

Abstract
Type 2 diabetes and obesity are associated with systemic inflammation, generalized enlargement of fat depots, and uncontrolled release of fatty acids (FA) into the circulation. These features support the occurrence of cardiac adiposity, which is characterized by an increase in intramyocardial triglyceride content and an enlargement of the volume of fat surrounding the heart and vessels. Both events may initially serve as protective mechanisms to portion energy, but their excessive expansion can lead to myocardial damage and heart disease. FA overload promotes FA oxidation and the accumulation of triglycerides and metabolic intermediates, which can impair calcium signaling, β-oxidation, and glucose utilization. This leads to damaged mitochondrial function and increased production of reactive oxygen species, pro-apoptotic, and inflammatory molecules, and finally to myocardial inflammation and dysfunction. Triglyceride accumulation is associated with left ventricular hypertrophy and dysfunction. The enlargement of epicardial fat in patients with metabolic disorders, and coronary artery disease, is associated with the release of proinflammatory and proatherogenic cytokines to the subtending tissues. In this review, we examine the evidence supporting a causal relationship linking FA overload and cardiac dysfunction. Also, we disentangle the separate roles of FA oxidation and triglyceride accumulation in causing cardiac damage. Finally, we focus on the mechanisms of inflammation development in the fatty heart, before summarizing the available evidence in humans. Current literature confirms the dual (protective and detrimental) role of cardiac fat, and suggests prospective studies to establish the pathogenetic (when and how) and possible prognostic value of this potential biomarker in humans.
Keywords: type 2 diabetes, obesity, cardiovascular disease, imaging, ectopic fat, epicardial fat, lipid, lipotoxicity
Abbreviations: ACS - acyl-CoA synthetase; Acyl-CoA - acyl-coenzym A; AGPAT - acylglycerolphosphate acyltransferase; AMP - adenosine 5'-monophosphate; AMPKα - AMP-activated protein kinase-alpha; ANP - atrial natriuretic factor; ATGL - adipose tissue triglyceride lipase; ATP - adenosine triphosphate; BMI - body mass index; BNP - brain natriuretic peptide; CAD - coronary artery disease; CCL2 - chemokine (C-C motif) ligand 2; CPT1 - carnitine palitoyl transferase 1; CTE-1 - cytosolic thioesterase-1; DAG - diacyglycerol; DGAT1- diacyglycerolacyltransferases isoform 1; eNOS - endothelial nitrix oxide sinthase; ER - endoplasmic reticulum; FA - fatty acids; FABPpm - FA binding protein; FAT - fatty acid translocase (also called CD36); FATP1/6 - FA transport protein 1 (and 6); GPAT - glycerolphosphate acyltransferase; GLUT4 - glucose transporter 4; GPAT - glycerol-3-phosphate acyltranspherase; HFD - high fat diet; HSL - hormone sensitive lipase; IKK - I kappaB kinase; IL - interleukine; JNK - C-Jun N-terminal kinase; KO - knock-out; LFD - low-fat diet; LPA - lysophosphatidic acid; LpL - lipoprotein lipase; LV - left ventricular; MAPK - mitogen-activated protein kinase; MCP1 - monocyte chemoattractant proein-1; MHC-a/b - myosin heavy chain isoform a/b; MMP - matrix metalloproteinases; MRS - magnetic resonance spectroscopy; MTE-1 - mitochondrial thioesterase-1; MTP - microsomal triglyceride transfer protein; MVO2 - myocardial oxygen consumption; NF-κB - nuclear factor kappa-light-chain-enhancer of activated B cells; PA - phosphatidic acid; PDH - pyruvate dehydrogenase; PDK-4 - pyruvate dehydrogenase kinase-4; PET - positron emission tomography; PKB/C - protein kinase B/C; PPARα - peroxisome proliferator activated receptor alpha; ROS - reactive oxygen species; SOD - superoxide dismutase; SPT - serine palmitolyltransferase; SR - sarcoplasmic reticulum; STZ - streptozoticin; T2D - type 2 diabetes; TG - triglyceride; TNFα - tumor necrosis factor alpha; Tpl2 - tumor progression locus 2; UCP-3 - uncoupling protein-3; WD - western diet
Introduction
Cardiac complications are the leading cause of mortality in patients suffering from type 2 diabetes (T2D) or obesity. In these patients, an increased supply of fatty acids is correlated with augmented cardiac adiposity and cardiac dysfunction. Indeed, the increased prevalence of metabolic and cardiovascular diseases has directed attention to the action of myocardial metabolic regulation mechanisms. Recent advances in imaging technology have allowed to explore, and partly support, the association between cardiac adiposity and systemic inflammation in patients with obesity, diabetes, and heart disease. However, correlative human studies have not yet established any causal mechanisms. Animal models of obesity and diabetes, and genetically modified (molecularly targeted) models, have been developed and used to characterize the relationship between intramyocardial lipid accumulation and ventricular dysfunction.
Cardiac adiposity refers to the presence of a large number of lipid droplets inside the myocytes that emerges because of excessive fat accumulation in the cardiac muscle. Intramyocardial triglyceride depots, which are increased in obesity and type 2 diabetes, are associated with greater left ventricular (LV) mass, impaired diastolic func left ventricular (LV) mass, impaired diastolic function, higher cardiac work, and oxygen consumption [1, 2]. In this context, cardiac dysfunction correlates with myocyte accumulation of lipotoxic intermediates and apoptosis. Another definition of cardiac adiposity is increased thickness or volume of adipose tissue layers surrounding the heart and vessels. These fat layers are described as epicardial (between myocardium and visceral pericardium), or pericardial (between visceral and parietal pericardium, or on the external surface of the pericardium).
Both fat depots inside and around the heart may initially serve as a fatty acid (FA) deposits and exert a protective role in energy portioning. However, their excessive expansion has been associated with inflammation, various kinds of myocardial damage, and heart disease. Increased FA availability is being considered as one of the major players in the development of myocardial damage and chronic inflammation. In fact, increased lipolysis leading to a higher FA supply to non-adipose tissues is a common feature in diabetes, insulin resistance, and obesity (once the adipocyte buffering capacity saturates), and may be associated with the adrenergic activation that correlates with heart failure [3]. In the healthy heart, FAs represent the main fuel, being responsible for 70% of the oxidative ATP production [4]. Intracellular FAs activate the transcription factor peroxisome proliferator activated receptor alpha (PPARα), which regulates several genes involved in FA metabolism [5], including those involved in FA uptake, esterification, transport into mitochondria, and β-oxidation. As cardiac FA influx increases, PPARα is activated in a feed-forward fashion. Thereby, in response to FA overload, both FA oxidation and esterification are increased. Eventually, TG accumulation occurs, and is further promoted by the exhaustion of oxidative capacity [6].
Increased FA oxidation is associated with production of reactive oxygen species (ROS) which leads to myocardial dysfunction and cardiomyopathy [7-9]. Moreover, FAs might be diverted to non-oxidative pathways, including the formation of lipotoxic intermediates and pro-apoptotic species. FA overload also decreases glucose oxidation by inhibiting pyruvate dehydrogenase (PDH) activity through a PPARα-regulated mechanism, and reduces the expression of sarcolemma glucose transporter 4 (GLUT4), thus deteriorating insulin sensitivity [8, 10].
In this review, we examine the evidence supporting a causal relationship linking FA overload with cardiac dysfunction. Also, we disentangle the separate roles of FA oxidation, and TG accumulation, in causing cardiac damage, and we focus on the mechanisms underlying the development of inflammation in the fatty heart. Finally, we summarize the available evidence for the fatty heart in humans.
