatory index determined after addition of LDL was significantly improved after treatment with apoA-I mimetic peptides. The values for the monocyte chemotactic activity obtained with the control LDL (diamonds) were normalized to 1.0 and used as the basis for expression of values obtained with the patients’ vehicle- (squares) or L-4F-treated (circles) LDL. P<0.005 ESRD versus control LDL.
High-density lipoprotein isolated from ERSD plasma was highly proinflammatory and was markedly less proinflammatory after treatment of the ERSD plasma with 4F in vitro (Figure 3). HDL isolated from ERSD plasma was proinflammatory in both diabetic and non-diabetic patients (data not shown), suggesting a dominant role of ESRD. To determine the effect of L-4F on HDL-inflammatory index of normal individuals, we prepared plasma from an additional group of 10 normal healthy individuals (five male and five female, aged 47±11.7 years). These samples were sham treated or treated with L-4F in vitro and the HDL-inflammatory index was determined as described in Materials and Methods. As shown in Figure 4, the untreated HDL in these normal healthy individuals was anti-inflammatory (HDL-inflammatory index=0.43±0.05) and was rendered more anti-inflammatory after treatment with L-4F in vitro (HDL-inflammatory index=0.20±0.04).
Figure 3.
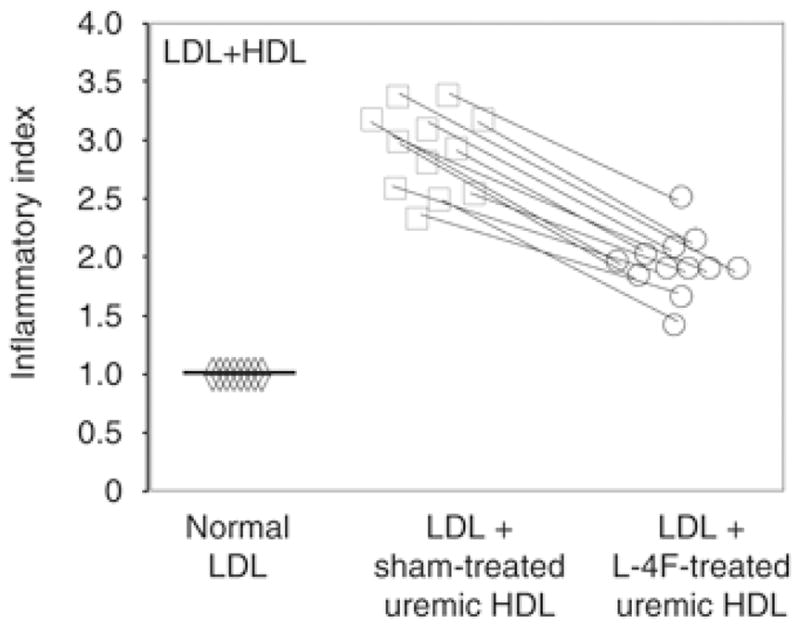
HDL-inflammatory index (HII) was significantly improved after treatment with ApoA-I mimetic peptides. The values for the monocyte chemotactic activity obtained with the control LDL (diamonds) were normalized to 1.0 and used as the basis for expression of activity obtained with LDL plus the patients’ vehicle- (squares) or L-4F-treated (circles) HDL. P<0.005 ESRD versus control HDL.
Figure 4.
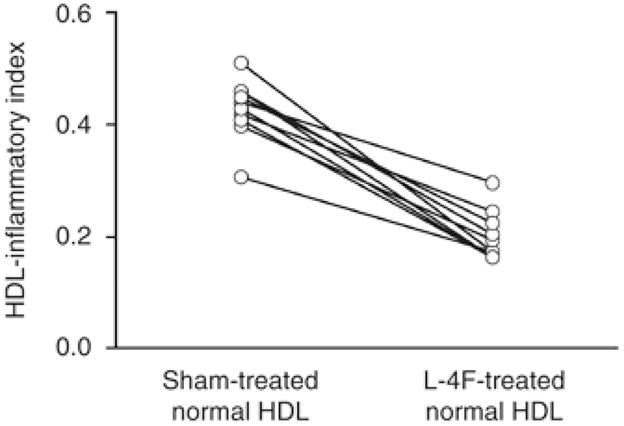
L-4F improves the HDL-inflammatory index of normal plasma. Plasma was obtained from 10 normal healthy individuals aged 47.2±11.7 years (five male and five female individuals) and was sham-treated or treated with L-4F and the HDL-inflammatory index was determined as described in the Materials and Methods section.
