

Abstract
Glucagon is the main secretory product of the pancreatic alpha-cells. The main function of this peptide hormone is to provide sustained glucose supply to the brain and other vital organs during fasting conditions. This is exerted by stimulation of hepatic glucose production via specific G protein-coupled receptors in the hepatocytes. Type 2 diabetic patients are characterized by elevated glucagon levels contributing decisively to hyperglycemia in these patients. Accumulating evidence demonstrates that targeting the pancreatic alpha-cell and its main secretory product glucagon is a possible treatment for type 2 diabetes. Several lines of preclinical evidence have paved the way for the development of drugs, which suppress glucagon secretion or antagonize the glucagon receptor. In this review, the physiological actions of glucagon and the role of glucagon in type 2 diabetic pathophysiology are outlined. Furthermore, potential advantages and limitations of antagonizing the glucagon receptor or suppressing glucagon secretion in the treatment of type 2 diabetes are discussed with a focus on already marketed drugs and drugs in clinical development. It is concluded that the development of novel glucagon receptor antagonists are confronted with several safety issues. At present, available pharmacological agents based on the glucose-dependent glucagonostatic effects of GLP-1 represent the most favorable way to apply constraints to the alpha-cell in type 2 diabetes.
Keywords: alpha-cell, DPP-4, GLP-1, glucagon receptor antagonist, glucagon-like peptide 1, incretin-based therapy, LY-2409021, MK-0893, pramlintide, type 2 diabetes
Abbreviations: AMP - adenosine monophosphate; DPP-4 - dipeptidyl peptidase 4; Gcgr - glucagon receptor; GI - gastrointestinal; GIP - glucose-dependent insulinotropic polypeptide; GLP-1 - glucagon-like peptide-1; HbA1c - glycated hemoglobin; IAPP - islet amyloid polypeptide; LDL - low-density lipoprotein
Introduction
Glucagon is a 29-amino acid peptide hormone, the main secretory product of the pancreatic alpha-cells, acting to increase plasma glucose levels. The secretion of glucagon from the alpha-cell is regulated by multiple physiological and pharmacological factors, with hypoglycemia constituting the most potent glucagonotropic stimulus [1]. Traditionally, glucagon has been viewed as a hormone that opposes the action of insulin in the liver. In the portal vein, the insulin-to-glucagon ratio (insulin:glucagon) is thought to control the rates of gluconeogenesis, glycogenesis, and glycogenolysis comprising pivotal mechanisms for the control of glucose metabolism; mechanisms which suffer from severe disruptions in diabetes.
Therefore, over the last decades, glucagon has attracted a lot of interest from a physiological perspective. However, from a pharmacological perspective, glucagon has not gained much attention until recently when drugs based on the naturally occurring glucagon-suppressive peptides glucagon-like peptide-1 (GLP-1) (released into the circulation from enteroendocrine L-cells upon food ingestion) and amylin (co-secreted with insulin from pancreatic beta-cells) were developed for the treatment of diabetes. Furthermore, the current investigation of several small molecule glucagon receptor (Gcgr) antagonists for clinical use centers glucagon as one of the key hormones in the pathophysiological phenotype of type 2 diabetes and in the treatment of type 2 diabetic hyperglycemia. In this review, the physiological actions of glucagon will be outlined and the role of glucagon in type 2 diabetic pathophysiology will be accounted for. Furthermore, potential pharmacological advantages (and disadvantages) of suppressing glucagon secretion and antagonizing the Gcgr, respectively, in the treatment of type 2 diabetes will be reviewed with a focus on drugs in clinical development and already marketed drugs.
Glucagon measurement and glucagon physiology
Glucagon is the result of posttranslational processing of proglucagon, a molecule of 160 amino acids produced in pancreatic alpha-cells. The pancreatic processing of proglucagon to glucagon involves cleavage by the enzyme prohormone convertase 2 resulting in liberation of glicentin-related pancreatic peptide (proglucagon 1-30), glucagon (proglucagon 33-61), and a major proglucagon fragment (proglucagon 72-158) from the proglucagon moiety. Apart from glucagon, these fragments have been shown to be biologically inactive. This is in contrast to the intestinal processing of proglucagon which occurs in the endocrine L-cells of the intestinal mucosa. In L-cells, prohormone convertase 1 cleaves proglucagon to glicentin (proglucagon 1-69), which is further cleaved to glicentin-related pancreatic peptide (proglucagon 1-30) and oxyntomodulin (proglucagon 33-69), and the glucagon-like peptides, GLP-1 (proglucagon 78-107) and GLP-2 (proglucagon 126-158) [2].
The concentrations of glucagon in the plasma are low, with fasting concentrations in healthy individuals around 6-10 pmol/l [3]. Being an essential part of glucose regulation, glucagon secretion is tightly linked to the plasma concentrations of glucose. Thus, when glucose levels are lowered to 2-3 mmol/l glucagon levels rise several times (up to 40 pmol/l), and when glucose levels are elevated (to around 10-12 mmol/l) glucagon levels in plasma decrease down to 1-2 pmol/l [3]. The dynamics of glucagon secretion are therefore displayed within a relative small concentration interval, which represents a technical challenge, since many assays have a lower limit of quantification around 10-15 pmol/l. An assay with a detection limit in the low picomolar range measuring the C-terminal end of fully processed glucagon is therefore essential when investigating glucagon physiology and evaluating therapies exerting suppressive effects on alpha-cells. This difficulty represents a major caveat when assessing trials reporting glucagon values.
As mentioned, the main stimulus for pancreatic glucagon secretion is hypoglycemia, but certain amino acids, and activity in the autonomic nervous system, stimulate the release of glucagon from pancreatic alpha-cells as well. Glucagon secretion is pulsatile with distinct secretory bursts occurring every 5 minutes, correlating inversely to the well-known pulses of insulin secretory bursts [4, 5]. Following secretion into the portal circulation glucagon targets its primary site of action, the liver. The actions of glucagon in the liver are complex. They involve binding to the Gcgr, a G protein-coupled receptor, located in the plasma membrane, which results in amplification of signal transduction cascades and regulation of transcription factors. Ultimately, this has a stimulatory effect on hepatic glucose production [1]. Gcgr primarily acts through activation of adenylate cyclase and subsequent increase in intracellular cyclic AMP concentrations. However, Gcgr is also coupled to a Ca2+-mediated signaling pathway [6].
In humans, glucagon-stimulated hepatic glucose production predominates glycogenesis during the fasting state, and ensures sufficient plasma glucose levels to accommodate the needs of glucose-consuming organs. In contrast, glucagon secretion is suppressed and insulin secretion increased following carbohydrate ingestion in healthy subjects. This changes the balance towards hepatic glycogen storage and peripheral glucose uptake, especially in muscle tissue. Thus, in healthy individuals, the balance of glucagon suppression and stimulation contributes vitally to maintain normoglycemia.
In addition to the liver, the Gcgr is expressed in other tissues, including adipose tissue, pancreas, the gastrointestinal tract, heart, brain, and kidney, suggesting a broader physiological role beyond glucose homeostasis. However the physiological significance of most of the human extrahepatic effects of glucagon are unknown, as exemplified by the measurable effects of glucagon on human isolated adipocytes [7], and lack thereof in in vivo studies [8]. Nevertheless, in supraphysiological doses, the extrahepatic effects of glucagon become clearer (Figure 1). Thus, glucagon has been used as a drug in emergency medicine to counteract hypoglycemia and for its inotropic and chronotropic cardiac effects as a part of the treatment against cardiodepressive drug overdoses [9, 10]. Furthermore, at supraphysiological levels, glucagon has been shown to decrease appetite and food intake in humans, possibly via centrally mediated Gcgr activation combined with inhibitory effects on gastrointestinal motility including gastric emptying [11-13] (Figure 1). Finally, indirect calorimetry studies in humans have demonstrated that glucagon may increase the rate of energy expenditure [14].
Figure 1. Organ-specific pharmacological effects of glucagon.