Fatty acid overload and cardiac function
In lipotoxic heart disease, increased plasma FA availability is a first and likely causative event correlated with cardiac hypertrophy and dysfunction. FA cellular uptake is mediated by sarcolemma proteins CD36, plasma membrane FA-binding protein (FABPpm), and FA transport protein 1 and 6 (FATP1/6). Also, intracellular proteins may independently enhance FA uptake by binding and modifying the intracellular FA pool, thereby creating an inward FA gradient. Among them, acyl-CoA synthetase (ACS) represents the first step in FA intracellular metabolism, since it converts FAs to acyl-CoA derivates, which may eventually be oxidized, or incorporated into lipids (Figure 1).
Figure 1. Enzymatic pathways regulating fatty acid (FA) metabolism in the heart.
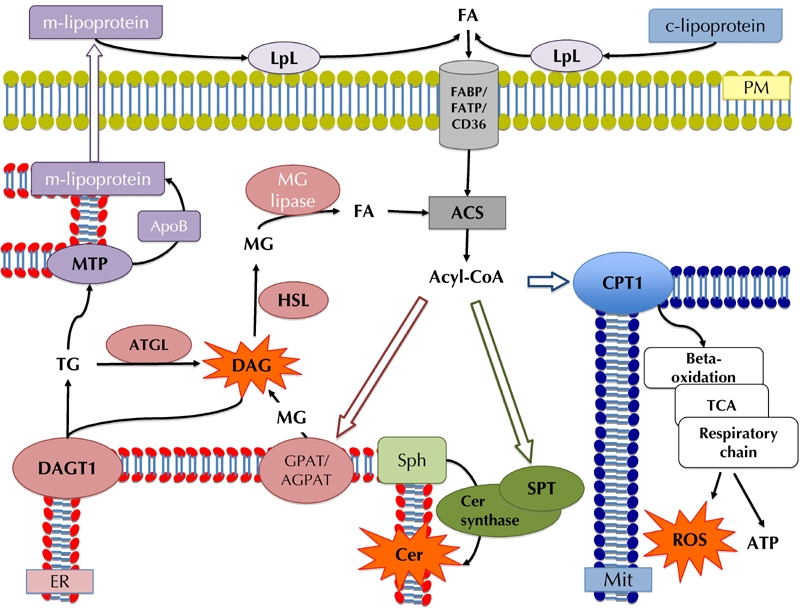
FAs enter the cardiomyocyte through specific transporters, and they are converted to acyl-CoA by acyl-CoA synthethase (ACS). Acyl-CoAs might be used for β-oxidation (blue pathway), or they can be diverted to non-oxidative pathways, including esterification and TG synthesis (pink pathway) and formation of lipotoxic intermediates, namely ceramides (Cer) (bottom, green pathway). Reactive oxygen species (ROS) and diacylglycerol (DAG), resulting from oxidation or esterification, are also toxic intermediates. Intracellular TG may accumulate in lipoproteins (top left, purple pathway) to be eventually released in the circulation, or be hydrolyzed by adipose tissue triglyceride lipase (ATGL), leading to intracellular release of FA. Intracellular FAs can follow one of the pathways mentioned above, with further accumulation of cytotoxic molecules. Potentially toxic products are shown in orange. Abbreviations: Acyl-CoA: acyl coenzyme A. ACS: acyl-CoA synthetase. AGPAT: acyl glycerolphosphate acyltransferase. ApoB: apolipoprotein B. ATGL: adipose tissue triglyceride lipase. Cer: ceramide. CPT1: carnitine palmitoyl transferase-1. DAG: diacyloglycerol. DGAT1: diacylglycerol-acyltransferase 1. ER: endoplasmic reticulum. FA: fatty acid. FABP: fatty acid binding protein. FATP: fatty acid transport protein. GPAT: glycerolphosphate acyltransferase. HSL: hormone sensitive lipase. LpL: lipoproein lipase. m-/c-lipoprotein: cardiomyocyte released / circulating lipoprotein. MG: monoacylglycerol. Mit: mitochondrion. MTP: microsomal triglyceride transfer protein. PM: plasma membrane. ROS: reactive oxygen species. Sph: sphingosine. SPT: serine palmitoyl-transferase. TCA: tricarboxylic acid cycle. TG: triglyceride.
Studies have been carried out in wild type and genetically modified rodent models, undergoing high-fat or control diet feeding, to elucidate the effect of an FA overload on cardiac function. Mutations in leptin, and leptin receptor, genes are responsible for augmented food intake observed in db/db, and ob/ob, mice models, in which diabetes and reduced cardiac power are observed at 5 and 15 weeks of age, respectively. Hearts of diabetic db/db mice develop a 2-3-fold increase in myocardial TG accumulation, which is associated with LV contractile dysfunction [8]. Isolated hearts of insulin-resistant ob/ob mice showed augmented FA oxidation and myocardial oxygen consumption (MVO2) at the expense of glucose utilization, in response to perfusion with increasing palmitate concentrations. This finding was independent of insulin administration. It indicates that ob/ob mice are unable to modulate substrate utilization [2].
In obese compared to lean Zucker rats, the higher number of lipid droplets found in cardiomyocytes is due to enhanced FA transport through FABPpm and CD36, which is not accompanied by an increase in the FA oxidation rate [11]. Moreover, Burgmaier et al. observed that obese Zucker rats fed a western diet (WD, with excess calories and lipids) showed a significant decrease in cardiac power, increased TG synthesis and accumulation after only 7 days of WD feeding [12]. These changes were not seen in animals fed a control-diet, or in lean rats, in which FA oxidation was upregulated after 7 days of high-calorie diet. The latter effect counteracted the initial FA overload. This response was observed with a delay of 4 weeks from the start of WD in the obese group, in which the mismatch between FA uptake and FA oxidation led to a massive myocardial TG accumulation and LV dysfunction at 7 days of WD. The subsequent (though late) activation of oxidation prevented a further impairment in cardiac function. Metabolic findings correlated with gene expression in the obese animal group, in which the levels of glycerol-3-phosphate acyltranspherase (GPAT), promoting TG synthesis, were increased after 7 days of WD. Whereas, the levels of oxidative enzymes (pyruvate dehydrogenase kinase-4 PDK-4, uncoupling protein-3 UCP-3, mitochondrial and cytosolic thioesterase-1 MTE-1 and CTE-1 respectively) did not increase until the fourth week of diet. Ouwens et al. reported that rats fed a high-fat diet (HFD) for 8 weeks exhibited increased TG accumulation myocyte hypertrophy, and a decreased LV fractional shortening and ejection fraction [13]. Also, they showed impaired systemic glucose tolerance, relative to rats fed an isocaloric low-fat diet (LFD).
A HFD induces cardiac hypertrophy, and alters the subcellular localization of CD36. Current evidence suggests that the sarcolemma relocalization of CD36 is induced by FA overload itself (through PPARα activation). It precedes the onset of cardiac dysfunction, and it is related to the phosphorylation and activation of protein kinase B (PKB/Akt) signaling, which regulates insulin sensitivity [8, 14]. In fact, CD36 deficiency prevents myocardial TG accumulation, and counteracts LV dysfunction in rats overexpressing cardiac PPARα. CD36 deficiency does not affect the FA oxidative pathway, but it enhances glucose utilization [5, 6]. Accordingly, in isolated working hearts of FAT/CD36-null mice perfused with either low or high concentrations of palmitate, the rate of FA oxidation is significantly lower than in hearts of wild type mice. Notably, this animal model displayed a compensatory increase in myocardial glucose utilization, and an improvement in cardiac output and coronary blood flow [15].
Although CD36 is responsible for 40-60% of FA uptake, there are other membrane-associated transporters that provide the heart with this substrate. For example, cardiac overexpression of FATP1 results in higher FA uptake, which leads to a 2-fold increase in FA oxidation, and a 50% decrease in glucose metabolism, as measured by radioactive tracers (11C-palmitate, 14C-glucose, 3H-palmitate) [16]. Enhanced FATP1-mediated FA uptake, similar to CD36 membrane translocation, is associated with greater LV mass and internal diameter, a reduction in heart rate, and an impairment in diastolic function [16]. Thus, FAs are specifically implicated in the pathogenesis of diastolic dysfunction, which is a typical finding in obesity and diabetes, and a recognized early predictor of systolic dysfunction and heart failure.