DISCUSSION
As expected plasma HDL-cholesterol concentration was significantly reduced in ESRD patients as compared with that found in the control group. This was associated with marked reductions of plasma apoA-1 and LCAT concentrations. LCAT is a key HDL-associated enzyme, which catalyzes conversion of cell-derived free cholesterol to cholesterol ester on the surface of HDL. This process depends on the dual function of LCAT as phopholipase-2, which catalyzes hydrolysis and release of the SN-2 fatty acid in the phospholipid molecule, and as Acyl-CoA cholesterol acyltransferase, which catalyzes esterification of free cholesterol with the fatty acid generated by the latter reaction.49 LCAT-mediated conversion of amphipathic-free cholesterol to hydrophobic cholesterol ester results in the shift of cholesterol from the surface into the core of HDL. This process is critical for maintaining favorable gradient for maximal uptake of cholesterol by HDL, maturation of lipid-poor nascent HDL to cholesterol ester-rich HDL-2, and efficiency of reverse cholesterol transport. Thus, the observed LCAT deficiency contributes to diminished HDL-cholesterol content, impaired HDL maturation, and defective reverse cholesterol transport in patients with advanced CKD. The reduction of plasma LCAT concentration found in the ESRD patients used in this study extends the findings of earlier studies, which showed diminished plasma LCAT enzymatic activity in ESRD patients50, 51, 52, 53 and downregulation of hepatic LCAT gene expression in rats with chronic renal failure.50
The reduction in HDL concentration in the ESRD group was compounded by severe reduction of its antioxidant capacity confirming recent studies.33, 34 This was associated with marked reduction of paraoxonase and GPX enzyme activity in the study population. Paraoxonase and GPX are major HDL-associated antioxidant enzymes that catalyze reduction of oxidized lipids and hydroperoxides, and as such their deficiency contributes to impaired antioxidant capacity of HDL in ESRD patients. In addition, acquired LCAT deficiency in ESRD can contribute to reduction of HDL antioxidant capacity. This is because by hydrolysis of SN-2 fatty acids, LCAT removes oxidized fatty acids in oxidatively modified phospholipids, which could otherwise initiate/sustain oxidation-chain reaction and exert proinflammatory and platelet-activating factor-like activity.
The ESRD group showed a marked reduction of HDL anti-inflammatory activity as evidenced by impaired HDL-mediated inhibition of LDL-induced monocyte chemotactic activity. The observed defect cannot be attributed to low plasma HDL level in the ESRD group, as equal amounts of isolated HDL were used in these in vitro experiments. Consequently, the observed HDL dysfunction must be due, primarily to qualitative abnormalities. Systemic inflammation has been shown to lower antioxidant and anti-inflammatory activity of HDL and transform HDL to a pro-oxidant, proinflammatory agent known as acute-phase HDL.24, 25, 26 Oxidative stress and inflammation are nearly constant features of advanced CKD,6, 8, 9, 10 as evidenced by increased levels of inflammatory mediators (IL-6, IL-8, and tumor necrosis factor-α) and markers of oxidative stress (Protein carbonyls and MDA) in the ESRD patients used here. The prevailing inflammatory state likely contributes to the observed reduction of HDL anti-inflammatory function in the ESRD population. This could lead to a vicious cycle in which the underlying inflammation and oxidative stress induce HDL dysfunction and are aggravated by it. In addition to uptake of surplus cholesterol, HDL removes intact and oxidized phospholipids from the macrophages and resident cells in the artery wall.53, 54 Consequently, HDL carries the bulk of oxidized phospholipids and lipoperoxides in the plasma55 for enzymatic processing and ultimate disposal in the liver. Oxidized phospholipids and lipoperoxides participate in a vicious cycle of oxidative stress and inflammation through lipid peroxidation chain reaction and activation of lectin-like oxidized LDL receptor, scavenger receptor A-1, and oxidized phospholipid receptor.56, 57, 58, 59 Consequently by uptake, processing and disposal of lipoperoxides, and oxidized phospholipids, HDL plays an important role in mitigating oxidative stress and inflammation. However when present, systemic inflammation leads to inactivation of the HDL-associated antioxidant enzymes and dramatic increase in the HDL burden of oxidized lipids and modified proteins. These modifications result in marked reduction of HDL antioxidant and anti-inflammatory capacity and even its conversion to a proinflammatory agent.24, 25, 26 In addition, modification of apoA-1 by reactive oxygen species impairs its interaction with ATP-binding cassette transporter A-1 (ABCA-1) and thereby diminishes HDL-mediated cholesterol efflux.60
