

Abstract
The fifteen known Fanconi Anemia (FA) proteins cooperate in a pathway which regulates DNA interstrand crosslink repair. Recent studies indicate that the FA pathway also controls Rev1-mediated translesion DNA synthesis (TLS). Here we identify a novel protein FAAP20, which is an integral subunit of the multisubunit FA core complex. FAAP20 binds to FANCA subunit and is required for complex stability and monoubiquitination of FANCD2. FAAP20 contains a UBZ4 (Ubiquitin Binding Zinc finger 4) domain and binds to the monoubiquitinated form of Rev1. FAAP20 binding stabilizes Rev1 nuclear foci and promotes the interaction of the FA core with PCNA/Rev1 DNA damage bypass complexes. FAAP20 therefore provides a critical link between the FA pathway and TLS polymerase activity. We propose that the FA core complex regulates crosslink repair, by channeling lesions to damage bypass pathways and preventing large DNA insertions and deletions.
Keywords: Fanconi Anemia, Ubiquitination, Translesion DNA Synthesis, Rev1
INTRODUCTION
Fanconi Anemia (FA) is an inherited chromosome instability syndrome characterized by congenital abnormalities, cancer susceptibility, and cellular hypersensitivity to DNA damaging agents. There are fifteen known FA genes, and the FA proteins cooperate in a DNA damage response (DDR) pathway required for DNA interstrand crosslink repair1,2. Loss of the FA pathway results in increased spontaneous chromosome breakage and decreased point mutation frequency.
Eight of the FA proteins comprise the FA core complex, a multisubunit complex required for DNA damage recognition at a stalled replication fork3. In response to DNA damage, the complex, an E3 ligase, monoubiquitinates the FANCD2/FANCI heterodimer4,5. The monoubiquitinated D2/I complex in turn is recruited to chromatin where it interacts with downstream FA proteins involved in homologous recombination (HR) repair (FANCD1/BRCA2, FANCJ, FANCN and FANCO). Monoubiquitinated FANCD2 also provides a binding site for the FANCP/SLX4 nuclease complex and for the FAN1 nuclease6–8. These nucleases contain a UBZ4 motif required for monoubiquitin binding9,10. Overall, the monoubiquitinated D2/I complex appears to coordinate nuclease events and the downstream TLS step required for DNA interstrand crosslink repair11.
An intriguing feature of Fanconi Anemia patient derived cells is their hypomutability for point mutations12. These observations have led to the supposition that the FA pathway normally facilitates an error-prone repair process that causes point mutations whilst protecting cells from gross chromosomal rearrangements through HR13. The predominant cellular mechanism that generates point mutations in DNA is translesion synthesis (TLS)14,15. TLS promotes the replicative bypass of DNA lesions that impede replication fork progression. TLS requires specialized DNA polymerases with unconstrained active sites that can recognize helix-distorting lesions as template nucleotides for DNA replication. Mammalian cells possess several distinct TLS polymerases, each with a penchant for bypassing a particular class of DNA lesions16. Depending on the TLS polymerase utilized and the type of lesion bypassed, this process can be error-free or error-prone. TLS polymerases are recruited to blocked replication forks, at least in part, through their interaction with monoubiquitinated PCNA9. Studies by Niedzwiedz et al.17 in chicken DT40 cells demonstrated an epistatic relationship between FANCC of the FA core complex and the error-prone TLS polymerase, Rev1, suggesting that the FA pathway regulates TLS through this mechanism. The Rev1 enzyme is a deoxycytidyl (dCMP) nucleotidyl transferase belonging to the Y family of TLS polymerases, and it is a major source of damage-inducible mutagenesis through error-prone TLS15. Moreover, FANCC-deficient DT40 cells display a reduced frequency of spontaneous point mutations, suggesting that the FA pathway is required for Rev1-mediated TLS of abasic sites or other spontaneously generated DNA lesions17,18.