In addition to membrane transporter proteins, FA uptake is regulated by the activity of ACS, which converts FA to acyl-CoA, and thus creates an intracellular deposit for FA. Lee et al. showed that in ACS transgenic mice, an augmented FA import caused heart hypertrophy, increased the LV anterior and posterior wall thicknesses, and compromised fractional shortening and systolic function [17, 18]. This cardiac dysfunction was associated with enlarged myocytes, accumulation of lipid droplets (FA uptake was increased by 59%, against a 16% increase in β-oxidation), myofiber disorganization, interstitial fibrosis, and a 3.3-fold rise in ceramide levels and apoptosis. [19].
FA overload resulting from overexpression of the membrane anchored lipoprotein lipase (LpL) in cardiomyocytes also induces lipid accumulation, involving TGs, ceramides, and cholesterol. LpL transgenic mice exhibit LV hypertrophy, increased PPARα expression, and mortality from dilated cardiomyopathy [20].
Molecular mechanisms underlying cardiac dysfunction caused by FA overload include alterations in cardiomyocyte excitation-contraction coupling and Ca2+ handling [13]. According to Haim et al., exposure to palmitate decreases contractility by reducing Ca2+ transients through an effect on sarcolemma membrane excitability in isolated ventricular myocytes from rodents [21]. Specifically, palmitate exposure increases the voltage-dependent K+ outward current resulting in potential action shortening, reduction in voltage-dependent Ca2+ influx, and Ca2+-dependent release of Ca2+ from intracellular stores. These effects reduce cell contractility. By comparing wild type with diabetic ob/ob mice, Fauconnier et al. suggested that in wild type mice, palmitate dissipates the membrane potential and increases mitochondrial ROS production, which are responsible for impaired Ca2+ handling [22]. According to this study, these effects are not observed in ob/ob mouse cardiomyocytes exposed to palmitate, suggesting that a prolonged exposure to high FA concentrations eventually results in cardiomyocyte adaptation.
Consequently, the available evidence indicates that FA overload exerts toxic effects on cardiac function, since it enhances metabolic pathways that alter intracellular metabolites concentrations and signaling molecules. Either FA oxidation, or FA accumulation, in TG are amplified in FA overload states, and it is difficult to quantify their individual contributions to cardiac damage. The following paragraphs address the evidence implicating each of these two pathways. Table 1 summarizes these studies.
Table 1. Summary of genetic and pharmacological animal models investigating the relation between fatty heart and cardiac function.
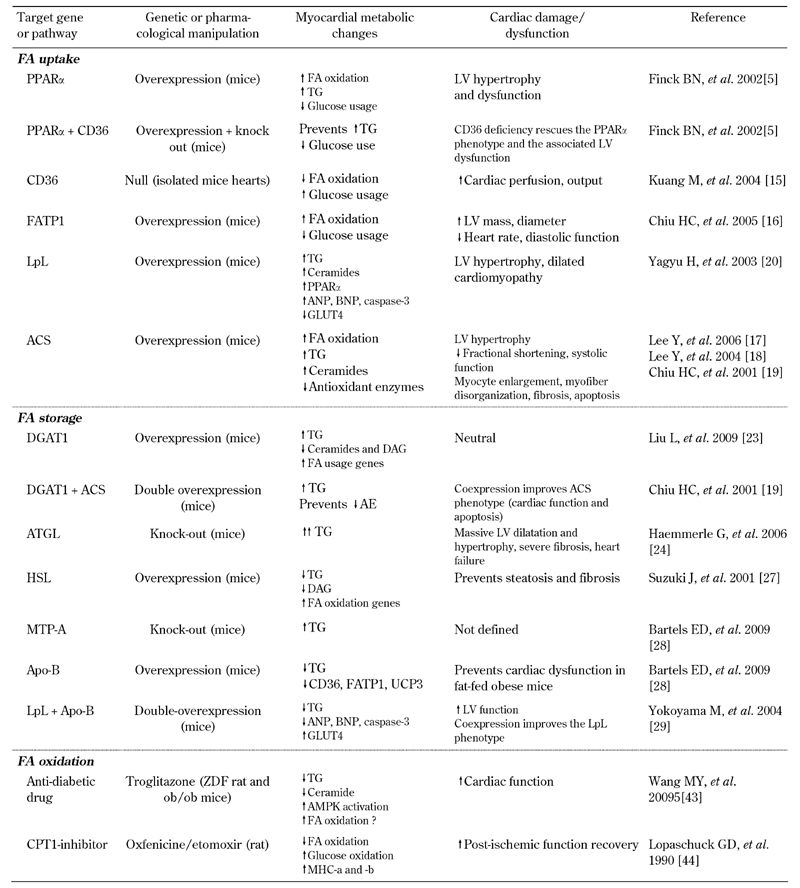
Legend: AE: antioxidant enzymes. AMPK: AMP-activated protein kinase. ANP: atrial natriuretic peptide. Apo-B: apolipoprotein B. ATGL: adipose tissue triglyceride lipase. BNP: brain natriuretic peptide. CD36: cluster of differentiation 36, also called fatty acid translocase (FAT). CPT1: carnitine palitoyl transferase 1. DAG: diacyglycerol. DGAT1: diacyglycerolacyltransferases isoform 1. HSL: hormone sensitive lipase. FA: fatty acid. FATP1: FA transport protein 1. GLUT4: glucose transporter 4. LpL: lipoprotein lipase. LV: left ventricle. MHC: myosin heavy chain. MTP-A: microsomal triglyceride transfer protein A. PPARα: peroxisome proliferator-activated receptor alpha. TG: triglyceride. UCP: uncoupling protein.
Triglyceride accumulation and cardiac function
Increased FA levels result in TG accumulation in cardiomyocytes. In fact, as FA oxidation becomes saturated, FA storage within TG depots provides a protective buffer mechanism, which prevents the incorporation of excessive FA (especially the long-chain saturated FAs like palmitate) into lipotoxic species, namely ceramides and diacyglycerols (DAGs). Nevertheless, massive intracellular TG accumulation correlates with ventricular hypertrophy, cardiac dysfunction, insulin resistance, and inflammatory markers. Esterification of intracellular FAs is catalyzed by acylglycerolphosphate acyltransferases (GPAT and AGPAT), which leads to formation of DAG. This is converted to TG by diacyglycerolacyltransferases, which is mostly represented by isoform 1 (DGAT1) in the heart. Intracellular TG lipolysis is catalyzed by adipose tissue triglyceride lipase (ATGL) and hormone sensitive lipase (HSL). Moreover, the microsomal triglyceride transfer protein (MTP), which is located in the endoplasmatic reticulum (ER), transfers neutral lipids onto the apoB polypeptide, contributing to triglyceride packaging and secretion in lipoproteins. Overall, the cardiac triglyceride pool is not inert, but rather depends on, and responds dynamically to, the intracellular fluxes of fatty acids. Figure 1 summarizes the pathways described above.
Cardiac overexpression of DGAT1 in mice increases TG accumulation, while decreasing ceramides, DAG, and FA by 35%, 26%, and 20% respectively, without altering cardiac function [23]. Interestingly, in mice which overexpress ACS and exhibit enlarged TG depots because of an imbalance between FA uptake and oxidation, overexpression of DGAT1 partially compensates for the impairment in fractional shortening, LV dysfunction, endoplasmic reticulum (ER) stress, and apoptosis [19]. When ACS and DGAT1 are increased simultaneously, gene expression patterns suggest that TG storage and FA oxidation are both enhanced, resulting in a protective metabolic milieu. DGAT1 overexpression induces expression of ATGL and carnitine palitoyl transferase-1 (CPT1), and partially reverts the downregulation of antioxidant genes catalase and glutathione peroxidase observed in ACS transgenic mice [23].