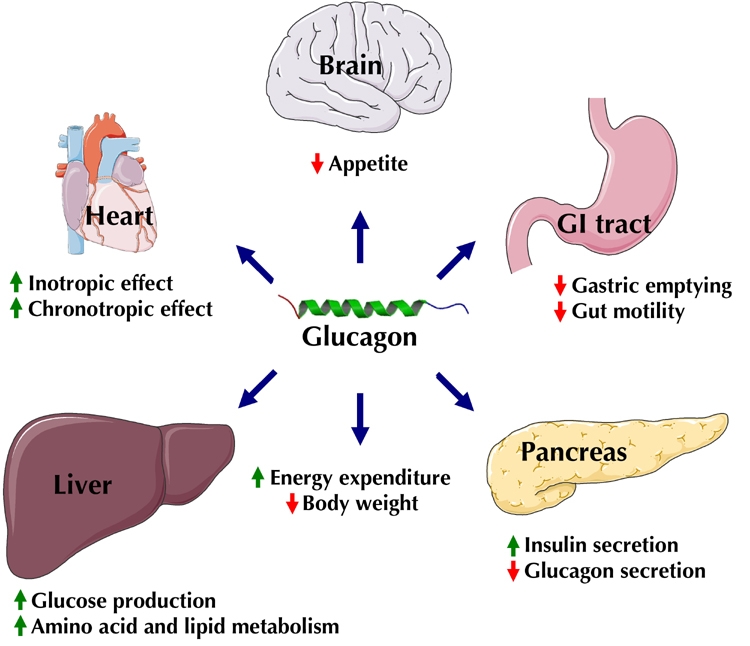
In the central nervous system, glucagon mediates satiety. Other possible central effects of glucagon are increased energy expenditure and, on the longer term, body weight reduction. In the gastrointestinal (GI) tract, glucagon reduces motility and may slow gastric emptying. In the pancreas, glucagon induces insulin release and exerts feedback inhibition of glucagon release. In the liver, glucagon increases hepatic glucose production and affects amino acid metabolism and lipid metabolism. In the heart, glucagon increases contractility and heart rate.
Diabetic hyperglucagonemia
The finely tuned balance of the two major pancreatic hormones, insulin and glucagon, is perturbed in type 2 diabetic subjects. These patients feature a bihormonal disorder where absolute insulin insufficiency or relative lack of insulin (in relation to prevailing insulin resistance) are present alongside fasting and postprandial hyperglucagonemia. It is important to note that the level of glucagon is undesirably high in the specific context of hyperglycemia and hyperinsulinemia, whereas in untreated type 2 diabetes the level is sometimes not elevated in absolute terms [15]. Interestingly, it has recently been reported that the well-known disturbed pulsatility of insulin secretion in type 2 diabetes [16] is present alongside a disturbed glucagon pulsatility (higher pulse mass in patients with type 2 diabetes), possibly contributing to the hyperglucagonemia in these patients [5].
The "bihormonal hypothesis", i.e. the notion that the combination of elevated glucagon and relative lack of insulin is a major determinant in diabetic hyperglycemia, was first proposed by Unger and Orci in 1975 [17], and has since then been a matter of controversy [15, 18]. Key arguments against the concept of glucagon as a major contributor to diabetic hyperglycemia are that hyperglycemia and ketoacidosis occurs despite pancreatectomy in man [19], and that most of the scientific evidence demonstrating hyperglycemic effects of glucagon have used the somatostatin clamp method. The somatostatin clamp technique consists of a somatostatin infusion to suppress endogenous glucagon and insulin secretion. This technique allows plasma concentrations of glucagon and insulin to be clamped at pre-specified levels by exogenous administration. However, beside suppression of glucagon, the clamp technique affects several non-glucagon-mediated mechanisms involved in glucose homeostasis [20]. Pancreatectomy as a model for diabetes without glucagon is still a matter of controversy, because of the unclear physiological role of extrapancreatic glucagon [21], and the limitations in determining the origin and exact size of the glucagon measured with the current glucagon assays. However, in past decades, increasing evidence, including various interventions targeting glucagon secretion, has emerged to unequivocally support the role of fasting and postprandial hyperglucagonemia as major contributing factors for the elevated levels of blood glucose that characterize diabetes [15].
It is well established now that elevated levels of glucagon lead to increased rates of hepatic glucose output, and thereby to the elevation of postabsorptive and postprandial blood glucose levels in type 2 diabetes. In fact, studies indicate that postabsorptive hyperglucagonemia can be regarded as responsible for ≈50% of the pathological increment in plasma glucose excursions following oral glucose ingestion in diabetics [22-24]. Interestingly, in the postprandial state, the prevailing hyperglucagonemia is aggravated by the oral intake of glucose (compared to intravenous administration producing the same plasma glucose excursions). This implies that gut-derived factors contribute to the derangement of postprandial glucagon responses [25, 26], or possibly that measurable glucagon is released from the intestine [27].
As previously mentioned, the derangement of glucagon secretion occurs not only in the postprandial state. Several studies have demonstrated that fasting glucagon levels are positively correlated with fasting blood glucose levels and basal rates of hepatic glucose production in patients with type 2 diabetes [28, 29]. This finding illustrates again the importance of the contribution of increased hepatic glucose production to diabetes. Whereas, the mechanisms for hyperglucagonemia in the fasting and the postprandial state are not entirely identical; both pathophysiological characteristics suggest suppression of glucagon secretion/action throughout the day as a therapeutic target.
Targeting the alpha-cell - preclinical data
Lack of glucagon receptors
A good model to examine complete absence of Gcgr activation is in transgenic mice with targeted disruption of the Gcgr gene (Gcgr-/- mice). Phenotypically, Gcgr-/- mice display significantly lower fasting and postprandial plasma glucose levels and improved oral glucose tolerance compared to wild-type mice [30-32]. Common features are also elevated circulating glucagon concentrations and, nonetheless, normal insulin levels and lipidemia relative to wild-type mice [30]. Interestingly, these mice without functional Gcgrs are not prone to hypoglycemia, although long-term fasting (24 hours) could provoke severe hypoglycemia in these animals compared to wild-type littermates [31]. It is also worthwhile to note that the hyperglucagonemia in Gcgr-/- mice is accompanied by hyperplasia of the pancreatic alpha-cells, and, to a lesser degree, also delta-cells [31].
Although not evident in all preclinical models, Gcgr signaling has been implicated in the regulation of circulating triglycerides and lipoprotein accumulation [33]. Gcgr-/- hepatocytes exhibit profound defects in lipid oxidation, and accumulate excessive lipid deposition in the liver during fasting. Transcriptional and proteomic profiling of liver tissue and metabolomic analysis of plasma from Gcgr-/- mice demonstrated that the animals feature downregulation of gluconeogenesis, amino acid catabolism, and fatty acid oxidation processes. In contrast, glycolysis, fatty acid synthesis, and cholesterol biosynthetic processes are significantly upregulated [34]. These changes at the level of the liver were manifested through an altered plasma metabolite profile in the receptor knock-out mice. This profile includes decreased glucose and glucose-derived metabolites, and increased amino acids, cholesterol, and bile acid levels [34]. A very recent and remarkable finding by Unger and co-workers was the observation that streptozotocin-induced beta-cell destruction did not cause the expected hyperglycemic and catabolic disorders in Gcgr-/- mice [35]. This study exemplifies the importance of glucagon signaling in the pathophysiology of streptozotocin-induced diabetes in mice, and raises questions regarding the possible implications for human diabetes [36].
Another artful way to reduce hepatic Gcgr signaling in strains of diabetic mice is to administer Gcgr antisense oligonucleotides. Repeated parenteral administration of Gcgr antisense oligonucleotides in various rodent obesity and/or diabetes models significantly reduced blood glucose levels, serum and liver triglycerides, and improved glucose tolerance [37, 38]. Despite decreased Gcgr expression and increased glucagon levels, there was no hypoglycemia [37]. However, elevated concentrations of plasma glucagon in Gcgr antisense oligonucleotide-treated rodents were accompanied by pancreatic alpha-cell hypertrophy in most models. Also, some models exhibited marked alpha-cell hyperplasia [38]. An interesting finding was that Gcgr antisense oligonucleotide treatment increased alpha-cell expression of active GLP-1 and insulin levels in pancreatic islets. Therefore, it is conceivable that the proglucagon processing exhibit some degree of plasticity, which may contribute to the regulation of glucose metabolism [38].
Neutralizing antibodies against glucagons
Neutralizing monoclonal antibodies against glucagon have also been used as an instrument to study the effect of selective glucagon deficiency [39, 40]. The applicability of this model was confirmed in non-diabetic rats by demonstrating that high-affinity monoclonal antibodies to glucagon completely abolished the hyperglycemic effect of exogenous glucagon [39]. In streptozotocin-treated diabetic rats, glucagon antibodies abolished the otherwise prominent increase in postprandial blood glucose [39]. Likewise, in rabbits with reduced or nonexistent beta-cells due to alloxan treatment, glucagon antibodies essentially normalized fasting glucose levels. Interestingly, the elevated glucagon levels were estimated (from clamp studies) to contribute to half of the total basal endogenous glucose production [40].
Deletion of alpha-cells
Another interesting model of reduced glucagon action is the transgenic mouse with a deletion of the alpha-cell transcription factor Arx, resulting in a complete loss of the glucagon-producing pancreatic alpha-cells [41]. These mice without alpha-cells have reduced fasting glucose, improved glucose tolerance, and showed resistance to hyperglycemia following destruction of beta-cells by streptozotocin treatment. These observations underline again the pivotal role of alpha-cells and glucagon in glucose metabolism and the potential pharmacological advantage of suppressing/blocking glucagon secretion in the treatment of type 2 diabetes.