The ESRD patients showed a marked increase in proinflammatory activity of LDL, compounding the effects of the associated HDL deficiency and dysfunction. This phenomenon is most likely due to previously demonstrated elevation of plasma level of oxidized LDL in this population.6, 8, 10 Increased LDL oxidation in ESRD maybe caused by the prevailing oxidative stress and triglyceride enrichment of LDL, which heightens its vulnerability to oxidation. Triglyceride enrichment of LDL in chronic renal failure is due to lipoprotein lipase and hepatic lipase deficiencies, which lead to impaired VLDL metabolism and formation of oxidation-prone, triglyceride-rich LDL particles in humans and experimental animals.61, 62 Increased lipoprotein oxidation and impaired HDL function and structure are coupled with upregulation of influx pathway and accumulation of lipids in the arterial wall of animals with CKD.63
Treatment of plasma samples from the ESRD patients with the potent apoA-1 mimetic peptide, 4F, resulted in a marked increase in HDL anti-inflammatory activity and significant reduction of LDL proinflammatory activity. Although the improvement following 4F treatment was highly significant, treatment of most samples failed to return the values completely to normal. As shown in Figure 4, HDL from normal healthy donors was anti-inflammatory and was even more anti-inflammatory after treatment with L-4F in vitro. The HDL-inflammatory index values in the sham-treated plasma from ERSD patients were on the order 2–3; after L-4F treatment the values for the ERSD patients were on the order of 1.5–2.0. This dramatically contrasts with the normal healthy individuals who had HDL-inflammatory index values of −0.4 with sham treatment and −0.2 with L-4F treatment. Thus, L-4F treatment in vitro was effective in both ERSD and normal plasma despite markedly different HDL-inflammatory properties. This suggests that L-4F treatment is removing or inactivating substances that are present in both plasmas but is likely present in higher concentrations in the ERSD patients. The most likely candidates for these substances are proinflammatory oxidized lipids.64 4F is an amphipathic 18-amino acid apoA-1 mimetic peptide, which is active at nanomolar concentrations in the presence of large molar concentrations of apoA-1 in vivo. Detailed study of the physical–chemical characteristics of the apoA-1 mimetic peptides has illustrated the critical role of the hydrophobic region of the peptide in its bioactivity. The favorable effects of the apoA-1 mimetic peptide are mediated by its remarkable ability to preferentially remove oxidation products from lipoproteins and cell membranes and thereby restore HDL and LDL function and structure, improve cell function, and attenuate inflammation.45, 64 ApoA-1 mimetic peptides have been shown to reduce atherosclerosis and attenuate inflammation in experimental animals without significantly changing plasma lipid levels.45 These observations illustrate that the anti-inflammatory properties of HDL are as important as the levels of HDL-cholesterol and that strategies aimed at improving HDL function may be effective in the treatment of atherosclerosis and other chronic inflammatory conditions.
In conclusion, oxidative stress and inflammation in ESRD are associated with marked reduction of HDL antioxidant/anti-inflammatory activities and heightened LDL proinflammatory properties. In vitro, these abnormalities were greatly improved by treatment with the apoA-1 mimetic peptide, 4F. Clinical studies are needed to explore the efficacy of 4F therapy in ESRD patients.
MATERIAL AND METHODS
Patients
The study protocol was approved by the Institutional Review Board of the University of California, Irvine and completed with the assistance of the University of California, Irvine General Clinical Research Center. Written informed consent was obtained from all participants.
Twelve stable patients with ESRD maintained on hemodialysis for a minimum of 3 months at the dialysis center of University of California, Irvine Medical Center were recruited for the study. Hemodialysis therapy was performed thrice weekly using cellulose triacetate dialyzers. Individuals with evidence of acute or chronic infection, or acute intercurrent illnesses were excluded. Among the study patients, three were receiving HMG-CoA reductase inhibitors and two were receiving either an AT1 receptor blocker or an ACE inhibitor. All patients were receiving phosphate binders and multivitamin preparations.
Control group
A group of 12 normal age-matched control individuals served as controls. Individuals exhibiting acute or chronic infection, acute intercurrent illnesses, hypertension, diabetes, malignancy, psychiatric disorders, or those requiring medications were excluded.
Blood collection
Blood samples were obtained by venipuncture in the control group and from the vascular access before initiation of dialysis procedure in the ESRD patients. Samples were collected in heparinized tubes and centrifuged immediately, plasma was separated and processed for various assays.