In the abovementioned model, TG accumulation due to DGAT1 overexpression appears to be a beneficial metabolic adaptation. Contrary to this, knock-out of the TG lipolytic enzyme ATGL results in heart enlargement (20-fold), and a generalized increase in all tissue fat depots. Haemmerle et al. showed that ATGL-KO mice have severe cardiac fibrosis, increased posterior LV wall thickness, and ventricular dilatation of both ventricles [24]. Thereby, mice die from cardiac insufficiency caused by a mechanical contraction defect which is due to massive lipid accumulation. This effect is consistent with the described disarrangement in contractile protein composition observed in obese Zucker rats [25, 26]. In the latter, FA overload and TG accumulation are associated with upregulation of isoform b of the myosin heavy chain (MHC-b), but not MHC-a, which leads to a depressed cardiac power. Thus, excessive intracellular lipid accumulation might represent a physical hindrance, which mechanically disturbs protein contraction and contributes to cell dysfunction. Whereas, in control and streptozoticin (STZ)-induced diabetic mice, cardiac-specific overexpression of the lipolytic enzyme HSL prevents TG accumulation under fasting conditions, increases expression of FA oxidative genes, and prevents interstitial fibrosis and mortality [27]. TG and DAG HSL-mediated hydrolysis contributes to intracellular FA production and upregulation of several genes involved in lipid metabolism.
Lipoprotein secretion also plays a role in modulating intra-cardiac TG accumulation. Fasting and fat-feeding conditions induce the expression of cardiac MTP and formation of Apo-B-containing lipoparticles, which promote TG release. Transgenic mice exhibit an expansion in TG content upon both fasting and fat feeding, when the cardiac-specific MTP-A isoform is deleted [28]. In contrast, cardiac Apo-B overexpression prevents TG accumulation and the expression of genes involved in FA uptake, cytosolic release, and mitochondrial oxidation uncoupling such as CD36, FATP1, UCP3. The latter molecules are implicated in the lipotoxic effect. As a result, cardiac function is more preserved in Apo-B transgenic mice than control animals during high fat-feeding [28]. Moreover, the double overexpression of Apo-B and LpL in mice reduces the severe lipotoxic cardiomyopathy of the LpL transgenic model by decreasing cardiac TG and FA content, atrial natriuretic factor (ANP) gene expression, and brain natriuretic peptide (BNP) gene expression, while increasing GLUT4 [29].
All considered, these results suggest that it remains unclear whether, and when, cardiac TG synthesis and accumulation are protective or detrimental. The available studies suggest that cardiac TG storage can be beneficial to cardiomyocyte function and survival, at least to some extend. A modest increase in TG may not be detrimental, and can be seen as a preferential pathway preventing FA from ceramide and toxic intermediate formation in situations of FA overload. Saturation of this storage capacity results in the formation of intermediate species. As the intracellular lipid pool grows, it may undergo peroxidation, and become a source of oxidative damage. Once it becomes excessively large, mechanical decoupling and disruption may occur.
Fatty acid oxidation and cardiac function
β-oxidation per se is a protective pathway. It provides energy for cell contraction and survival, and it deters FA from excessive TG accumulation and formation of toxic species. However, an increase in FA oxidation lowers myocardial glucose utilization and enhances myocardial oxygen consumption (MVO2). Moreover, increased FA oxidation―especially that of long-chain saturated FA, e.g. palmitate―can induce apoptotic signaling through dissipation of the mitochondrial membrane potential and overproduction of ROS [30, 31]. Most likely, this occurs at the level of complexes I and III of the mitochondrial respiratory chain.
ROS overproduction damages mitochondrial structure, which results in the release of pro-apoptotic mitochondrial proteins, citochrome c leakage, and activation of caspase 3 and 9 [32, 33]. Besides programmed cell death, ROS activate kinase pathways, which are associated with cardiac hypertrophy and loss of function. For instance, induction of BNP promoter depends on NF-κB, which can be activated by ROS and plays a role in cardiac hypertrophy [34]. ROS may also activate the mitogen-activated protein kinase (MAPK) signaling pathway [35] and matrix metalloproteinases (MMP), which may in turn contribute to cardiac growth and hypertrophic response. MMP-2 in particular is emerging as an important signaling protease implicated in the proteolytic regulation of intracellular proteins in myocardial oxidative stress injury [36]. ROS can damage excitation-contraction coupling, which impairs Ca2+ homeostasis in isolated cadiomyocytes treated with palmitate. In fact, increased ROS production can decrease L-type Ca2+ flow amplitude. This increases the open probability of sarcoplasmic reticulum (SR) Ca2+ release channels, and slows down SR Ca2+ uptake. At the same time it increases sarcolemma Na+/Ca2+ exchange activity, which eventually leads to reduced SR Ca2+ content and depressed myocyte shortening [22].
Increased FA oxidation is associated with a reduced energetic efficiency in ATP production. Compromised ATP production suggests an FA-mediated activation of uncoupling proteins (UCP). Again, ROS are also responsible for the oxidation of lipids and proteins to produce highly reactive species, which may activate UCPs [37, 38]. UCPs represent a mechanism by which protons can re-enter the mitochondrial matrix, which lowers the membrane potential and bypasses ATP synthesis. UCP-2 and UCP-3 are expressed in the heart. Their content is elevated in db/db mice [39], but it is reduced in hypoxic hearts, which use glucose as main energy source [40]. Upregulation in UCP3 expression has not been uniformly found by all authors. This suggests that mitochondrial uncoupling may reflect the allosteric activation of uncoupling mechanisms independent of changes in gene expression. The occurrence of uncoupling seems to be in agreement with observations in hearts of obese ob/ob mice. Despite an increase in mitochondrial mass, these hearts manifest an impaired expression in complexes I, III, and IV of the electron transport chain, and a defect in oxidative phosphorylation in response to increased FA uptake and oxidation [8, 41].
Whether modulating β-oxidation has a beneficial effect on ventricular function is still controversial. Treatment with the anti-diabetic drug troglitazone lowers cardiac TG and ceramide accumulation, and improves cardiac function [42]. The mechanism mediating these effects of troglitazone has been identified in the activation of AMP-activated protein kinase alpha (AMPKα) in ZDF rats or ob/ob mice [43].
On the other hand, the pharmacological inhibition of FA oxidation has been widely studied as a therapeutic strategy against ischemic cardiomyopathy to prevent the superoxide-related consequences. CPT1 inhibitors, such as oxfenicine and etomoxir, have been shown to benefit post-ischemic cardiac function recovery. In the reperfused ischemic myocardium, FAs are the preferred substrate, providing up to 90% of the required ATP. In this model, etomoxir administration can enhance glucose oxidation and increase the expression of both the a and the b isoform of MHC, which improves heart functional recovery [44]. Conversely, etomoxir was recently reported to fail in reversing heart failure in a rat model with mechanical-induced hypertrophy [45]. This suggests that only the ischemic, i.e. poorly oxygenated heart, may benefit from the metabolic shift from FA oxidation to the utilization of glucose, which is an oxygen-sparing substrate.
Trimetazidine is another anti-ischemic drug acting as a metabolic modulator, which shifts cardiac energy metabolism from FA to glucose oxidation [46]. Recently, a positron emission tomography (PET) study showed that trimetazedine administration preferentially reduces the oxidation of TG-released FA, while activating glucose metabolism in the myocardium of obese, otherwise healthy human subjects [47]. This drug has been shown to improve LV ejection fraction and cardiac function in patients with heart failure, with and without diabetes [48-50].
Genetic and pharmacological interventions affect lipid uptake and accumulation. Therefore, it has been difficult to isolate the effect of modulation of FA oxidation. Generally, the beneficial outcome of FA oxidation seems to be counterbalanced by the proportional rate of ROS production, which contributes to tissue damage and oxygen consumption. Therefore, the suppression of FA oxidation, leading to the utilization of glucose as energy source by the myocardium, seems to be beneficial in conditions of oxygen deficiency such as ischemia.