Antagonizing the glucagon receptor
An approach to reduce glucagon signaling with a larger therapeutic applicability is to administer Gcgr antagonising agents. Initial studies employing a peptide antagonist administered to diabetic (streptozotocin-treated) rats provided proof of concept by demonstrating clear-cut dose-dependent blood glucose reductions by up to two-thirds [42]. Similarly, several other Gcgr antagonists have demonstrated suppression of blood glucose levels in various animal models [43-55]. Some of these diverse Gcgr antagonists differ regarding their mechanism of action, oral bioavailability, and selectivity towards Gcgr compared with closely related class II family G protein-coupled receptors (e.g. receptors for glucose-dependent insulinotropic polypeptide (GIP), GLP-1 and GLP-2) [43-55].
Thus, combined data from preclinical observations suggest that glucagon plays an important physiological role in determining fasting and postprandial glucose levels, with an estimated contribution of glucagon to post-absorptive rates of endogenous glucose production by fifty percent. These preclinical models confirm the observations from human studies (employing somatostatin clamps), and indicate that the absence of glucagon to some degree reduces the effects of insulinopenia or insulin resistance by decreasing endogenous hepatic glucose production without overt safety issues.
Targeting the glucagon receptor - clinical data
Interestingly, reports of human mutations in the Gcgr gene suggest common phenotypical features with extreme hyperglucagonemia and marked pancreatic hyperplasia presenting with an abdominal tumor [56-58]. Despite the absence of glucagon signaling, the patients did not exhibit disrupted glycemic control or dyslipidemia and only vague clinical signs and symptoms, mostly ascribed to the abdominal mass [56]. Because of the benign clinical course described in these patients, the development of pharmacological agents blocking the glucagons receptor was not discontinued.
Glucagon receptor antagonism
Based on the encouraging preclinical data (as outlined above) demonstrating improved metabolic outcome measures, several Gcgr antagonists have been put forth into clinical development. The first of this kind, the orally available glucagon antagonist, BAY 27-995, blocked the effects of exogenous glucagon during a somatostatin clamp experiment in healthy males [59]. Without BAY 27-995, plasma glucose concentrations increased from basal values of about 5 mmol/l to about 10 mmol/l during the hyperglucagonemic period (due to an increase in hepatic glucose production). With administration of BAY 27-995, blood glucose concentrations decreased dose-dependently (stimulated by exogenous glucagon infusion during a somatostatin clamp) from 10 mmol/l to mean levels of 7.6 mmol/l by lowering the rates of endogenous glucose production. However, BAY 27-995 was abandoned in clinical development without a publicly available rationale. Nevertheless, the development of Gcgr antagonists have remained a research focus of the pharmaceutical industry [60]. Very recently, human experimental data on two putative Gcgr antagonists in clinical development were presented at scientific meetings [61-68].
The glucagon receptor antagonist MK-0893
In a phase II study, 342 type 2 diabetic patients were randomized to once-daily administration of the glucagon antagonist MK-0893 in four different dosages, metformin, or placebo [65]. At 12 weeks, treatment with MK-0893 resulted in significant, dose-dependent reductions in fasting and postprandial plasma glucose. From baseline, fasting values (10-11 mmol/l) were reduced by 3.5 mmol/l with MK-0893 compared to 2 mmol/l with metformin and 0.1 mmol/l with placebo. The resulting reductions in HbA1c at 12 weeks ranged from 0.6 to 1.5% with MK-0893, compared to 0.8% with metformin and 0.5% with placebo. Similarly, low incidences of hypoglycemia were observed across all groups. However, at the same time, LDL cholesterol was dose-dependently increased from baseline, and MK-0893 also increased liver transaminases. Furthermore, MK-0893 dose-dependently increased body weight (+2.3 kg (80 mg) versus -1.0/-0.9 kg (metformin/placebo). One trial has reported that treatment with MK-0893 resulted in increased blood pressure in patients with type 2 diabetes [66].
Another phase II study investigated the efficacy and safety of MK-0893 in combination with metformin or the dipeptidyl peptidase 4 (DPP-4) inhibitors (sitagliptin) in 146 patients with type 2 diabetes [64]. MK-0893 plus metformin was superior to both sitagliptin plus metformin and MK-0893 plus sitagliptin in lowering fasting plasma glucose and 24-hour weighted mean glucose (reductions of 6.5 mmol/l, 5.6 mmol/l, and 4.7 mmol/l, respectively). The results could suggest some overlap in effect when combining the glucagon antagonist with sitagliptin, which is known to suppress glucagon. The liver transminases were elevated in groups treated with combinations of MK-0893 and another drug. Also, total cholesterol and LDL cholesterol were increased from baseline with MK-0893 plus sitagliptin, relative to reductions observed with MK-0893 plus metformin and sitagliptin plus metformin.
The potentially increased risk of hypoglycemia and the compromised ability to recover from hypoglycemia was specifically addressed in other studies [67, 68]. In order to determine whether MK-0893 increased the time to recovery after hypoglycemia, insulin-induced 30-min hypoglycemic clamps (2.8 mmol/l) were applied in 13 healthy males after single dose of placebo, low dose MK-0893 (≈60% blockade of the Gcgr signaling), or high dose MK-0893 (≈90% blockade of the Gcgr signaling) [67]. Recovery times to a plasma glucose level of 3.9 mmol/l were 33 min (placebo), 45 min (low-dose MK-0893), and 59 min (high-dose MK-0893). The recovery times during Gcgr blockade were influenced by adrenergic signaling (i.e. beta-adrenergic blockade), but compensatory increments in the secretion of cortisol, glucagon, epinephrine, and/or growth hormone could be observed as well [68]. Very recently, the development of the MK-0893 compound was discontinued.
The glucagon receptor antagonist LY-2409021
In a phase I/II study, pharmacokinetic and pharmacodynamic parameters of 6 different single doses of LY-2409021 were assessed in 23 healthy subjects and 9 patients with type 2 diabetes [61]. Postprandial plasma glucose values were lowered by up to 0.6 mmol/l in the healthy subjects compared to 1.7 mmol/l in patients. No safety or tolerability issues were described. In another study, the ability of LY-2409021 to block endogenous glucose production was assessed during a somatostatin clamp experiment with exogenously induced hyperglucagonemia. LY-2409021 attenuated increases in both hepatic glucose production and blood glucose concentration by up to 84% and 81%, respectively. The lowering of plasma glucose levels was highly correlated to the reduction in hepatic glucose production, suggesting this to be the main causal mechanism.
Further studies were performed in 47 patients with type 2 diabetes randomized to either one of four doses of LY-2409021 once daily or metformin [62]. Patients had a mean baseline fasting blood glucose of 8.2 mmol/l, and a baseline HbA1c of 8%. By day 28, mean reductions in HbA1c were dose-dependent and statistically significant compared with baseline in all treatment groups (0.7-1% with LY-2409021 compared to 0.5% with placebo). Reversible increases in fasting glucagon (up to 4.2-fold) and fasting active GLP-1 (up to 1.6-fold) compared with baseline were observed across all LY-2409021 dose levels. The agent was generally well tolerated, with infrequent reports of mild hypoglycemia (four mild to moderate hypoglycemic events were reported in the highest-dose group consisting of nine patients). Reversible elevations in liver transaminases (up to 3 times upper normal values) were seen in more than half of the patients in the highest-dose group, with no clinical signs or significant elevations in bilirubin or alkaline phosphatase.
Thus, when looking at the Gcgr antagonists as a drug class, some safety issues seem to exist concerning hypoglycemia, as expected, and concerning increases in liver enzymes and plasma lipids. The increases in lipids and liver enzymes could be associated with the recent observation that glucagon stimulates oxidation of fatty acids in cryopreserved human hepatocytes (possibly fueling hepatic glucose production), and that Gcgr antagonism antagonizes this effect dose-dependently [69]. As aforementioned, the clinical developments of both BAY 27-995 and MK-0893 were discontinued as a probable consequence of safety issues, including elevated liver enzymes. LY-2409021 is still in clinical development.
Marketed drugs affecting alpha-cells
While drugs directly targeting glucagon signaling by Gcgr antagonism or Gcgr uncoupling are still in clinical development, several pharmacological compounds with profound effects on pancreatic alpha-cells are already in the market. Notably, these agents are not primarily developed to correct diabetic hyperglucagonemia. Therefore, a common feature is that the alpha-cell is not the only point of action, and suppression of glucagon could somehow be regarded as an added benefit of these agents. This may enable the glucagonostatic effects to be fully acclaimed and exploited for therapeutic purposes.
Incretin-based therapy
Incretin-based therapy comprises two major classes of drugs, namely the GLP-1 receptor agonists and the DPP-4 inhibitors (reviewed elsewhere in this issue [70, 71]). These agents act by increasing GLP-1 receptor signaling either by administration of an exogenous GLP-1 analogue or enhancement of endogenous GLP-1 levels [72]. Activation of GLP-1 receptors effectively inhibits glucagon secretion in humans together with deceleration of gastric emptying, inhibition of food intake, and elevation of insulin secretion [72].