HDL isolation for assays without cells
High-density lipoprotein-containing supernatants were prepared by removal of the apolipoprotein B-containing proteins as described earlier.22, 33, 34 Briefly, 200 l of plasma were incubated with 40 l of the Magnetic Bead Reagent (Polymedco Inc., Chicago, IL) at room temperature for 5 min with gentle mixing. The mixture was placed on the magnetic particle concentrator for 3 min and the HDL-containing supernatant was removed. The HDL-cholesterol content was measured by a colorimetric assay (Thermo Electron Inc., Waltham, MA) using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Paraoxonase activity assay
Serum paraoxonase activity was measured using an Arylesterase/paraxonase assay kit purchased from ZeptoMetrix Inc. (Buffalo, NY) according to the manufacturer’s specifications. Arylesterase activity was measured at 25°C with a cuvette path length of 1 cm. Change in absorbance was observed on a SpectraMax M5 for a total of 80 s. Data from the first 20 s were not included in the calculations.
Glutathione peroxidase activity
Serum GPX activity was measured using a kit purchased from ZeptoMetrix Inc., according to the manufacturer’s specifications. GPX activity was measured at 25°C with a cuvette path length of 1 cm. Change in absorbance was observed on a SpectraMax M5 for a total of 120 s. Data from the first 40 s were not included in the calculations.
Apolipoprotein A-1 concentration
Apolipoprotein A-1 concentration was determined using an ELISA kit purchased from Alpco Diagnostics (Salem, NH) and a SpectraMax M5 plate reader as specified in the manufacturer’s protocol.
LCAT concentration
Plasma LCAT protein concentration was determined using an EIA kit from Alpco Diagnostics, following the manufacturer’s protocol and a SpectraMax M5 plate reader.
HDL antioxidant activity assay
This assay is based on the ability of HDL to reverse oxidation of LDL using 2,7-dichlorofluoresceindiacetate (H2DCFDA; Invitrogen Inc., Carlsbad, CA, USA) and a copper-oxidized LDL preparation (Kalen Biomedical Inc., Savage, MD). DCFH-DA was dissolved in fresh methanol at 2.0 mg/ml and incubated at room temperature in dark for 30 min to release DCFH. Oxidized LDL (25 μl of a 250 μg/ml solution) alone and a mixture of oxidized LDL plus 6.4 μl of individual HDL samples (normalized at 100 μlg/ml) were placed in a round-bottom, black polypropylene microtiter plate and incubated at 37°C on nutating mixer (Fisher Scientific, Pittsburgh, PA) set at 24 r.p.m. for 1 h. DCFH solution was then diluted 10-fold and 25 μl of this working solution was added to each well (including to wells without lipoproteins), mixed and incubated in dark on a nutating mixer set at 24 r.p.m 37°C for 2 h. Interaction with lipid oxidation products converts DCFH to DCF, which produces intense fluorescence. Fluorescence intensity was determined using a Fluorescent Plate Reader (SpectraMax M5 plate reader, Molecular Devices) set at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. A sensitivity level of 0.1 and slit widths of 2.5 and 10 nm were used for excitation and emission, respectively.
Chemotactic activity assays
These assays were performed using the methods described earlier.64, 65 Plasma from ESRD patients was treated with 1 μg/ml of L-4F or vehicle for 15 min at 37°C with gentle mixing. This concentration of L-4F (0.43 nM) was earlier found to produce a maximal effect in vitro.64 The samples were then filtered through 100 k spin filters. Plasma was fractionated by fast performance liquid chromatography and LDL and HDL containing fractions were pooled. Cultured human aortic endothelial cells were treated with normal LDL alone, with vehicle- or L-4F-treated patient LDL, and with normal LDL plus vehicle- or L-4F-treated patient HDL. Briefly, a standard control human LDL prepared by ultracentrifugation of the plasma of a healthy volunteer was added as an internal standard to all cultures at a concentration of 100 μg/ml cholesterol. After 8 h, the supernatants were collected and the monocyte chemotactic activity (which is largely due to the activity of monocyte chemoattractant protein-1) in the supernatant was determined as described earlier.64, 65 The values for the control internal standard LDL were normalized to 1.0. For determination of the HDL-inflammatory index, a standard control human HDL prepared by ultracentrifugation of the plasma of a healthy volunteer or mouse HDL prepared by fast performance liquid chromatography was added at 50 μg/ml cholesterol together with the control human internal standard LDL at 100 μg/ml cholesterol. Monocyte chemotactic activity was measured as migrated monocytes per high-powered field, in triplicates in six separate fields, after incubation of the endothelial cells with the lipoproteins. The value obtained by addition of the control human internal standard LDL together with the test HDL was divided by the monocyte chemotactic activity obtained after adding this LDL to the endothelial cells without HDL. In this assay, anti-inflammatory HDL results in inflammatory index values <1.0 and proinflammatory HDL results in inflammatory index values >1.0. For determination of the LDL-inflammatory index, the test LDL was added to the cells at 100 μg/ml cholesterol without added HDL and the resulting monocyte chemotactic activity was divided by the monocyte chemotactic activity obtained after addition of the control human internal standard LDL at 100 μg/ml cholesterol without added HDL. In this assay, if the test LDL induces more monocyte chemotactic activity than the control human internal standard LDL, the inflammatory index value will be >1.0. Conversely, if the test LDL produces less monocyte chemotactic activity than the control human internal standard LDL, the inflammatory index value will be <1.0.