Cardiac adiposity and dysfunction in humans
Cardiac complications represent the major cause of mortality in diabetic and severely obese patients. These conditions are characterized by an elevated release of FA from adipose tissue, likely representing the major contributor to the development of heart adiposity. Both intramyocardial stores and epicardial fat depots are augmented in these patients compared to respective control populations. Table 2 summarizes the results from studies carried out in diabetic and obese patients, which are designed to clarify the relationship between cardiac fat and function.
Table 2. Relationship between intramyocardial or epicardial fat and obesity, T2D, cardiac metabolism, and cardiac function in humans.
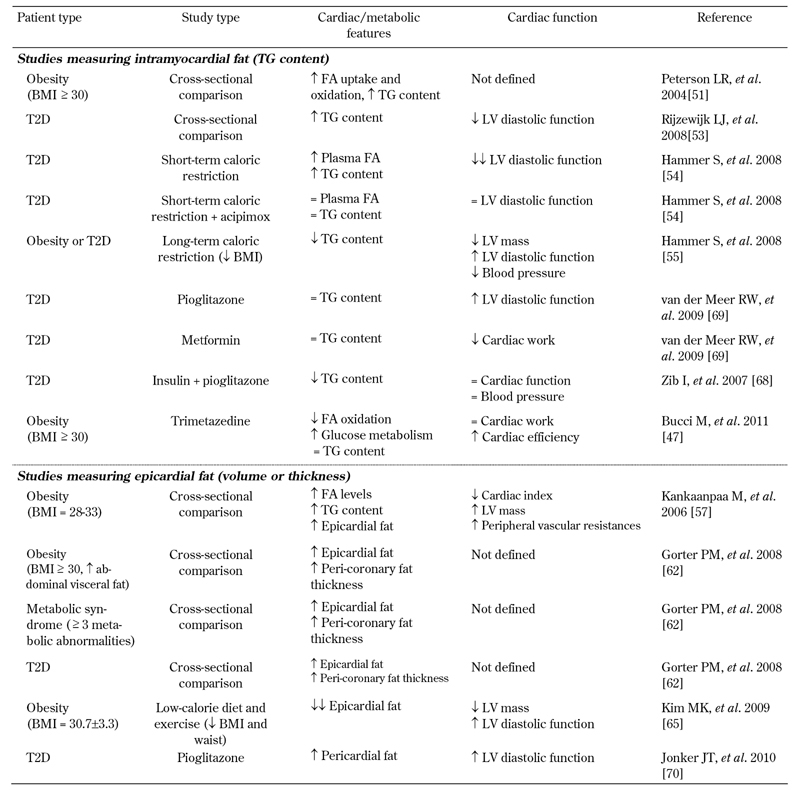
Legend: =: unchanged. BMI: body mass index. FA: fatty acid. LV: left ventricle. T2D: type 2 diabetes. TG: triglyceride.
Intramyocardial fat
Consistent with animal studies, in non-diabetic obese [51] or type 2 diabetic patients, myocardial FA uptake and oxidation (as measured with PET) and TG content (determined by MRS) are increased compared to those in lean individuals [52]. In type 2 diabetic patients, myocardial TG content is associated with impaired left ventricular diastolic function, independent of age, body mass index (BMI), heart rate, visceral fat, and diastolic blood pressure [53]. In both type 2 diabetic and healthy individuals, a short-term caloric restriction leads to an elevation in FA levels, myocardial TG accumulation, and deterioration of LV function, unless FA overload is prevented, e.g. by the pharmacologic suppression of lipolysis [54]. However, when obese or type 2 diabetic patients undergo a very low calories diet for a longer term (6-16 weeks), a reduction in myocardial TG content and LV mass combined with a significant improvement in cardiac diastolic function beyond BMI and blood pressure are observed [55]. These results suggest that intracellular TG content is highly flexible, and rapidly adapts to the changing metabolic milieu.
Epicardial fat
In addition to cardiac steatosis, diabetes and obesity have been positively correlated with the amount of fat surrounding heart and vessels [56]. Consistently, a positive correlation between epicardial fat volume and intramyocardial lipid content has been found [57]. Epicardial fat accounts for about 20% of the total ventricular mass, and is significantly greater in the hypertrophied heart. It is characterized by smaller adipocyte size, different fatty acid composition, lower glucose utilization rates, and higher fatty acid synthesis and metabolism compared to other fat depots [56]. Because of its close anatomical relationship to the heart, and the absence of physical fascial boundaries, epicardial fat may modulate vessels and heart metabolism via paracrine, or local direct secretion. Several protective roles have been ascribed to epicardial fat. Firstly, it serves as local energy supplier and buffer against excessive FA. Secondly, it is a source of protective cytokines and adipokines, such as adiponectin and anti-inflammatory interleukine-10 (IL-10). Finally, it mechanically attenuates vascular tension. Conversely, under pathological circumstances, such as obesity and diabetes, it may contribute to local inflammatory cell infiltration [56] and secretion of pro-inflammatory cytokines [58, 59]. These events are associated with a deterioration in coronary artery disease (CAD).
Epicardial fat thickness [60] and volume [61] are considered to be independent predictors of metabolic syndrome and pro-inflammatory citokine level. Its expansion is associated with lipotoxic heart disease. Epicardial adipose tissue volume correlates with abdominal visceral fat and BMI, and increases with the number of metabolic syndrome components [62]. This means that epicardial adipose tissue progressively increases in patient classes, from lean to obese individuals with normal glucose tolerance to those having impaired glucose tolerance and type 2 diabetes [63]. Also, its thickness is positively correlated with several circulating proatherogenic and proinflammatory adipokine levels [64], while it is inversely related to adiponectin concentrations. Kankaanpaa et al. have shown that in moderately obese men, epicardial fat is positively related to peripheral vascular resistances, and negatively associated with the cardiac index [57]. Low calorie diet and exercise have been shown to decrease epicardial fat thickness in obese men, resulting in changes in LV mass and diastolic function [65]. This is consistent with data suggesting that there is a relationship between epicardial fat mass and LV or RV mass [66].
Imaging studies have documented that patients with CAD have larger depots of pericardial fat compared to healthy individuals. Also, these studies have shown that epicardial fat thickness, more than any other fat depots or the waist circumference, correlates with vascular aging and subclinical atherosclerosis [67]. In fact, the enlarged and hypoxic epicardial fat becomes an active supplier of pro-inflammatory and pro-atherogenic cytokines to the subtending myocardium and vasculature. A high variability across populations and parameters considered in different studies makes it difficult to define a unique mechanistic association between epicardial fat thickness and cardiometabolic risk. Nevertheless, it is likely that epicardial fat has an influence on CAD disease, plaque composition, and lipid core, which are markers of plaque vulnerability.
Effects of diabetes and hypoglycemic drugs on cardiac fat and function
Cardiac fat is more extensive in type 2 diabetic than in similarly obese non-diabetic patients. This suggests a role of progressive insulin resistance, hyperinsulinemia, and hyperglycemia in the deterioration of cardiac adiposity. Consistently, the association of LV dysfunction and the amount of fat inside, or around, the heart has been described more frequently in diabetic than non-diabetic obese subjects. Hyperglycemia may provide an increased amount of glycerol, which is a substrate of TG synthesis, and may competitively impair the catabolism of FAs, thereby promoting their storage.