Whether all FA proteins, or only a subset, participate in TLS regulation has remained unknown. We recently investigated the role of the FA pathway in error-prone TLS, by deploying a shuttle vector-based mutagenesis assay to measure the mutation frequencies of FA patient-derived human cells of several genetic complementation groups19. Our results demonstrated that FA core complex subunits, such as FANCA and FANCG, are required for efficient point mutagenesis, whereas FANCD2, the downstream substrate for monoubiquitination, is not. Indeed, FANCA-deficient and FANCG-deficient cells exhibited a decrease in spontaneous and DNA damage-inducible point mutagenesis while FANCD2-deficient cells exhibited an increase. Moreover, the recruitment of the error-prone TLS polymerase Rev1 into nuclear foci depended on an intact FA core complex but was independent of FANCD2-Ub19,20. Taken together, these results support a branched model of the FA pathway in which the FA core complex could coordinate two independent functions-TLS and HR. The mechanism by which the FA core complex interacts with the Rev1 polymerase has remained unknown.
In the current study, we have identified a novel 20 kD subunit of the FA core complex, referred to as FAAP20, which provides the missing link between the FA core and TLS polymerase complexes. FAAP20 binds directly to the FANCA subunit of the FA core complex. The carboxyl terminal region of FAAP20 contains a UBZ4 (Ubiquitin Binding Zinc finger 4) domain which binds to monoubiquitinated Rev1 polymerase. Interestingly, DNA damage activates the UBZ4-mediated colocalization of the FA core complex with monoubiquitinated Rev1 complexes, providing a functional linkage between the FA pathway and TLS. Disruption of these events, by downregulation of FA core complex or downregulation of Rev1, results in decreased point mutagenesis (ie, loss of TLS) and increased chromosome breakage (ie, loss of HR).
RESULTS
C1orf86 binds to ubiquitin and functions in the FA pathway
Ubiquitin mediates a variety of DNA damage response pathways21,22. To identify new ubiquitin-binding proteins that participate in DNA repair pathways, we performed a bioinformatic search with the Hidden Markov Model (HMM) using a conserved sequence signature of UBZ4 as bait (Supplementary method). UBZ4 is a ubiquitin binding domain which is commonly found on DNA repair proteins, especially proteins involved in TLS repair and crosslink repair10. One of the hits, an uncharacterized 20 kD C1orf86 (Chromosome 1 open reading frame 86; NCBI ID NP_872339) protein conserved in higher vertebrates, contains a conserved UBZ4 domain at the C-terminus and binds to ubiquitin in vitro (Fig. 1a and Supplementary Fig. 1a,b). Mutation of two conserved cysteine residues abolished this interaction, confirming that C1orf86 is a bona fide ubiquitin-binding protein (Supplementary Fig. 1b). Since other UBZ4-containing proteins, including RAD18, SLX4, FAN1, and SNM1A, interact with the FA pathway7,8,23, we hypothesized that C1orf86 may also be involved in regulating DNA interstrand crosslink (ICL) repair. Intriguingly, depletion of C1orf86 in HeLa cells, using two independent siRNAs, impaired monoubiquitination of FANCD2 upon treatment with various DNA damage agents (Fig. 1b and Supplementary Fig. 1c,d). Consequently, C1orf86 knockdown prevented chromatin targeting and damage-induced FANCD2 foci formation following genotoxic stress (Fig. 1c,d). Knockdown also caused an increase in mitomycin C (MMC) sensitivity comparable to FANCA depletion (Fig. 1e) and resulted in dramatic elevation in chromosome radial formation, the hallmark of the FA phenotype (Fig. 1f and Supplementary Fig. 1e). Taken together, these data suggest that C1orf86 is a novel protein required for FANCD2 activation and ICL repair. We refer to this protein as FAAP20 (Fanconi anemia-associated protein, 20 kD).
Figure 1. C1orf86 is required for the FA pathway activation.