Protein carbonyl assay
Protein carbonyl content of plasma was measured using a kit purchased from Cayman Chemical Inc. (Ann Arbor, MI), following the manufacturer’s protocol. The assay is based on the reaction between 2,4-dinitrophenylhydrazine (DNPH) and protein carbonyls, which leads to formation of an Schiff base. This produces the corresponding hydrazone, which can be quantified spectrophotometrically at 360–385 nm using a SpectraMax M5 plate reader.
Malondialdehyde assay
Plasma lipoperoxides were determined by measuring malondialdehyde-thiobarbituric acid (MDA-TBA) using high-performance liquid chromatography. Briefly, a 50 μl aliquot of plasma was mixed with 750 μl of 0.44 M H3PO4, 25 μl of aqueous 42 μmM TBA and 45 μl distilled water. The mixture was heated in a boiling water bath for 60 min. After cooling on ice, an equal volume, 1.5 ml of alkaline methanol (made using 50 ml methanol+4.5 ml 1 N NaOH) was added and centrifuged in a microcentrifuge for 5 min. Supernatant (50 μl) was then removed and injected into a high-performance liquid chromatography (SP8700; Spectra-Physics, San Jose, CA, USA) using a manually operated injector. An Econosphere C18 column 5 μm particle, 4.6×250 mm (Alltech Inc., Deerfield, IL, USA), and a guard column, 3.9×2.3 mm, packed with Bondapak Corasil C18 (37–50 μm particle diameter) were used. Mobile phase was a mixture of 50 mM phosphate buffer, pH 6.8, (600 ml) and methanol (400 ml). The flow rate was set at 1 ml/min and was monitored with a ultraviolet detector at 532 nm. The peak retention time was 2.9±0.15 min. The concentration of plasma MDA was determined by interpolation of peak area into an SP4270 integrator (Spectra-Physics), which was calibrated with a tetramethoxypropane standard.
Measurements of plasma cytokine concentrations
Plasma concentration of IL-6, IL-8, and tumor necrosis factor- were determined using Millipore 13-plex inflammatory cytokine panel (Billerica, MA, USA) run on Luminex 100 IS system (Austin, TX, USA). The data were analyzed using MiraiBio MasterPlex QT Version: 0.1.171 software (San Francisco, CA, USA).
Data analysis
Data were analyzed with Student’s t-test using SPSS 13.5 software (Chicago, IL, USA) and expressed as mean±s.e.; P-values less than 0.05 were considered significant.
Acknowledgments
This study was conducted in part at the General Clinical Research Center, School of Medicine, University of California, Irvine (NCRR 5M01RR 00827-29, US Public Health Service) and supported in part by US Public Health Service Grant HL-30568 and the Laubisch, Castera, and M.K. Grey Funds at UCLA.
Footnotes
DISCLOSURE
Drs Alan M Fogelman and Mohamad Navab are principals and shareholders in Bruins Pharma. The other authors have no conflict of interest to report.