The addition of the antidiabetic drug pioglitazone to insulin treatment in T2D patients was shown to reduce intramyocardial triglyceride content without affecting heart function [68]. Moreover, in men with well-controlled T2D, the evidence is that hypoglycemic drugs pioglitazone and metformin improved LV diastolic function, and decreased cardiac work respectively, without changing cardiac TG content [69]. This markedly mitigates the role of intramyocellular fat in the pathogenesis of heart dysfunction. The pathophysiological implication of enlarged pericardial fat is not clear, since a correlation with cardiac function has not yet been demonstrated. In fact, treatment with pioglitazone for 24 weeks has been found to improve LV diastolic function in T2D patients, despite increasing pericardial fat volume, whereas the administration of metformin did not result in any effect [70]. Direct drug influences on the metabolic and hemodynamic profiles, both of which have a recognized effect on cardiac function, may have masked the relationship between TG storage and cardiac outcome in these studies.
The controversial findings described above suggest a need for further studies to find out:
1. Whether differences in cardiac adiposity (in the range observed in humans with and without diabetes) have any clinically significant impact on heart function. Also, it may help to explain the high prevalence of cardiovascular disease associated with obesity and diabetes.
2. If hyperglycemia is implicated in the regulation of intramyocardial and epicardial fat.
3. If 2. is valid, then whether this form of glucose toxicity can be alleviated by hypoglycemic drugs.
Cardiac adiposity and inflammation
Intramyocardial fat
Cardiac adiposity and its related LV hypertrophy and dysfunction are positively associated with inflammatory markers. Myocardial FA intermediates, lysophosphatidic acid (LPA), phosphatidic acid (PA), diacylglycerol (DAG), and ceramides, resulting from impaired/saturated FA oxidation and/or biosynthetic pathways, are responsible for the inflammatory response caused by a prolonged cardiac FA overload. LPA, PA, and DAG are intermediate products that emerge during the synthesis of triglycerides. Ceramides are produced by the hydrolysis of sphyngomyelin sphingomielinase (SMase), or they are synthesized de novo by serine palmitolyltransferase (SPT) and ceramide synthase resulting from the condensation of palmitoyl-CoA with serine.
Ceramide has been shown to inhibit the mitochondrial respiratory chain by interacting with complex III in isolated cardiomyocytes [71]. In programmed cell death of neonatal rat cardiomyocytes, a progression has been observed from release of citochrome c and loss of membrane potential to ceramide accumulation, which finally results in the activation of caspase-3 like protease. All of these are documented as hallmarks of impending cell death [27]. Myriocin administration (SPT inhibitor) to a normal and lipotoxic heart of mice overexpressing LpL reduces ceramide concentration and improves cardiac function simultaneously with the normalization of substrate utilization [72].
Also, FA intermediates (e.g. DAG) can activate different serine/threonine-specific protein kinases which belong to the superfamily of mitogen-activated protein kinases (MAPK), IkB (IKK), and protein kinase C (PKC). These kinases play a critical role in apoptosis, inflammation, T cell differentiation, and insulin action. MAPK direct and integrate a complex signaling network which regulates the growth of myocytes and activates C-Jun N-terminal kinases (JNK). The network controls important cell function including cell growth, differentiation, and apoptosis [73]. Ceramides can inhibit Akt/PKB, another serine/threonine protein kinase, specially involved in glucose and protein metabolism and survival pathways inhibiting apoptosis [74]. Activation of serine kinases by FA intermediates might be significant, representing the mechanism of convergence between dysfunctional lipid overload and inflammation. In fact, activation of JNK and IKK promotes the nuclear factor κB (NF-κB) pathway, which induces transcription of several proteins causing proliferation, inflammation, and apoptosis.
Epicardial fat
All the damage mechanisms occurring in the fatty myocardium, once exposed to an FA overflow, are amplified by the consensual expansion of epicardial adipose tissue. Lipid accumulation in epicardial adipocytes causes cell enlargement, which decreases oxygen delivery. The combination of local hypoxia and lipid overload triggers hypoxia-sensitive pathways in adipose tissue with ROS production, activation of JNK1 and IKK/NF-κB pathways, and induction of genes involved in the inflammatory response [75].
The hypoxic and inflammatory status is characterized by increased production and release of proinflammatory cytokines, such as TNFα, IL-6, and C-reactive protein. These cytokines activate the NF-κB pathway and a series of complex intracellular signals, which contribute to macrophage, and T cell, recruitment and tissue infiltration. Macrophages are responsible for the majority of TNFα and a significant part of IL6 expression in adipose tissue [76]. They also release other proinflammatory cytokines, resulting in a feed-forward cycle, which exacerbates the inflammatory status and its propagation to subtending tissues.
Adipokine, cytokine, and growth factors may propagate to the myocardium via simple diffusion. Data from the Framingham study document that epicardial, but not pericardial (intrathoracic), fat volume is associated with CAD [77]. Recently, Sacks et al. found that the expression of 39 genes was increased in epicardial fat of patients with severe CAD compared with controls [78]. Seventeen mRNAs were linked to inflammatory processes. These included interleukin 8 (IL-8), mitogen-activated protein kinase 8 (Tpl2), lipocalin-2, monocyte chemoattractant proein-1 (MCP1/CCL2), p50 subunit of nuclear factor κB1 (NF-κB1), PAI-1, and TNFa. Seven mRNAs were related to oxidative stress, including GPX3, heme oxigenase 1, endothelial nitrix oxide sinthase (eNOS), and superoxide dismutase (SOD). Fifteen were involved in metabolism and angiogenesis, or they were specific to fat cells. These results support the hypothesis that expression of pro-inflammatory, redox, and angiogenic genes in epicardial fat may contribute locally to CAD and/or correlate with its severity.
Conclusions
The studies summarized in this review indicate that fatty heart is a typical finding in subjects with obesity and type 2 diabetes. Fatty heart correlates with LV dysfunction and systemic or local inflammation in several studies. However, not all studies confirm these associations. The level of association between severity of cardiac adiposity and cardiac damage is stronger in animal studies, in which myocardial metabolism can be manipulated in a more extreme (though unphysiological) fashion. Therefore, it is important to explore whether intra-myocardial TG stores are altered in human patients with heart disease, independent of diabetes and obesity.
Prospective studies are required to assess the prognostic role of a fatty heart in changing the risk, or the progression, of cardiovascular disease. It is important to recognize that the fatty heart likely reflects a dysregulation in the FA-trapping capacity of peripheral adipose tissue. The latter is a more likely the primary culprit, and possibly the best target for prevention and treatment related to the toxic and inflammatory consequences of cardiac adiposity.
Disclosures: The authors report no conflict of interests in relation to this work.