The mechanism causing inhibition of glucagon secretion is not entirely clear. Insulin is generally thought to inhibit glucagon secretion. Local increments in insulin levels and other beta-cell products might inhibit alpha-cell secretion in a paracrine manner. However, the preserved and pronounced inhibitory effect of GLP-1 in type 1 diabetic patients without residual beta-cell function suggests that other mechanisms must also be involved [73]. A possibility is that GLP-1 via enhanced pancreatic somatostatin secretion from neighboring pancreatic delta-cells could inhibit glucagon secretion in a paracrine manner. This concept is supported by studies in the perfused rat pancreas, where the otherwise strong inhibition of glucagon secretion by GLP-1 is abolished by somatostatin antibodies and a somatostatin receptor 2 antagonist, respectively [74]. Regardless of the precise mechanism, the inhibitory effect of GLP-1 on glucagon secretion in vivo is only observed at glucose levels at or above fasting levels. In graded hypoglycemic clamp studies (in humans), the inhibitory effect of GLP-1 is lost at glucose levels just below normal fasting levels, and the normal stimulation of glucagon secretion at hypoglycemic levels is unimpeded by GLP-1 [75]. In contrast, glucagon suppression is progressively enhanced at higher glucose levels [72].
GLP-1 receptor agonists
In agreement with the effects of native GLP-1 on glucagon secretion described above, treatment with GLP-1 receptor agonists provides clear-cut reductions in glucagon [76-85]. Repeated administration in diabetic patients leads to reductions in glucagon in both the fasting and postprandial state. Notably, insulin and C-peptide responses during long-term treatment are relatively unchanged or even lowered, athough these drugs have originally been developed because of their powerful insulinotropic effects [76]. This has been ascribed to the lower levels of glycemia induced by the treatment and a consequent normalization of glucose-to-insulin ratio. Also, the slowing effect of GLP-1 on gastric emptying seems to decrease over time [86, 87].
Therefore, the glucose-lowering effects of GLP-1, in the setting of unchanged insulin levels and gastric emptying, points to a substantial role of the glucagonostatic effect in this treatment modality. The relative contribution of these separate mechanisms during long-term treatment with GLP-1 receptor agonists has not been reliably quantified. However, in a recent study, the relative contributions of the insulinotropic and glucagonostatic effects of short-term intravenously administered native GLP-1 was quantified in patients with type 2 diabetes [88]. This was elegantly done by measuring the glucose response to a pharmacological dose of GLP-1, and comparing this measure to situations with either elevated insulin (without glucagon suppression) or glucagon suppression (without elevated insulin) using somatostatin clamps on separate days. The insulinotropic effect and the glucagonostic effect were found to contribute equally to the overall glucose-lowering effect of GLP-1 [88]. Possibly, this reflects the equal contribution of decreased endogenous glucose production and increased peripheral disposal to the glucose-lowering effect of GLP-1 in patients with type 2 diabetes.
Dipeptidyl peptidase 4 inhibitors
In addition to increasing intact levels of GLP-1, inhibition of DPP-4 was also expected to affect glucagon levels through elevated levels of the other incretin hormone, GIP, which has renowned glucagon-releasing effects [3, 89, 90]. However, the glucagonostatic effects of GLP-1 prevails. Besides the improvements in plasma glucose and HbA1c, trials of DPP-4 inhibition in patients with type 2 diabetes convincingly demonstrate that glucagon levels are reduced [77, 91-102].
The suppression of glucagon is observed with both acute [103, 104] and chronic treatment [77, 91-102]. Some studies have demonstrated clear dose-dependency with regard to restraining postprandial glucagon responses [101, 104, 105]. One study has even demonstrated lowering of fasting glucagon levels as a dose-dependent effect [97]. Others did not report clear dose-dependent effect (probably as a consequence of low assay sensitivity) [96, 102], but reported clearly lower glucagon levels compared to placebo. Thus, the glucagonostatic effect is a consistent finding across the various applications of DPP-4 inhibitors, and could be considered a class effect.
Considering the alpha-cell-related effects of DPP-4 inhibition, it has recently been demonstrated that treatment with a DPP-4 inhibitor increases the glucagon response to a hypoglycemic clamp, besides providing the expected suppression of postprandial glucagon levels [95]. Thus, DPP-4 inhibition could help to enhance alpha-cell responsiveness to both the suppressive effects of hyperglycemia and the stimulatory effects of hypoglycemia [95]. The latter effect could be explained by increased levels of GIP contributing to enhanced glucagon secretion and thereby the low risk of hypoglycemia associated with DPP-4 inhibitor treatment.
Pramlintide
Amylin, also called islet amyloid polypeptide (IAPP), is a 37-amino acid peptide hormone co-secreted with insulin from the beta-cells of the pancreatic islets (in concentrations 100 fold less than insulin). Pramlintide is a synthetic analogue of IAPP and differs by replacement with proline at positions 25 (alanine), 28 (serine), and 29 (serine). Pramlintide was approved in the United States in 2005 for treatment of both type 1 diabetes and type 2 diabetes as an adjunct treatment in patients who are using mealtime insulin and have failed to achieve desired glucose control despite optimal insulin therapy. Currently, amylin is not approved for use in the European Union.
In humans, IAPP inhibits meal-related glucagon secretion without effects on hypoglycemic stimulation of glucagon secretion [106]. Also, amylin suppresses gastrointestinal motility and food intake [107, 108], which contributes to improved postprandial glycemia. The mechanism of glucagon suppression has been reported to be extrinsic to the pancreatic islet as it does not occur in isolated islets or isolated perfused pancreata [109]. However, others have reported a direct intra-islet inhibitory effect of IAPP on glucagon secretion [110]. Nevertheless, evidence suggests that the pivotal receptors conveying the actions of amylin are situated in the area postrema [111], a periventricular structure in the hindbrain, unprotected by the blood-brain barrier and critical for sensing and integrating peripheral and central meal-related signals.
Clinical studies with pramlintide have shown similar glucagon-suppressive effects caused by native amylin and pramlintide [112]. Several clinical trials of pramlintide in patients with type l diabetes [113-117] and in patients with type 2 diabetes [118-121] have demonstrated robust postprandial glucagon-suppressive effects in combination with dose-dependent reductions in HbA1c of around 0.4-0.7% and weight loss of around 1 kg, with treatment durations of 3-12 months. Thus, the efficacy of pramlintide seems to be clinically relevant. However, the relative contribution of the positive effects, i.e. inhibitory effects on glucagon release, gastric motility, and appetite, are largely unrevealed.
Perspectives
Convincing evidence from preclinical and human physiological studies has demonstrated the applicability of targeting the pancreatic alpha-cell and its main secretory product, glucagon, in the treatment of type 2 diabetes. With many promising data in preclinical and early clinical studies, Gcgr antagonists now have to prove their efficacy and safety in larger randomized controlled trials. Several notes of caution have been raised, including the increase in recovery time from hypoglycemia in combination with elevated liver transaminases and LDL cholesterol observed in clinical trials (not foreseen in preclinical studies). The data on body weight gain and increased blood pressure, although insufficient, call for caution as well.
Furthermore, concerns raised on preclinical models regarding uncontrolled alpha-cell growth and development of pancreatic neuroendocrine tumors in mice lacking functional Gcgrs are supported by some clinical data. The casuistic reports of patients with defective Gcgrs presenting with abdominal tumors are concerning and suggest that complete and glucose-independent antagonism of glucagon signaling is not an optimal way to dampen endogenous glucose production. Recently, the fact that the clinical development of some of the Gcgr antagonists has been abandoned because of safety issues raised new concerns for other Gcgrs in development.
Meanwhile, readily available pharmacological agents targeting the alpha-cell in a glucose-dependent fashion together with tackling some of the other pathophysiological traits of type 2 diabetes could be a better alternative. In this regard, incretin-based therapy, as well as inhibiting endogenous glucose production and possibly providing improved alpha-cell response to hypoglycemia, could also provide cardiovascular risk reductions in several ongoing trials. The placement of the glucagonostatic effect and provision of long-term outcomes and durability of pramlintide in treating of type 2 diabetes could benefit from further clarification. Thus, for now, the inappropriate glucagon secretion in type 2 diabetes seems most favorably addressed by employing incretin-based treatment. Moreover, these agents have the greatest potential to provide improvements in quality of life and life span of patients with type 2 diabetes, which is the prerequisite modern standard for new antidiabetic treatment options.
Disclosures: No financial support was received for the preparation of this review. TV has received fees for giving lectures and for consultation from Novo Nordisk, Bristol-Myers Squibb, AstraZeneca, Novartis, MSD, Boehringer Ingelheim, and Eli Lilly & Co. TV is on the advisory boards of Novo Nordisk, MSD, and Bristol-Myers Squibb. FKK has received fees for giving lectures from Novo Nordisk, Bristol-Myers Squibb, AstraZeneca, Novartis, MSD, Gilead, Ono Pharmaceutical Co, and Eli Lilly & Co, and has consulted for AstraZeneca, BMS, and Gilead.