(a) Sequence alignment of the C1orf86 UBZ4 domain with known UBZ4 domains. Stars indicate the conserved residues that form a short mononucleate zinc finger, and arrows point to cysteine residues (Cys147 and 150) important for ubiquitin interaction. (b) FANCA and FANCD2 were analyzed by immunoblot in cell lysates from HeLa cells, transfected with control or C1orf86 siRNA and treated with DNA damage-inducing agents. (c) FACND2 was analyzed by immunoblot in cytosolic (S) and chromatin-containing (P) fractions of HeLa cells, transfected with control or C1orf86 siRNA and treated with 50 ng ml−1 MMC for 17 h. (d) Immunostaining of FANCD2 in HeLa cells, transfected with control or C1orf86 siRNA and treated with 2 mM HU for 6 h. Representative images are shown, and at least 150 cells were counted for quantification. Data shown are mean ± s.d. from three independent experiments. * p < 0.01. (e) Clonogenic survival of HeLa cells transfected with siRNA control, C1orf86, or FANCA treated with increasing doses of MMC and plated for 12 days. (f) Quantification of chromosomal aberrations and radial chromosomes of 293T cells transfected with siRNAs and exposed to 20 ng ml−1 MMC.
FAAP20 is an integral part of the FA core complex
Since many subunits of the FA core complex are required for FANCD2 monoubiquitination, we next asked whether FAAP20 is a component of this complex. Flag-tagged FAAP20 co-immunoprecipitated with FANCA, FANCE, and FANCC, indicating that FAAP20 associates with the FA core complex (Fig. 2a). Additionally, in vitro translated FANCA, but not FANCG, co-immunoprecipitated with Flag-FAAP20 suggesting that there is a direct interaction between FANCA and FAAP20 in vitro (Fig. 2b and Supplementary Fig. 2a). Next, we determined whether the UBZ4 domain of FAAP20 is required for the interaction with FANCA. Deletion of the N-terminus, but not the C-terminal UBZ4 domain, of FAAP20 abolished the interaction with FANCA, indicating that the ubiquitin-binding UBZ4 domain does not mediate the association with the FA core complex (Fig. 2c). Strikingly, reduction of FAAP20 expression significantly decreased the level of other FA core subunits as well as FANCA (Fig. 2d), and the inhibition of proteasomal degradation partially rescued these protein levels (Fig. 2e). Importantly, siRNA-resistant wild-type and UBZ4 deletion mutants, but not the N-terminal deletion mutant, could stabilize the FANCA protein which had been decreased by endogenous FAAP20 depletion (Fig. 2f and Supplementary Fig. 2b,c). Taken together, these data support the idea that FAAP20 plays a crucial role in stabilizing the FA core complex by directly interacting with FANCA via its N-terminus and preventing its degradation. Thus, FAAP20 is a new subunit of the FA core complex.
Figure 2. FAAP20 is required for the FA core complex stability.

(a) Immunoblot of anti-Flag immunoprecipitates (IP) of cell lysates from Flag-FAAP20 expressing 293T cells. (b) Direct interaction between myc-FAAP20 and FANCA in vitro analyzed by anti-myc IP of in vitro translated protein mixture. (c) Anti-Flag IP and immunoblot analysis of 293T cell lysates expressing Flag-tagged FAAP20 (F20) wild-type, ΔN (FAAP2048–180) or ΔC (FAAP201–163). (d) Immunoblot of cell lysates from HeLa cells transfected with siRNA control or FAAP20 for 72 h. (e) FANCA and FANCE were analyzed by immunoblot of HeLa cells, transfected with siRNA oligos and treated with 20 μM MG132 for 4 h. (*) denotes nonspecific band. (f) FANCA was analyzed by immunoblot of HeLa cells, pretreated with siRNA that targets 3′ UTR of FAAP20 mRNA and transfected with Flag-tagged wild-type or indicated mutants for 48 h.