References
- 1.Excerpts from the United States Renal Data system 2005 Annual Data Report. Atlas of end-stage renal disease in the United States. Am J Kidney Dis. 2006;47:S1–S286. [Google Scholar]
- 2.Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 3.Kovesdy CP, Trivedi BK, Anderson JE. Associations of kidney function with mortality in patients with chronic kidney disease not yet on dialysis: a historical prospective cohort study. Adv Chronic Kidney Dis. 2006;13:183–188. doi: 10.1053/j.ackd.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Muntner P, He J, Astor BC, et al. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: results from the atherosclerosis risk in communities study. J Am Soc Nephrol. 2005;16:529–538. doi: 10.1681/ASN.2004080656. [DOI] [PubMed] [Google Scholar]
- 5.Vaziri ND. Effect of chronic renal failure on nitric oxide metabolism. Am J Kidney Dis. 2001;38:S74–S79. doi: 10.1053/ajkd.2001.27409. [DOI] [PubMed] [Google Scholar]
- 6.Vaziri ND. Oxidative stress in chronic renal failure: The nature, mechanism and consequences. Semin Nephrol. 2004;24:469–473. doi: 10.1016/j.semnephrol.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri ND, Ni Z, Wang XQ, et al. Downregulation of nitric oxide synthase in chronic renal insufficiency: Role of excess PTH. Am J Physiol, Renal Physiol. 1998;274:F642–F649. doi: 10.1152/ajprenal.1998.274.4.F642. [DOI] [PubMed] [Google Scholar]
- 8.Himmelfarb J, Stenvinkel P, Ikizler TA, et al. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002;62:1524–1538. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- 9.McCullough PA. Why is chronic kidney disease the ‘spoiler’ for cardiovascular outcomes? J Am Coll Cardiol. 2003;41:725–728. doi: 10.1016/s0735-1097(02)02955-8. [DOI] [PubMed] [Google Scholar]
- 10.Stenvinkel P, Alvestrand A. Inflammation in end-stage renal disease: Sources, consequences, and therapy. Semin Dial. 2002;15:329–337. doi: 10.1046/j.1525-139x.2002.00083.x. [DOI] [PubMed] [Google Scholar]
- 11.Vaziri ND. Dyslipidemia of chronic renal failure: The nature, mechanisms and potential consequences. Am J Physiol, Renal Physiol. 2006;290:262–272. doi: 10.1152/ajprenal.00099.2005. [DOI] [PubMed] [Google Scholar]
- 12.Hansson GK, Robertson AK, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 13.Wilensky RL, Hamamdzic D. The molecular basis of vulnerable plaque: potential therapeutic role for immunomodulation. Curr Opin Cardiol. 2007;22:545–551. doi: 10.1097/HCO.0b013e3282f028fe. [DOI] [PubMed] [Google Scholar]
- 14.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- 15.Obryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37:208–222. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Botham KM, Moore EH, De Pascale C, et al. The induction of macrophage foam cell formation by chylomicron remnants. Biochem Soc Trans. 2007;35:454–458. doi: 10.1042/BST0350454. [DOI] [PubMed] [Google Scholar]
- 17.Gleissner CA, Leitinger N, Ley K. Effects of native and modified low-density lipoproteins on monocyte recruitment in atherosclerosis. Hypertension. 2007;50:276–283. doi: 10.1161/HYPERTENSIONAHA.107.089854. [DOI] [PubMed] [Google Scholar]
- 18.Davidson MH, Toth PP. High-density lipoprotein metabolism: potential therapeutic targets. Am J Cardio. 2007;100:n32–n40. doi: 10.1016/j.amjcard.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 19.Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol. 2003;23:1724–1731. doi: 10.1161/01.ATV.0000094961.74697.54. [DOI] [PubMed] [Google Scholar]
- 20.Navab M, Imes SS, Hama SY, et al. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest. 1991;88:2039–2046. doi: 10.1172/JCI115532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson AD, Berliner JA, Hama SY, et al. Protective effect of high density lipoprotein associated paraxonase: inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995;96:2882–2891. doi: 10.1172/JCI118359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ansell BJ, Navab M, Hama S, et al. Inflammatory/anti-inflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-denisty lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–2756. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 23.Feig JE, Shamir R, Fisher EA. Atheroprotective effects of HDL: beyond reverse cholesterol transport. Curr Drug Targets. 2008;9:196–203. doi: 10.2174/138945008783755557. [DOI] [PubMed] [Google Scholar]