References
- 1.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88(2):389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53(9):2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 3.Lommi J, Kupari M, Yki-Jarvinen H. Free fatty acid kinetics and oxidation in congestive heart failure. Am J Cardiol. 1998;81(1):45–50. doi: 10.1016/s0002-9149(97)00804-7. [DOI] [PubMed] [Google Scholar]
- 4.Park TS, Yamashita H, Blaner WS, Goldberg IJ. Lipids in the heart: a source of fuel and a source of toxins. Curr Opin Lipidol. 2007;18(3):277–282. doi: 10.1097/MOL.0b013e32814a57db. [DOI] [PubMed] [Google Scholar]
- 5.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD. et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109(1):121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iozzo P. Myocardial, perivascular, and epicardial fat. Diabetes Care. 2011;34(Suppl 2):S371–S379. doi: 10.2337/dc11-s250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146(12):5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 9.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115(25):3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 10.Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. 2007;101(4):335–347. doi: 10.1161/CIRCRESAHA.107.150417. [DOI] [PubMed] [Google Scholar]
- 11.Holloway GP, Snook LA, Harris RJ, Glatz JF, Luiken JJ, Bonen A. In obese Zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J Physiol. 2011;589(Pt 1):169–180. doi: 10.1113/jphysiol.2010.198663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgmaier M, Sen S, Philip F, Wilson CR, Miller CC 3rd, Young ME, Taegtmeyer H. Metabolic adaptation follows contractile dysfunction in the heart of obese Zucker rats fed a high-fat “western” diet. Obesity (Silver Spring) 2010;18(10):1895–1901. doi: 10.1038/oby.2009.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouwens DM, Diamant M, Fodor M, Habets DD, Pelsers MM, El Hasnaoui M, Dang ZC, van den Brom CE, Vlasblom R, Rietdijk A. et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia. 2007;50(9):1938–1948. doi: 10.1007/s00125-007-0735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A. et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54(12):3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- 15.Kuang M, Febbraio M, Wagg C, Lopaschuk GD, Dyck JR. Fatty acid translocase/CD36 deficiency does not energetically or functionally compromise hearts before or after ischemia. Circulation. 2004;109(12):1550–1557. doi: 10.1161/01.CIR.0000121730.41801.12. [DOI] [PubMed] [Google Scholar]
- 16.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM. et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96(2):225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y, Naseem RH, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Alpha-lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem Biophys Res Commun. 2006;344(1):446–452. doi: 10.1016/j.bbrc.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y, Naseem RH, Duplomb L, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci U S A. 2004;101(37):13624–13629. doi: 10.1073/pnas.0405499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107(7):813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M. et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111(3):419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haim TE, Wang W, Flagg TP, Tones MA, Bahinski A, Numann RE, Nichols CG, Nerbonne JM. Palmitate attenuates myocardial contractility through augmentation of repolarizing Kv currents. J Mol Cell Cardiol. 2010;48(2):395–405. doi: 10.1016/j.yjmcc.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fauconnier J, Andersson DC, Zhang SJ, Lanner JT, Wibom R, Katz A, Bruton JD, Westerblad H. Effects of palmitate on Ca(2+) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes. 2007;56(4):1136–1142. doi: 10.2337/db06-0739. [DOI] [PubMed] [Google Scholar]
- 23.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284(52):36312–36323. doi: 10.1074/jbc.M109.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S. et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 25.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51(8):2587–2595. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 26.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18(14):1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki J, Shen WJ, Nelson BD, Patel S, Veerkamp JH, Selwood SP, Murphy GM Jr, Reaven E, Kraemer FB. Absence of cardiac lipid accumulation in transgenic mice with heart-specific HSL overexpression. Am J Physiol Endocrinol Metab. 2001;281(4):E857–E866. doi: 10.1152/ajpendo.2001.281.4.E857. [DOI] [PubMed] [Google Scholar]
- 28.Bartels ED, Nielsen JM, Hellgren LI, Ploug T, Nielsen LB. Cardiac expression of microsomal triglyceride transfer protein is increased in obesity and serves to attenuate cardiac triglyceride accumulation. PLoS One. 2009;4(4):e5300. doi: 10.1371/journal.pone.0005300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokoyama M, Yagyu H, Hu Y, Seo T, Hirata K, Homma S, Goldberg IJ. Apolipoprotein B production reduces lipotoxic cardiomyopathy: studies in heart-specific lipoprotein lipase transgenic mouse. J Biol Chem. 2004;279(6):4204–4211. doi: 10.1074/jbc.M311995200. [DOI] [PubMed] [Google Scholar]
- 30.Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB. A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2000;279(5):H2124–H2132. doi: 10.1152/ajpheart.2000.279.5.H2124. [DOI] [PubMed] [Google Scholar]
- 31.Listenberger LL, Schaffer JE. Mechanisms of lipoapoptosis: implications for human heart disease. Trends Cardiovasc Med. 2002;12(3):134–138. doi: 10.1016/s1050-1738(02)00152-4. [DOI] [PubMed] [Google Scholar]
- 32.Li CJ, Zhang QM, Li MZ, Zhang JY, Yu P, Yu DM. Attenuation of myocardial apoptosis by alpha-lipoic acid through suppression of mitochondrial oxidative stress to reduce diabetic cardiomyopathy. Chin Med J (Engl) 2009;122(21):2580–2586. [PubMed] [Google Scholar]
- 33.Chen L, Knowlton AA. Mitochondria and heart failure: new insights into an energetic problem. Minerva Cardioangiol. 2010;58(2):213–229. [PMC free article] [PubMed] [Google Scholar]
- 34.Hirotani S, Otsu K, Nishida K, Higuchi Y, Morita T, Nakayama H, Yamaguchi O, Mano T, Matsumura Y, Ueno H. et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105(4):509–515. doi: 10.1161/hc0402.102863. [DOI] [PubMed] [Google Scholar]
- 35.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63(3):467–475. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 36.Kandasamy AD, Chow AK, Ali MA, Schulz R. Matrix metalloproteinase-2 and myocardial oxidative stress injury: beyond the matrix. Cardiovasc Res. 2010;85(3):413–423. doi: 10.1093/cvr/cvp268. [DOI] [PubMed] [Google Scholar]
- 37.Boudina S, Abel ED. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology (Bethesda) 2006;21:250–258. doi: 10.1152/physiol.00008.2006. [DOI] [PubMed] [Google Scholar]
- 38.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG. et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56(10):2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 39.Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes. 2005;54(12):3496–3502. doi: 10.2337/diabetes.54.12.3496. [DOI] [PubMed] [Google Scholar]
- 40.Essop MF, Razeghi P, McLeod C, Young ME, Taegtmeyer H, Sack MN. Hypoxia-induced decrease of UCP3 gene expression in rat heart parallels metabolic gene switching but fails to affect mitochondrial respiratory coupling. Biochem Biophys Res Commun. 2004;314(2):561–564. doi: 10.1016/j.bbrc.2003.12.121. [DOI] [PubMed] [Google Scholar]
- 41.Boudina S, Sena S, O'Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112(17):2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 42.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97(4):1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang MY, Unger RH. Role of PP2C in cardiac lipid accumulation in obese rodents and its prevention by troglitazone. Am J Physiol Endocrinol Metab. 2005;288(1):E216–E221. doi: 10.1152/ajpendo.00004.2004. [DOI] [PubMed] [Google Scholar]
- 44.Lopaschuk GD, Spafford MA, Davies NJ, Wall SR. Glucose and palmitate oxidation in isolated working rat hearts reperfused after a period of transient global ischemia. Circ Res. 1990;66(2):546–553. doi: 10.1161/01.res.66.2.546. [DOI] [PubMed] [Google Scholar]
- 45.Schwarzer M, Faerber G, Rueckauer T, Blum D, Pytel G, Mohr FW, Doenst T. The metabolic modulators, Etomoxir and NVP-LAB121, fail to reverse pressure overload induced heart failure in vivo. Basic Res Cardiol. 2009;104(5):547–557. doi: 10.1007/s00395-009-0015-5. [DOI] [PubMed] [Google Scholar]