References
- 1.Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. 2007;28(1):84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 2.Holst JJ. Glucagon and glucagon-like peptides 1 and 2. Results Probl Cell Differ. 2010;50:121–135. doi: 10.1007/400_2009_35. [DOI] [PubMed] [Google Scholar]
- 3.Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes. 2011 doi: 10.2337/db11-0979. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porksen N, Munn S, Steers J, Veldhuis JD, Butler PC. Impact of sampling technique on appraisal of pulsatile insulin secretion by deconvolution and cluster analysis. Am J Physiol. 1995;269(6 Pt 1):E1106–E1114. doi: 10.1152/ajpendo.1995.269.6.E1106. [DOI] [PubMed] [Google Scholar]
- 5.Menge BA, Grüber L, Jorgensen SM, Deacon CF, Schmidt WE, Veldhuis JD. et al. Loss of inverse relationship between pulsatile insulin and glucagon secretion in patients with type 2 diabetes. Diabetes. 2011;60(8):2160–2168. doi: 10.2337/db11-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamatani K, Saito K, Ikezawa Y, Ohnuma H, Sugiyama K, Manaka H. et al. Relative contribution of Ca2+-dependent mechanism in glucagon-induced glucose output from the liver. Arch Biochem Biophys. 1998;355(2):175–180. doi: 10.1006/abbi.1998.0710. [DOI] [PubMed] [Google Scholar]
- 7.Perea A, Clemente F, Martinell J, Villanueva-Penacarrillo ML, Valverde I. Physiological effect of glucagon in human isolated adipocytes. Horm Metab Res. 1995;27(8):372–375. doi: 10.1055/s-2007-979981. [DOI] [PubMed] [Google Scholar]
- 8.Gravholt CH, Moller N, Jensen MD, Christiansen JS, Schmitz O. Physiological levels of glucagon do not influence lipolysis in abdominal adipose tissue as assessed by microdialysis. J Clin Endocrinol Metab. 2001;86(5):2085–2089. doi: 10.1210/jcem.86.5.7460. [DOI] [PubMed] [Google Scholar]
- 9.White CM. A review of potential cardiovascular uses of intravenous glucagon administration. J Clin Pharmacol. 1999;39(5):442–447. [PubMed] [Google Scholar]
- 10.Bailey B. Glucagon in beta-blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol. 2003;41(5):595–602. doi: 10.1081/clt-120023761. [DOI] [PubMed] [Google Scholar]
- 11.Schulman JL, Carleton JL, Whitney G, Whitehorn JC. Effect of glucagon on food intake and body weight in man. J Appl Physiol. 1957;11(3):419–421. doi: 10.1152/jappl.1957.11.3.419. [DOI] [PubMed] [Google Scholar]
- 12.Penich SB, Hinkle LE Jr. Depression of food intake induced in healthy subjects by glucagon. N Engl J Med. 1961;264:893–897. doi: 10.1056/NEJM196105042641801. [DOI] [PubMed] [Google Scholar]
- 13.Geary N, Kissileff HR, Pi-Sunyer FX, Hinton V. Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am J Physiol. 1992;262(6 Pt 2):R975–R980. doi: 10.1152/ajpregu.1992.262.6.R975. [DOI] [PubMed] [Google Scholar]
- 14.Calles-Escandon J. Insulin dissociates hepatic glucose cycling and glucagon-induced thermogenesis in man. Metab Clin Exp. 1994;43(8):1000–1005. doi: 10.1016/0026-0495(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 15.Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. 2007;28(3):253–283. doi: 10.1210/er.2006-0026. [DOI] [PubMed] [Google Scholar]
- 16.Porksen N, Hollingdal M, Juhl C, Butler P, Veldhuis JD, Schmitz O. Pulsatile insulin secretion: detection, regulation, and role in diabetes. Diabetes. 2002;51(Suppl 1):S245–S254. doi: 10.2337/diabetes.51.2007.s245. [DOI] [PubMed] [Google Scholar]
- 17.Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet. 1975;1(7897):14–16. doi: 10.1016/s0140-6736(75)92375-2. [DOI] [PubMed] [Google Scholar]
- 18.Raju B, Cryer PE. Maintenance of the postabsorptive plasma glucose concentration: insulin or insulin plus glucagon? Am J Physiol Endocrinol Metab. 2005;289(2):E181–E186. doi: 10.1152/ajpendo.00460.2004. [DOI] [PubMed] [Google Scholar]
- 19.Barnes AJ, Bloom SR, Goerge K, Alberti GM, Smythe P, Alford FP. et al. Ketoacidosis in pancreatectomized man. N Engl J Med. 1977;296(22):1250–1253. doi: 10.1056/NEJM197706022962202. [DOI] [PubMed] [Google Scholar]
- 20.Moller LN, Stidsen CE, Hartmann B, Holst JJ. Somatostatin receptors. Biochim Biophys Acta. 2003;1616(1):1–84. doi: 10.1016/s0005-2736(03)00235-9. [DOI] [PubMed] [Google Scholar]
- 21.Knop FK, Hare KJ, Pedersen J, Hendel J, Holst J, Vilsboll T. Prohormone convertase 2 positive enteroendocrine cells are more abundant in patients with type 2 diabetes - a potential source of gut-derived glucagon. EASD, 47th Annual Meeting; 2011. [Google Scholar]
- 22.Dinneen S, Alzaid A, Turk D, Rizza R. Failure of glucagon suppression contributes to postprandial hyperglycaemia in IDDM. Diabetologia. 1995;38(3):337–343. doi: 10.1007/BF00400639. [DOI] [PubMed] [Google Scholar]
- 23.Shah P, Basu A, Basu R, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol. 1999;277(2 Pt 1):E283–E290. doi: 10.1152/ajpendo.1999.277.2.E283. [DOI] [PubMed] [Google Scholar]
- 24.Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2000;85(11):4053–4059. doi: 10.1210/jcem.85.11.6993. [DOI] [PubMed] [Google Scholar]
- 25.Knop FK, Vilsboll T, Madsbad S, Holst JJ, Krarup T. Inappropriate suppression of glucagon during OGTT but not during isoglycaemic i.v. glucose infusion contributes to the reduced incretin effect in type 2 diabetes mellitus. Diabetologia. 2007;50(4):797–805. doi: 10.1007/s00125-006-0566-z. [DOI] [PubMed] [Google Scholar]
- 26.Hare KJ, Vilsboll T, Holst JJ, Knop FK. Inappropriate glucagon response after oral compared with isoglycemic intravenous glucose administration in patients with type 1 diabetes. Am J Physiol Endocrinol Metab. 2010;298(4):E832–E837. doi: 10.1152/ajpendo.00700.2009. [DOI] [PubMed] [Google Scholar]
- 27.Holst JJ, Christensen M, Lund A, de Heer J, Svendsen B, Kielgast U. et al. Regulation of glucagon secretion by incretins. Diabetes Obes Metab. 2011;13(Suppl 1):89–94. doi: 10.1111/j.1463-1326.2011.01452.x. [DOI] [PubMed] [Google Scholar]
- 28.Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64(1):106–110. doi: 10.1210/jcem-64-1-106. [DOI] [PubMed] [Google Scholar]
- 29.Consoli A, Nurjhan N, Capani F, Gerich J. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes. 1989;38(5):550–557. doi: 10.2337/diab.38.5.550. [DOI] [PubMed] [Google Scholar]
- 30.Parker JC, Andrews KM, Allen MR, Stock JL, McNeish JD. Glycemic control in mice with targeted disruption of the glucagon receptor gene. Biochem Biophys Res Commun. 2002;290(2):839–843. doi: 10.1006/bbrc.2001.6265. [DOI] [PubMed] [Google Scholar]
- 31.Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L. et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA. 2003;100(3):1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorensen H, Winzell MS, Brand CL, Fosgerau K, Gelling RW, Nishimura E. et al. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes. 2006;55(12):3463–3469. doi: 10.2337/db06-0307. [DOI] [PubMed] [Google Scholar]
- 33.Guettet C, Rostaqui N, Mathe D, Lecuyer B, Navarro N, Jacotot B. Effect of chronic glucagon administration on lipoprotein composition in normally fed, fasted and cholesterol-fed rats. Lipids. 1991;26(6):451–458. doi: 10.1007/BF02536072. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, MacDougall ML, McDowell MT, Xi L, Wei R, Zavadoski WJ. et al. Polyomic profiling reveals significant hepatic metabolic alterations in glucagon-receptor (GCGR) knockout mice: implications on anti-glucagon therapies for diabetes. BMC Genomics. 2011;12:281. doi: 10.1186/1471-2164-12-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60(2):391–397. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgerton DS, Cherrington AD. Glucagon as a critical factor in the pathology of diabetes. Diabetes. 2011;60(2):377–380. doi: 10.2337/db10-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C. et al. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes. 2004;53(2):410–417. doi: 10.2337/diabetes.53.2.410. [DOI] [PubMed] [Google Scholar]