- 24.Yu R, Yekta B, Vakili L, et al. Proatherogenic high- density lipoprotein, vascular inflammation, and mimetic peptides. Curr Atheroscler Rep. 2008;10:171–176. doi: 10.1007/s11883-008-0025-z. [DOI] [PubMed] [Google Scholar]
- 25.Haque S, Mirjafari H, Bruce IN. Atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. Curr Opin Lipidol. 2008;19:338–343. doi: 10.1097/MOL.0b013e328304b65f. [DOI] [PubMed] [Google Scholar]
- 26.Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high-density lipoprotein. Curr Opin Lipidol. 2007;18:427–434. doi: 10.1097/MOL.0b013e3282364a17. [DOI] [PubMed] [Google Scholar]
- 27.Attman PO, Alaupovic P, Gustafson A. Serum apolipoprotein profile of patients with chronic renal failure. Kidney Int. 1987;32:368–375. doi: 10.1038/ki.1987.219. [DOI] [PubMed] [Google Scholar]
- 28.Attman PO, Alaupovic P. Lipid and apolipoprotein profiles of uremic dyslipoproteinemia--relation to renal function and dialysis. Nephron. 1991;57:401–410. doi: 10.1159/000186303. [DOI] [PubMed] [Google Scholar]
- 29.Muntner P, Hamm LL, Kusek JW, et al. The prevalence of nontraditional risk factors for coronary heart disease in patients with chronic kidney disease. Ann Intern Med. 2004;140:9–17. doi: 10.7326/0003-4819-140-1-200401060-00006. [DOI] [PubMed] [Google Scholar]
- 30.Kamanna VS, Kashyap ML, Pai R, et al. Uremic serum subfraction inhibits apolipoprotein A-I production by a human hepatoma cell line. J Am Soc Nephrol. 1994;5:193–200. doi: 10.1681/ASN.V52193. [DOI] [PubMed] [Google Scholar]
- 31.Shah GM, Lin ZL, Kamanna VS, et al. Effect of serum subfractions from peritoneal dialysis patients on Hep-G2 cell apolipoprotein A-I and B metabolism. Kidney Int. 1996;50:2079–2087. doi: 10.1038/ki.1996.532. [DOI] [PubMed] [Google Scholar]
- 32.Vaziri ND, Deng G, Liang K. Hepatic HDL receptor, SR-B1 and Apo A-I expression in chronic renal failure. Nephrol Dial Transplant. 1999;14:1462–1466. doi: 10.1093/ndt/14.6.1462. [DOI] [PubMed] [Google Scholar]
- 33.Moradi H, Pahl MV, Elahimehr R, et al. Impaired Antioxidant Activity of HDL in Chronic Kidney Disease. Translational Res. 2009;153:77–85. doi: 10.1016/j.trsl.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Kalantar-Zadeh K, Kopple JD, Kamranpour N, et al. HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int. 2007;72:1149–1156. doi: 10.1038/sj.ki.5002491. [DOI] [PubMed] [Google Scholar]
- 35.Belalcazar LM, Merched A, Carr B, et al. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- 36.Tangirala RK, Tsukamoto K, Chun SH, et al. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. 1999;100:816–822. doi: 10.1161/01.cir.100.17.1816. [DOI] [PubMed] [Google Scholar]
- 37.Calabresi L, Sirtori CR, Paoletti R, et al. Recombinant apolipoprotein A-IMilano for the treatment of cardiovascular diseases. Curr Atheroscler Rep. 2006;8:163–167. doi: 10.1007/s11883-006-0054-4. [DOI] [PubMed] [Google Scholar]
- 38.Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant apoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 39.Navab M, Anantharamaiah GM, Hama S, et al. D-4F and statins synergize to render HDL anti-inflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1426–1432. doi: 10.1161/01.ATV.0000167412.98221.1a. [DOI] [PubMed] [Google Scholar]
- 40.Navab M, Anantharamaiah GM, Reddy ST, et al. Oral D-4F causes formation of pre-b high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215–3220. doi: 10.1161/01.CIR.0000134275.90823.87. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Chyu KY, Faria Neto JR, et al. Differential effects of apolipoprotein A-I mimetic peptide on evolving and established atherosclerosis in apolipoprotein E-null mice. Circulation. 2004;110:1701–1705. doi: 10.1161/01.CIR.0000142857.79401.69. [DOI] [PubMed] [Google Scholar]
- 42.Shah PK, Chyu KY. Apolipoprotein A-I mimetic peptides: potential role in atherosclerosis management. Trends Cardiovasc Med. 2005;15:291–296. doi: 10.1016/j.tcm.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Ou J, Ou Z, Jones DW, et al. L-4F, an apolipoprotein A-I mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation. 2003;107:2337–2341. doi: 10.1161/01.CIR.0000070589.61860.A9. [DOI] [PubMed] [Google Scholar]