FAAP20 promotes Rev1 foci formation
The C-terminal UBZ4 domain of FAAP20 may therefore bind to another ubiquitinated target. Given that the FA core complex stabilized by FAAP20 functions as an E3 ligase to monoubiquitinate FANCD2, the C-terminal UBZ4 domain of FAAP20 may function independently of FANCD2 activation. Several lines of evidence suggest that the FA core complex regulates error-prone translesion synthesis independently of FANCD2 and that Rev1 may mediate this DNA damage bypass. First, Rev1 is recruited to stalled replication forks through its interaction with monoubiquitinated PCNA via a C-terminal ubiquitin-binding motif (UBM)9,24–26. Second, Rev1 is self-monoubiquitinated, but the functional significance of this modification has remained elusive19,24,25. Third, we previously demonstrated that the FA core complex is necessary for recruiting Rev1 to replication foci containing a monoubiquitinated PCNA19.
Thus, we next analyzed damage-inducible Rev1 foci formation in FAAP20-depleted cells. Due to the low cellular expression of Rev1 and the lack of an antibody capable of detecting endogenous Rev1 by immunofluorescence, we monitored Rev1 foci formation of an exogenously expressed GFP-tagged Rev1 protein20. Following UVC irradiation, cells with GFP-Rev1 foci increased to around 50%, reflecting the recruitment of Rev1 to the DNA lesion consistent with previous results (Fig. 3a). Of note, siRNA-mediated FAAP20 knockdown resulted in diminished Rev1 foci formation, comparable to FANCA depletion, whereas FANCD2 knockdown itself did not prevent Rev1 foci formation. Taken together, these results suggest that FAAP20 is essential for efficient Rev1 recruitment to DNA lesions (Fig. 3a).
Figure 3. FAAP20 interacts with Rev1 and promotes efficient Rev1 foci formation.

(a) Fluorescence microscopy of GFP-Rev1 in U2OS cells, serially transfected with indicated siRNA oligos and GFP-Rev1 followed by treatment with 15 J m−2 UVC for 14 h. Representative images of GFP-Rev1 foci are shown, and the right panel represents the quantification of cells displaying more than five GFP-Rev1 foci. Data shown are mean ± s.d. from three independent experiments. * p < 0.01 compared with UVC-treated siRNA control. The immunoblots show the knockdown efficiency. (**) denotes nonspecific band. (b) Anti-Flag immunoprecipitates were analyzed by immunoblot from 293T cells transiently coexpressing Flag-FAAP20 and GFP-Rev1 or GFP-Polη. (c) Immunoblot of anti-Flag immunoprecipitates of 293T cells transiently coexpressing GFP-Rev1 and Flag-tagged FAAP20 wild-type, UBZ4-point mutant (C147A C150A; CA), or UBZ4-deletion mutant (aa 1–163; Δ).
FAAP20 interacts with Rev1 through its UBZ4 domain
Since FAAP20 contains a UBZ4 domain and since the FA core complex is required for efficient Rev1 foci assembly, we next tested whether Rev1 is a ubiquitinated target of FAAP20. We first examined if FAAP20 and Rev1 interact in vivo. Flag-tagged FAAP20 co-immunoprecipitated with Rev1 (Fig. 3b) but failed to coimmunoprecipitate with another TLS polymerase, Polη. Consistent with this result, Polη is known to form damage-inducible foci independently of the FA core complex19. The interaction of FAAP20 and Rev1 was recapitulated by co-immunoprecipitation using different tagged proteins and cell line (Supplementary Fig. 3a,b). Next, we examined whether the UBZ4 domain of FAAP20 is required for this interaction. Neither point mutation in two conserved cysteines nor deletion of UBZ4 domain affected the interaction with FANCA, confirming that the UBZ4 domain is dispensable for association with the FA core complex (Fig. 3c). In contrast, significantly less Rev1 co-immunoprecipitated with FAAP20 UBZ4 mutants, arguing for the importance of the intact UBZ4 domain in the interaction with Rev1 (Fig. 3c). Reciprocally, Flag-Rev1 coimmunoprecipitated with wild-type GFP-FAAP20, but not with the UBZ4 mutant of FAAP20 (Supplementary Fig. 3c). Collectively, the ubiquitin-binding domain of FAAP20 is essential for the interaction with Rev1.