- 38.Sloop KW, Cao JX-C, Siesky AM, Zhang HY, Bodenmiller DM, Cox AL. et al. Hepatic and glucagon-like peptide-1-mediated reversal of diabetes by glucagon receptor antisense oligonucleotide inhibitors. J Clin Invest. 2004;113(11):1571–1581. doi: 10.1172/JCI20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brand CL, Rolin B, Jørgensen PN, Svendsen I, Kristensen JS, Holst JJ. Immunoneutralization of endogenous glucagon with monoclonal glucagon antibody normalizes hyperglycaemia in moderately streptozotocin-diabetic rats. Diabetologia. 1994;37(10):985–993. doi: 10.1007/BF00400461. [DOI] [PubMed] [Google Scholar]
- 40.Brand CL, Jorgensen PN, Svendsen I, Holst JJ. Evidence for a major role for glucagon in regulation of plasma glucose in conscious, nondiabetic, and alloxan-induced diabetic rabbits. Diabetes. 1996;45(8):1076–1083. doi: 10.2337/diab.45.8.1076. [DOI] [PubMed] [Google Scholar]
- 41.Hancock AS, Du A, Liu J, Miller M, May CL. Glucagon deficiency reduces hepatic glucose production and improves glucose tolerance in adult mice. Mol Endocrinol. 2010;24(8):1605–1614. doi: 10.1210/me.2010-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D. Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science. 1982;215(4536):1115–1116. doi: 10.1126/science.6278587. [DOI] [PubMed] [Google Scholar]
- 43.Gysin B, Johnson DG, Trivedi D, Hruby VJ. Synthesis of two glucagon antagonists: receptor binding, adenylate cyclase, and effects on blood plasma glucose levels. J Med Chem. 1987;30(8):1409–1415. doi: 10.1021/jm00391a024. [DOI] [PubMed] [Google Scholar]
- 44.Van Tine BA, Azizeh BY, Trivedi D, Phelps JR, Houslay MD, Johnson DG. et al. Low level cyclic adenosine 3',5'-monophosphate accumulation analysis of (des-His1, des- Phe6, Glu9) glucagon-NH2 identifies glucagon antagonists from weak partial agonists/antagonists. Endocrinology. 1996;137(8):3316–3322. doi: 10.1210/endo.137.8.8754757. [DOI] [PubMed] [Google Scholar]
- 45.Parker JC, McPherson RK, Andrews KM, Levy CB, Dubins JS, Chin JE. et al. Effects of skyrin, a receptor-selective glucagon antagonist, in rat and human hepatocytes. Diabetes. 2000;49(12):2079–2086. doi: 10.2337/diabetes.49.12.2079. [DOI] [PubMed] [Google Scholar]
- 46.Madsen P, Ling A, Plewe M, Sams CK, Knudsen LB, Sidelmann UG. et al. Optimization of alkylidene hydrazide based human glucagon receptor antagonists. Discovery of the highly potent and orally available 3-cyano-4-hydroxybenzoic acid (1-(2,3,5,6-tetramethylbenzyl)-1H-indol-4-ylmethylene)hydrazide. J Med Chem. 2002;45(26):5755–5775. doi: 10.1021/jm0208572. [DOI] [PubMed] [Google Scholar]
- 47.Qureshi SA, Rios Candelore M, Xie D, Yang X, Tota LM, Ding VD. et al. A novel glucagon receptor antagonist inhibits glucagon-mediated biological effects. Diabetes. 2004;53(12):3267–3273. doi: 10.2337/diabetes.53.12.3267. [DOI] [PubMed] [Google Scholar]
- 48.Shen DM, Zhang F, Brady EJ, Candelore MR, Dallas-Yang Q, Ding VD. et al. Discovery of novel, potent, and orally active spiro-urea human glucagon receptor antagonists. Bioorg Med Chem Lett. 2005;15(20):4564–4569. doi: 10.1016/j.bmcl.2005.06.101. [DOI] [PubMed] [Google Scholar]
- 49.Ladouceur GH, Cook JH, Doherty EM, Schoen WR, MacDougall ML, Livingston JN. Discovery of 5-hydroxyalkyl-4-phenylpyridines as a new class of glucagon receptor antagonists. Bioorg Med Chem Lett. 2002;12(3):461–464. doi: 10.1016/s0960-894x(01)00766-1. [DOI] [PubMed] [Google Scholar]
- 50.Lau J, Behrens C, Sidelmann UG, Knudsen LB, Lundt B, Sams C. et al. New beta-alanine derivatives are orally available glucagon receptor antagonists. J Med Chem. 2007;50(1):113–128. doi: 10.1021/jm058026u. [DOI] [PubMed] [Google Scholar]
- 51.Chang LL, Sidler KL, Cascieri MA, de Laszlo S, Koch G, Li B. et al. Substituted imidazoles as glucagon receptor antagonists. Bioorg Med Chem Lett. 2001;11(18):2549–2553. doi: 10.1016/s0960-894x(01)00498-x. [DOI] [PubMed] [Google Scholar]
- 52.Potterat O, Wagner K, Gemmecker G, Mack J, Puder C, Vettermann R. et al. BI-32169, a bicyclic 19-peptide with strong glucagon receptor antagonist activity from Streptomyces sp. J Nat Prod. 2004;67(9):1528–1531. doi: 10.1021/np040093o. [DOI] [PubMed] [Google Scholar]
- 53.Knappe TA, Linne U, Xie X, Marahiel MA. The glucagon receptor antagonist BI-32169 constitutes a new class of lasso peptides. FEBS Lett. 2010;584(4):785–789. doi: 10.1016/j.febslet.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 54.Madsen P, Kodra JT, Behrens C, Nishimura E, Jeppesen CB, Pridal L. et al. Human glucagon receptor antagonists with thiazole cores. A novel series with superior pharmacokinetic properties. J Med Chem. 2009;52(9):2989–3000. doi: 10.1021/jm8016249. [DOI] [PubMed] [Google Scholar]
- 55.Madsen P, Knudsen LB, Wiberg FC, Carr RD. Discovery and structure-activity relationship of the first non-peptide competitive human glucagon receptor antagonists. J Med Chem. 1998;41(26):5150–5157. doi: 10.1021/jm9810304. [DOI] [PubMed] [Google Scholar]
- 56.Yu R, Nissen NN, Dhall D, Heaney AP. Nesidioblastosis and hyperplasia of alpha cells, microglucagonoma, and nonfunctioning islet cell tumor of the pancreas: review of the literature. Pancreas. 2008;36(4):428–431. doi: 10.1097/MPA.0b013e31815ceb23. [DOI] [PubMed] [Google Scholar]
- 57.Zhou C, Dhall D, Nissen NN, Chen CR, Yu R. Homozygous P86S mutation of the human glucagon receptor is associated with hyperglucagonemia, alpha cell hyperplasia, and islet cell tumor. Pancreas. 2009;38(8):941–946. doi: 10.1097/MPA.0b013e3181b2bb03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen LH, Larger E, Chaput JC. Identification of a glucagon receptor gene deletion mutation in a patient with hyperglucagonemia and pseudo-adenomatous hyperplasia of pancreatic alpha-cells. J Clin Endocrinol Metab; Anual Meeting of the Endocrine Society; 1999. Abstract. [Google Scholar]
- 59.Petersen KF, Sullivan JT. Effects of a novel glucagon receptor antagonist (Bay 27-9955) on glucagon-stimulated glucose production in humans. Diabetologia. 2001;44(11):2018–2024. doi: 10.1007/s001250100006. [DOI] [PubMed] [Google Scholar]
- 60.Sloop KW, Michael MD, Moyers JS. Glucagon as a target for the treatment of type 2 diabetes. Exp Opin Ther Targets. 2005;9(3):593–600. doi: 10.1517/14728222.9.3.593. [DOI] [PubMed] [Google Scholar]
- 61.Deeg M, Abu-Raddad E, Tham L, Fu H, Pinaire J, Kelly R. Single doses of the glucagon receptor antagonist LY2409021 reduce blood glucose in healthy subjects and patients with type 2 diabetes mellitus. 47th EASD Annual Meeting; 12-16 September 2011; Lisbon, Portugal. Presentation Number 886. [Google Scholar]
- 62.Prince M, Garhyan P, Abu-Raddad E, Fu H, Lim C, Pinaire J. Short-term treatment with glucagon receptor antagonist LY2409021 effectively reduces fasting blood glucose (FBG) and HbA1c in patients with type 2 diabetes mellitus. 47th EASD Annual Meeting; 12-16 September 2011; Lisbon, Portugal. Presentation Number 190. [Google Scholar]
- 63.Kelly R, Tham L, Abu-Raddad E, Lim C, Loh M, Pinaire J. The glucagon receptor antagonist LY2409021 attenuates increases in hepatic glucose output (HGO) and blood glucose during hyperglucagonaemia in healthy male subjects. 47th EASD Annual Meeting; 12-16 September 2011; Lisbon, Portugal. Presentation Number 887. [Google Scholar]
- 64.Engel S, Teng R, Edwards R, Davies M, Kaufman K, Goldstein B. Efficacy and safety of the glucagon receptor antagonist, MK-0893, in combination with metformin or sitagliptin in patients with type 2 diabetes mellitus. 47th EASD Annual Meeting; 12-16 September 2011; Lisbon, Portugal. Presentation Number 191. [Google Scholar]