- 44.Ou J, Wang J, Xu H, et al. Effects of D-4F on vasodilation and vessel wall thickness in hypercholesterolemic LDL receptor-null and LDL receptor/apolipoprotein A-I double-knockout mice on Western diet. Circ Res. 2005;97:1190–1197. doi: 10.1161/01.RES.0000190634.60042.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navab M, Anantharamaiah GM, Reddy ST, et al. Apolipoprotein A-I mimetic peptides and their role in atherosclerosis prevention. Nat Clin Pract Cardiovasc Med. 2006;3:540–547. doi: 10.1038/ncpcardio0661. [DOI] [PubMed] [Google Scholar]
- 46.Van Lenten BJ, Wagner AC, Nayak DP, et al. High-density lipoprotein loses its anti-inflammatory properties during acute influenza A infection. Circulation. 2001;103:2283–2288. doi: 10.1161/01.cir.103.18.2283. [DOI] [PubMed] [Google Scholar]
- 47.Charles-Schoeman C, Banquerigo ML, Hama S, et al. Treatment with an apolipoprotein A-1 mimetic peptide in combination with pravastatin inhibits collagen-induced arthritis. Clin Immunol. 2008;127:234–244. doi: 10.1016/j.clim.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 48.Navab M, Anantharamaiah GM, Fogelman AM. The effect of apolipoprotein mimetic peptides in inflammatory disorders other than atherosclerosis. Trends Cardiovasc Med. 2008;18:61–66. doi: 10.1016/j.tcm.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 50.Vaziri ND, Liang K, Parks JS. Downregulation of hepatic lecithin:cholesterol acyltransferase (LCAT) gene expression in chronic renal failure. Kidney Int. 2001;59:2192–2196. doi: 10.1046/j.1523-1755.2001.00734.x. [DOI] [PubMed] [Google Scholar]
- 51.Vaziri ND, Liang K. ACAT inhibition reverses LCAT deficiency and improves plasma HDL in chronic renal failure. Am J Physiol Renal Physiol. 2004;287:F1038–F1043. doi: 10.1152/ajprenal.00150.2004. [DOI] [PubMed] [Google Scholar]
- 52.Bories PC, Subbaiah PV, Bagdade JD. Lecithin: cholesterol acyltransferase activity in dialyzed and undialyzed chronic uremic patients. Nephron. 1982;32:22–27. doi: 10.1159/000182726. [DOI] [PubMed] [Google Scholar]
- 53.Guarnieri GF, Moracchiello M, Campanacci L, et al. Lecithin-cholesterol acyltransferase (LCAT) activity in chronic uremia. Kidney Int Suppl. 1978;8:S26–S30. [PubMed] [Google Scholar]
- 54.Boadu E, Bilbey NJ, Francis GA. Cellular cholesterol substrate pools for adenosine-triphosphate cassette transporter A1-dependent high-density lipoprotein formation. Curr Opin Lipidol. 2008;19:270–276. doi: 10.1097/MOL.0b013e3282feea99. [DOI] [PubMed] [Google Scholar]
- 55.Bowry VW, Stanley KK, Stocker R. High density lipoprotein is the major carrier of lipid hydroperoxides in human blood plasma from fasting donors. Proc Natl Acad Sci USA. 1992;89:10316–10320. doi: 10.1073/pnas.89.21.10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deigner HP, Hermetter A. Oxidized phospholipids: emerging lipid mediators in pathophysiology. Curr Opin Lipidol. 2008;19:289–294. doi: 10.1097/MOL.0b013e3282fe1d0e. [DOI] [PubMed] [Google Scholar]
- 57.Mehta JL, Chen J, Hermonat PL, et al. Lectin-like, oxidized low-density lipoprotein receptor-1 (LOX-1): a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res. 2006;69:36–45. doi: 10.1016/j.cardiores.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Cominacini L, Pasini AF, Garbin U, et al. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem. 2000;275:12633–12638. doi: 10.1074/jbc.275.17.12633. [DOI] [PubMed] [Google Scholar]
- 59.Cominacini L, Rigoni A, Pasini AF, et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem. 2001;276:13750–13755. doi: 10.1074/jbc.M010612200. [DOI] [PubMed] [Google Scholar]
- 60.Navab M, Anantharamaiah GM, Reddy ST, et al. Mechanisms of disease: proatherogenic HDL--an evolving field. Nat Clin Pract Endocrinol Metab. 2006;2:504–511. doi: 10.1038/ncpendmet0245. [DOI] [PubMed] [Google Scholar]
- 61.Vaziri ND, Liang K. Down-regulation of tissue lipoprotein lipase expression in experimental chronic renal failure. Kidney Int. 1996;50:1928–1935. doi: 10.1038/ki.1996.515. [DOI] [PubMed] [Google Scholar]
- 62.Kaysen GA. Hyperlipidemia in chronic kidney disease. Int J Artif Organs. 2007;30:987–992. doi: 10.1177/039139880703001107. [DOI] [PubMed] [Google Scholar]
- 63.Moradi H, Yuan J, Ni Z, et al. Reverse cholesterol transport pathway in experimental chronic kidney disease. Am J Nephrol. 2009;30:147–154. doi: 10.1159/000210020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Lenten BJ, Wagner AC, Jung C-L, et al. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Navab M, Hama SY, Cooke CJ, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res. 2000;41:1481–1494. [PubMed] [Google Scholar]