- 46.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86(5):580–588. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 47.Bucci M, Borra R, Nagren K, Parkka JP, Del Ry S, Maggio R, Tuunanen H, Viljanen T, Cabiati M, Rigazio S. et al. Trimetazidine reduces endogenous free fatty acid oxidation and improves myocardial efficiency in obese humans. Cardiovasc Ther. 2011 doi: 10.1111/j.1755-5922.2011.00275.x. In press. [DOI] [PubMed] [Google Scholar]
- 48.Tuunanen H, Engblom E, Naum A, Nagren K, Scheinin M, Hesse B, Juhani Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H. et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118(12):1250–1258. doi: 10.1161/CIRCULATIONAHA.108.778019. [DOI] [PubMed] [Google Scholar]
- 49.Vitale C, Wajngaten M, Sposato B, Gebara O, Rossini P, Fini M, Volterrani M, Rosano GM. Trimetazidine improves left ventricular function and quality of life in elderly patients with coronary artery disease. Eur Heart J. 2004;25(20):1814–1821. doi: 10.1016/j.ehj.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 50.Fragasso G, Piatti Md PM, Monti L, Palloshi A, Setola E, Puccetti P, Calori G, Lopaschuk GD, Margonato A. Short- and long-term beneficial effects of trimetazidine in patients with diabetes and ischemic cardiomyopathy. Am Heart J. 2003;146(5):E18. doi: 10.1016/S0002-8703(03)00415-0. [DOI] [PubMed] [Google Scholar]
- 51.Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T. et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. 2004;109(18):2191–2196. doi: 10.1161/01.CIR.0000127959.28627.F8. [DOI] [PubMed] [Google Scholar]
- 52.Rijzewijk LJ, van der Meer RW, Lamb HJ, de Jong HW, Lubberink M, Romijn JA, Bax JJ, de Roos A, Twisk JW, Heine RJ. et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: studies with cardiac positron emission tomography and magnetic resonance imaging. J Am Coll Cardiol. 2009;54(16):1524–1532. doi: 10.1016/j.jacc.2009.04.074. [DOI] [PubMed] [Google Scholar]
- 53.Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52(22):1793–1799. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 54.Hammer S, van der Meer RW, Lamb HJ, de Boer HH, Bax JJ, de Roos A, Romijn JA, Smit JW. Short-term flexibility of myocardial triglycerides and diastolic function in patients with type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2008;295(3):E714–E718. doi: 10.1152/ajpendo.90413.2008. [DOI] [PubMed] [Google Scholar]
- 55.Hammer S, Snel M, Lamb HJ, Jazet IM, van der Meer RW, Pijl H, Meinders EA, Romijn JA, de Roos A, Smit JW. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J Am Coll Cardiol. 2008;52(12):1006–1012. doi: 10.1016/j.jacc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 56.Iacobellis G, Willens HJ. Echocardiographic epicardial fat: a review of research and clinical applications. J Am Soc Echocardiogr. 2009;22(12):1311–1319. doi: 10.1016/j.echo.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 57.Kankaanpaa M, Lehto HR, Parkka JP, Komu M, Viljanen A, Ferrannini E, Knuuti J, Nuutila P, Parkkola R, Iozzo P. Myocardial triglyceride content and epicardial fat mass in human obesity: relationship to left ventricular function and serum free fatty acid levels. J Clin Endocrinol Metab. 2006;91(11):4689–4695. doi: 10.1210/jc.2006-0584. [DOI] [PubMed] [Google Scholar]
- 58.Zhang H, Zhang C. Regulation of microvascular function by adipose tissue in obesity and type 2 diabetes: evidence of an adipose-vascular loop. Am J Biomed Sci. 2009;1(2):133–142. doi: 10.5099/aj090200133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA. et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119(12):1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- 60.Mazur A, Ostanski M, Telega G, Malecka-Tendera E. Is epicardial fat tissue a marker of metabolic syndrome in obese children? Atherosclerosis. 211(2):596–600. doi: 10.1016/j.atherosclerosis.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 61.Wang TD, Lee WJ, Shih FY, Huang CH, Chang YC, Chen WJ, Lee YT, Chen MF. Relations of epicardial adipose tissue measured by multidetector computed tomography to components of the metabolic syndrome are region-specific and independent of anthropometric indexes and intraabdominal visceral fat. J Clin Endocrinol Metab. 2009;94(2):662–669. doi: 10.1210/jc.2008-0834. [DOI] [PubMed] [Google Scholar]
- 62.Gorter PM, van Lindert AS, de Vos AM, Meijs MF, van der Graaf Y, Doevendans PA, Prokop M, Visseren FL. Quantification of epicardial and peri-coronary fat using cardiac computed tomography; reproducibility and relation with obesity and metabolic syndrome in patients suspected of coronary artery disease. Atherosclerosis. 2008;197(2):896–903. doi: 10.1016/j.atherosclerosis.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 63.Iozzo P, Lautamaki R, Borra R, Lehto HR, Bucci M, Viljanen A, Parkka J, Lepomaki V, Maggio R, Parkkola R. et al. Contribution of glucose tolerance and gender to cardiac adiposity. J Clin Endocrinol Metab. 2009;94(11):4472–4482. doi: 10.1210/jc.2009-0436. [DOI] [PubMed] [Google Scholar]
- 64.Kremen J, Dolinkova M, Krajickova J, Blaha J, Anderlova K, Lacinova Z, Haluzikova D, Bosanska L, Vokurka M, Svacina S. et al. Increased subcutaneous and epicardial adipose tissue production of proinflammatory cytokines in cardiac surgery patients: possible role in postoperative insulin resistance. J Clin Endocrinol Metab. 2006;91(11):4620–4627. doi: 10.1210/jc.2006-1044. [DOI] [PubMed] [Google Scholar]
- 65.Kim MK, Tomita T, Kim MJ, Sasai H, Maeda S, Tanaka K. Aerobic exercise training reduces epicardial fat in obese men. J Appl Physiol. 2009;106(1):5–11. doi: 10.1152/japplphysiol.90756.2008. [DOI] [PubMed] [Google Scholar]
- 66.Corradi D, Maestri R, Callegari S, Pastori P, Goldoni M, Luong TV, Bordi C. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc Pathol. 2004;13(6):313–316. doi: 10.1016/j.carpath.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 67.Natale F, Tedesco MA, Mocerino R, de Simone V, Di Marco GM, Aronne L, Credendino M, Siniscalchi C, Calabro P, Cotrufo M. et al. Visceral adiposity and arterial stiffness: echocardiographic epicardial fat thickness reflects, better than waist circumference, carotid arterial stiffness in a large population of hypertensives. Eur J Echocardiogr. 2009;10(4):549–555. doi: 10.1093/ejechocard/jep002. [DOI] [PubMed] [Google Scholar]
- 68.Zib I, Jacob AN, Lingvay I, Salinas K, McGavock JM, Raskin P, Szczepaniak LS. Effect of pioglitazone therapy on myocardial and hepatic steatosis in insulin-treated patients with type 2 diabetes. J Investig Med. 2007;55(5):230–236. doi: 10.2310/6650.2007.00003. [DOI] [PubMed] [Google Scholar]
- 69.van der Meer RW, Rijzewijk LJ, de Jong HW, Lamb HJ, Lubberink M, Romijn JA, Bax JJ, de Roos A, Kamp O, Paulus WJ. et al. Pioglitazone improves cardiac function and alters myocardial substrate metabolism without affecting cardiac triglyceride accumulation and high-energy phosphate metabolism in patients with well-controlled type 2 diabetes mellitus. Circulation. 2009;119(15):2069–2077. doi: 10.1161/CIRCULATIONAHA.108.803916. [DOI] [PubMed] [Google Scholar]
- 70.Jonker JT, Lamb HJ, van der Meer RW, Rijzewijk LJ, Menting LJ, Diamant M, Bax JJ, de Roos A, Romijn JA, Smit JW. Pioglitazone compared with metformin increases pericardial fat volume in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95(1):456–460. doi: 10.1210/jc.2009-1441. [DOI] [PubMed] [Google Scholar]
- 71.Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272(39):24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- 72.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED. et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49(10):2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118(9):2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stratford S, DeWald DB, Summers SA. Ceramide dissociates 3'-phosphoinositide production from pleckstrin homology domain translocation. Biochem J. 2001;354(Pt 2):359–368. doi: 10.1042/0264-6021:3540359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M. et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 76.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mahabadi AA, Massaro JM, Rosito GA, Levy D, Murabito JM, Wolf PA, O'Donnell CJ, Fox CS, Hoffmann U. Association of pericardial fat, intrathoracic fat, and visceral abdominal fat with cardiovascular disease burden: the Framingham Heart Study. Eur Heart J. 2009;30(7):850–856. doi: 10.1093/eurheartj/ehn573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sacks HS, Fain JN, Cheema P, Bahouth SW, Garrett E, Wolf RY, Wolford D, Samaha J. Depot-specific overexpression of proinflammatory, redox, endothelial cell, and angiogenic genes in epicardial fat adjacent to severe stable coronary atherosclerosis. Metab Syndr Relat Disord. 2011 doi: 10.1089/met.2011.0024. In press. [DOI] [PubMed] [Google Scholar]