- 65.Engel S, Xu L, Andryuk P, Davies M, Amatruda J, Kaufman K. Efficacy and tolerability of MK-0893, a glucagon receptor antagonist (GRA), in patients with type 2 diabetes (T2DM). American Diabetes Association 71st Scientific Sessions; 24-28 June 2011; San Diego, California. Presentation 309-OR. [Google Scholar]
- 66.Ruddy M, Pramanik B, Lunceford J, Li S, Cilissen C, Stoch A. Inhibition of glucagon-induced hyperglycemia predicts glucose lowering efficacy of a glucagon receptor antagonist, MK-0893, in type 2 diabetes (T2DM). American Diabetes Association 71st Scientific Sessions; 24-28 June 2011; San Diego, California. Presentation 311-OR. [Google Scholar]
- 67.Troyer M, Hompesch M, Pramanik B, Zheng W, Win K, Dunbar S. Recovery from hypoglycemia in healthy subjects is preserved despite glucagon receptor blockade by MK-0893. American Diabetes Association 71st Scientific Sessions; 24-28 June 2011; San Diego, California. Presentation 494-P. [Google Scholar]
- 68.Troyer M, Hompesch M, Jax T, Pramanik B, Liu F, Morrow L. Recovery from hypoglycemic clamp in type 2 diabetes (T2DM) patients during beta-adrenergic blockade plus glucagon receptor blockade with MK-0893. American Diabetes Association 71st Scientific Sessions; 24-28 June 2011; San Diego, California. Presentation 495-P. [Google Scholar]
- 69.Li X, Chen Y, Zhang A, Zhou G, Li C. A glucagon receptor antagonist regulates glucose and lipid metabolism in human hepatocytes. American Diabetes Association 71st Scientific Sessions; 24-28 June 2011; San Diego, California. Presentation 103-LB. [Google Scholar]
- 70.Deacon CF, Ahren B. Physiology of incretins in health and disease. Rev Diabet Stud. 2011;8(3):293–306. doi: 10.1900/RDS.2011.8.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garber AJ. Incretin therapy - present and future. Rev Diabet Stud. 2011;8(3):307–322. doi: 10.1900/RDS.2011.8.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 73.Creutzfeldt WO, Kleine N, Willms B, Orskov C, Holst JJ, Nauck MA. Glucagonostatic actions and reduction of fasting hyperglycemia by exogenous glucagon-like peptide I(7-36) amide in type I diabetic patients. Diabetes Care. 1996;19(6):580–586. doi: 10.2337/diacare.19.6.580. [DOI] [PubMed] [Google Scholar]
- 74.de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia. 2008;51(12):2263–2270. doi: 10.1007/s00125-008-1149-y. [DOI] [PubMed] [Google Scholar]
- 75.Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R. et al. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87(3):1239–1246. doi: 10.1210/jcem.87.3.8355. [DOI] [PubMed] [Google Scholar]
- 76.Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA. et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2003;88(7):3082–3089. doi: 10.1210/jc.2002-021545. [DOI] [PubMed] [Google Scholar]
- 77.DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross-over study. Curr Med Res Opin. 2008;24(10):2943–2952. doi: 10.1185/03007990802418851. [DOI] [PubMed] [Google Scholar]
- 78.Dupre J, Behme MT, McDonald TJ. Exendin-4 normalized postcibal glycemic excursions in type 1 diabetes. J Clin Endocrinol Metab. 2004;89(7):3469–3473. doi: 10.1210/jc.2003-032001. [DOI] [PubMed] [Google Scholar]
- 79.Degn KB, Juhl CB, Sturis J, Jakobsen G, Brock B, Chandramouli V. et al. One week’s treatment with the long-acting glucagon-like peptide 1 derivative liraglutide (NN2211) markedly improves 24-h glycemia and alpha- and beta-cell function and reduces endogenous glucose release in patients with type 2 diabetes. Diabetes. 2004;53(5):1187–1194. doi: 10.2337/diabetes.53.5.1187. [DOI] [PubMed] [Google Scholar]
- 80.Juhl CB, Hollingdal M, Sturis J, Jakobsen G, Agerso H, Veldhuis J. et al. Bedtime administration of NN2211, a long-acting GLP-1 derivative, substantially reduces fasting and postprandial glycemia in type 2 diabetes. Diabetes. 2002;51(2):424–429. doi: 10.2337/diabetes.51.2.424. [DOI] [PubMed] [Google Scholar]
- 81.Garber A, Henry R, Ratner R, Garcia-Hernandez PA, Rodriguez-Pattzi H, Olvera-Alvarez I. et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet. 2009;373(9662):473–481. doi: 10.1016/S0140-6736(08)61246-5. [DOI] [PubMed] [Google Scholar]
- 82.Zinman B, Gerich J, Buse JB, Lewin A, Schwartz S, Raskin P. et al. Efficacy and safety of the human glucagon-like peptide-1 analog liraglutide in combination with metformin and thiazolidinedione in patients with type 2 diabetes (LEAD-4 Met+TZD) Diabetes Care. 2009;32(7):1224–1230. doi: 10.2337/dc08-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH. et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6) Lancet. 2009;374(9683):39–47. doi: 10.1016/S0140-6736(09)60659-0. [DOI] [PubMed] [Google Scholar]
- 84.Kielgast U, Holst JJ, Madsbad S. Antidiabetic actions of endogenous and exogenous GLP-1 in type 1 diabetic patients with and without residual beta-cell function. Diabetes. 2011;60(5):1599–1607. doi: 10.2337/db10-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Malloy J, Capparelli E, Gottschalk M, Guan X, Kothare P, Fineman M. Pharmacology and tolerability of a single dose of exenatide in adolescent patients with type 2 diabetes mellitus being treated with metformin: a randomized, placebo-controlled, single-blind, dose-escalation, crossover study. Clin Ther. 2009;31(4):806–815. doi: 10.1016/j.clinthera.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 86.Nauck MA, Kemmeries G, Holst JJ, Meier JJ. Rapid tachyphylaxis of the glucagon-like peptide 1-induced deceleration of gastric emptying in humans. Diabetes. 2011;60(5):1561–1565. doi: 10.2337/db10-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kapitza C, Zdravkovic M, Hindsberger C, Flint A. The effect of the once-daily human glucagon-like peptide 1 analog liraglutide on the pharmacokinetics of acetaminophen. Adv Ther. 2011;28(8):650–660. doi: 10.1007/s12325-011-0044-y. [DOI] [PubMed] [Google Scholar]
- 88.Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes. 2010;59(7):1765–1770. doi: 10.2337/db09-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS. et al. Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes. 2009;58(6):1342–1349. doi: 10.2337/db08-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lund A, Vilsboll T, Bagger JI, Holst JJ, Knop FK. The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes. Am J Physiol Endocrinol Metab. 2011;300(6):E1038–E1046. doi: 10.1152/ajpendo.00665.2010. [DOI] [PubMed] [Google Scholar]
- 91.Ahren B, Landin-Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J Clin Endocrinol Metab. 2004;89(5):2078–2084. doi: 10.1210/jc.2003-031907. [DOI] [PubMed] [Google Scholar]
- 92.Azuma K, Radikova Z, Mancino J, Toledo FG, Thomas E, Kangani C. et al. Measurements of islet function and glucose metabolism with the dipeptidyl peptidase 4 inhibitor vildagliptin in patients with type 2 diabetes. J Clin Endocrinol Metab. 2008;93(2):459–464. doi: 10.1210/jc.2007-1369. [DOI] [PubMed] [Google Scholar]
- 93.Rosenstock J, Foley JE, Rendell M, Landin-Olsson M, Holst JJ, Deacon CF. et al. Effects of the dipeptidyl peptidase-IV inhibitor vildagliptin on incretin hormones, islet function, and postprandial glycemia in subjects with impaired glucose tolerance. Diabetes Care. 2008;31(1):30–35. doi: 10.2337/dc07-1616. [DOI] [PubMed] [Google Scholar]
- 94.Ahren B, Foley JE, Ferrannini E, Matthews DR, Zinman B, Dejager S. et al. Changes in prandial glucagon levels after a 2-year treatment with vildagliptin or glimepiride in patients with type 2 diabetes inadequately controlled with metformin monotherapy. Diabetes Care. 2010;33(4):730–732. doi: 10.2337/dc09-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ahren B, Schweizer A, Dejager S, Dunning BE, Nilsson PM, Persson M. et al. Vildagliptin enhances islet responsiveness to both hyper- and hypoglycemia in patients with type 2 diabetes. J Clin Endocrinol Metab. 2009;94(4):1236–1243. doi: 10.1210/jc.2008-2152. [DOI] [PubMed] [Google Scholar]
- 96.Iwamoto Y, Tajima N, Kadowaki T, Nonaka K, Taniguchi T, Nishii M. et al. Efficacy and safety of sitagliptin monotherapy compared with voglibose in Japanese patients with type 2 diabetes: a randomized, double-blind trial. Diabetes Obes Metab. 2010;12(7):613–622. doi: 10.1111/j.1463-1326.2010.01197.x. [DOI] [PubMed] [Google Scholar]
- 97.Chacra AR, Tan GH, Apanovitch A, Ravichandran S, List J, Chen R. Saxagliptin added to a submaximal dose of sulphonylurea improves glycaemic control compared with uptitration of sulphonylurea in patients with type 2 diabetes: a randomised controlled trial. Int J Clin Pract. 2009;63(9):1395–1406. doi: 10.1111/j.1742-1241.2009.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.D'Alessio DA, Denney AM, Hermiller LM, Prigeon RL, Martin JM, Tharp WG. et al. Treatment with the dipeptidyl peptidase-4 inhibitor vildagliptin improves fasting islet-cell function in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2009;94(1):81–88. doi: 10.1210/jc.2008-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jadzinsky M, Pfützner A, Paz-Pacheco E, Xu Z, Allen E, Chen R. Saxagliptin given in combination with metformin as initial therapy improves glycaemic control in patients with type 2 diabetes compared with either monotherapy: a randomized controlled trial. Diabetes Obes Metab. 2009;11(6):611–622. doi: 10.1111/j.1463-1326.2009.01056.x. [DOI] [PubMed] [Google Scholar]
- 100.Rosenstock J, Aguilar-Salinas C, Klein E, Nepal S, List J, Chen R. Effect of saxagliptin monotherapy in treatment-naïve patients with type 2 diabetes. Curr Med Res Opin. 2009;25(10):2401–2411. doi: 10.1185/03007990903178735. [DOI] [PubMed] [Google Scholar]
- 101.Hollander P, Li J, Allen E, Chen R. Saxagliptin added to a thiazolidinedione improves glycemic control in patients with type 2 diabetes and inadequate control on thiazolidinedione alone. J Clin Endocrinol Metab. 2009;94(12):4810–4819. doi: 10.1210/jc.2009-0550. [DOI] [PubMed] [Google Scholar]
- 102.DeFronzo RA, Hissa MN, Garber AJ, Luiz Gross J, Yuyan Duan R, Ravichandran S. et al. The efficacy and safety of saxagliptin when added to metformin therapy in patients with inadequately controlled type 2 diabetes with metformin alone. Diabetes Care. 2009;32(9):1649–1655. doi: 10.2337/dc08-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Balas B, Baig MR, Watson C, Dunning BE, Ligueros-Saylan M, Wang Y. et al. The dipeptidyl peptidase IV inhibitor vildagliptin suppresses endogenous glucose production and enhances islet function after single-dose administration in type 2 diabetic patients. J Clin Endocrinol Metab. 2007;92(4):1249–1255. doi: 10.1210/jc.2006-1882. [DOI] [PubMed] [Google Scholar]
- 104.Herman GA, Bergman A, Stevens C, Kotey P, Yi B, Zhao P. et al. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91(11):4612–4619. doi: 10.1210/jc.2006-1009. [DOI] [PubMed] [Google Scholar]
- 105.He YL, Serra D, Wang Y, Campestrini J, Riviere GJ, Deacon CF. et al. Pharmacokinetics and pharmacodynamics of vildagliptin in patients with type 2 diabetes mellitus. Clin Pharmacokinet. 2007;46(7):577–588. doi: 10.2165/00003088-200746070-00003. [DOI] [PubMed] [Google Scholar]
- 106.Young A. Inhibition of glucagon secretion. Adv Pharmacol. 2005;52:151–171. doi: 10.1016/S1054-3589(05)52008-8. [DOI] [PubMed] [Google Scholar]
- 107.Young A. Inhibition of gastric emptying. Adv Pharmacol. 2005;52:99–121. doi: 10.1016/S1054-3589(05)52006-4. [DOI] [PubMed] [Google Scholar]
- 108.Young A. Inhibition of food intake. Adv Pharmacol. 2005;52:79–98. doi: 10.1016/S1054-3589(05)52005-2. [DOI] [PubMed] [Google Scholar]
- 109.Silvestre RA, Rodriguez-Gallardo J, Jodka C, Parkes DG, Pittner RA, Young AA. et al. Selective amylin inhibition of the glucagon response to arginine is extrinsic to the pancreas. Am J Physiol Endocrinol Metab. 2001;280(3):E443–E449. doi: 10.1152/ajpendo.2001.280.3.E443. [DOI] [PubMed] [Google Scholar]
- 110.Wang F, Adrian TE, Westermark GT, Ding X, Gasslander T, Permert J. Islet amyloid polypeptide tonally inhibits beta-, alpha-, and delta-cell secretion in isolated rat pancreatic islets. Am J Physiol. 1999;276(1 Pt 1):E19–E24. doi: 10.1152/ajpendo.1999.276.1.E19. [DOI] [PubMed] [Google Scholar]
- 111.Roth JD, Maier H, Chen S, Roland BL. Implications of amylin receptor agonism: integrated neurohormonal mechanisms and therapeutic applications. Arch Neurol. 2009;66(3):306–310. doi: 10.1001/archneurol.2008.581. [DOI] [PubMed] [Google Scholar]
- 112.Asmar M, Bache M, Knop FK, Madsbad S, Holst JJ. Do the actions of glucagon-like peptide-1 on gastric emptying, appetite, and food intake involve release of amylin in humans? J Clin Endocrinol Metab. 2010;95(5):2367–2375. doi: 10.1210/jc.2009-2133. [DOI] [PubMed] [Google Scholar]
- 113.Whitehouse F, Kruger DF, Fineman M, Shen L, Ruggles JA, Maggs DG. et al. A randomized study and open-label extension evaluating the long-term efficacy of pramlintide as an adjunct to insulin therapy in type 1 diabetes. Diabetes Care. 2002;25(4):724–730. doi: 10.2337/diacare.25.4.724. [DOI] [PubMed] [Google Scholar]
- 114.Ratner RE, Dickey R, Fineman M, Maggs DG, Shen L, Strobel SA. et al. Amylin replacement with pramlintide as an adjunct to insulin therapy improves long-term glycaemic and weight control in type 1 diabetes mellitus: a 1-year, randomized controlled trial. Diabet Med. 2004;21(11):1204–1212. doi: 10.1111/j.1464-5491.2004.01319.x. [DOI] [PubMed] [Google Scholar]
- 115.Edelman S, Garg S, Frias J, Maggs D, Wang Y, Zhang B. et al. A double-blind, placebo-controlled trial assessing pramlintide treatment in the setting of intensive insulin therapy in type 1 diabetes. Diabetes Care. 2006;29(10):2189–2195. doi: 10.2337/dc06-0042. [DOI] [PubMed] [Google Scholar]
- 116.Nyholm B, Orskov L, Hove KY, Gravholt CH, Moller N, Alberti KG. et al. The amylin analog pramlintide improves glycemic control and reduces postprandial glucagon concentrations in patients with type 1 diabetes mellitus. Metab Clin Exp. 1999;48(7):935–941. doi: 10.1016/s0026-0495(99)90232-9. [DOI] [PubMed] [Google Scholar]
- 117.Fineman MS, Koda JE, Shen LZ, Strobel SA, Maggs DG, Weyer C. et al. The human amylin analog, pramlintide, corrects postprandial hyperglucagonemia in patients with type 1 diabetes. Metab Clin Exp. 2002;51(5):636–641. doi: 10.1053/meta.2002.32022. [DOI] [PubMed] [Google Scholar]
- 118.Hollander P, Ratner R, Fineman M, Strobel S, Shen L, Maggs D. et al. Addition of pramlintide to insulin therapy lowers HbA1c in conjunction with weight loss in patients with type 2 diabetes approaching glycaemic targets. Diabetes Obes Metab. 2003;5(6):408–414. doi: 10.1046/j.1463-1326.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- 119.Riddle M, Frias J, Zhang B, Maier H, Brown C, Lutz K. et al. Pramlintide improved glycemic control and reduced weight in patients with type 2 diabetes using basal insulin. Diabetes Care. 2007;30(11):2794–2799. doi: 10.2337/dc07-0589. [DOI] [PubMed] [Google Scholar]
- 120.Riddle M, Pencek R, Charenkavanich S, Lutz K, Wilhelm K, Porter L. Randomized comparison of pramlintide or mealtime insulin added to basal insulin treatment for patients with type 2 diabetes. Diabetes Care. 2009;32(9):1577–1582. doi: 10.2337/dc09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fineman M, Weyer C, Maggs DG, Strobel S, Kolterman OG. The human amylin analog, pramlintide, reduces postprandial hyperglucagonemia in patients with type 2 diabetes mellitus. Horm Metab Res. 2002;34(9):504–508. doi: 10.1055/s-2002-34790. [DOI] [PubMed] [Google Scholar]