Monoubiquitination of Rev1 enhances the FAAP20 interaction
The observation that the UBZ4 domain of FAAP20 mediates the interaction with Rev1 prompted us to test whether FAAP20 selectively binds ubiquitinated Rev1. To detect monoubiquitinated Rev1, we initially cotransfected 293T cells with the cDNA encoding Rev1 and the cDNA encoding HA-tagged ubiquitin. Rev1 monoubiquitination was confirmed by anti-Flag immunoprecipitation of Rev1 and anti-HA immunoblotting (Fig. 4a and Supplementary Fig. 3d). Also, treatment with ubiquitin aldehyde, which inhibits the deubiquitination process27, led to the appearance of monoubiquitinated Rev1, indicating that Rev1 undergoes a ubiquitination-deubiquitination cycle (Supplementary Fig. 3e).
Figure 4. Monoubiquitination of Rev1 enhances the interaction with FAAP20.

(a) Monoubiquitin of Rev1 was visualized by anti-Flag immunoprecipitation and immunoblot of 293T cells transfected with cDNAs encoding Flag-tagged Rev1 with or without HA-tagged ubiquitin. The slower-migrating band, above Flag-Rev1, represents the monoubiquitinated form of Rev1, which was confirmed by anti-HA immunoblot. (b) (Top) schematic of Rev1 mutants. (Bottom) anti-myc immunoprecipitation of 293T cell lysates coexpressing HA-ubiquitin and GFP-tagged Rev1 wild-type, UBM* (L946A P947A L1024A P1025A), or ΔC (aa1–892) mutant, mixed with the lysates expressing myc-tagged FAAP20. (c) Anti-myc immunoprecipitation of 293T cell lysates coexpressing HA-ubiquitin and GFP-tagged Rev1, mixed with lysates expressing myc-tagged FAAP20 wild-type or CA mutant (d) Anti-Flag immunoprecipitation of 293T cell lysates coexpressing HA-ubiquitin and Flag-tagged Rev1, mixed with lysates expressing either myc-tagged FAAP20 wild-type or CA mutant.
Mutation of the UBM domain of Rev1 has previously been shown to abolish DNA-damage inducible Rev1 foci formation and to confer hypersensitivity to UV and cisplatin by disrupting the interaction with monoubiquitinated PCNA24,25. Interestingly, monoubiquitination of Rev1 was disrupted when the UBM domain was mutated, suggesting that the UBM domain either promotes Rev1 monoubiquitination per se or stabilizes the conjugated ubiquitin (Fig. 4b). We next co-expressed myc-tagged FAAP20, containing a functional UBZ4 domain with either the wild-type or UBM mutant Rev1. FAAP20 coimmunoprecipitated with both the unubiquitinated and ubiquitinated isoforms of Rev1 as shown by both anti-GFP and anti-HA immunoblots, indicating that ubiquitination of Rev1 is not absolutely required for FAAP20 binding (Fig. 4b and Supplementary Fig. 4a).
We next compared the ability of wild-type FAAP20 versus the FAAP20 UBZ4 mutant to bind to the ubiquitinated and unubiquitinated isoforms of Rev1. Both wild-type and UBZ4 FAAP20 mutants coimmunoprecipated with the unubiquitinated isoform of Rev1 (Fig. 4c). Interestingly, only the wild-type FAAP20, with its functional UBZ4 domain, coimmunoprecipitated with the ubiquitinated isoform of Rev1 (Fig. 4c and Supplementary Fig. 4b). Taken together, these results indicate that the monoubiquitination of Rev1 is not required for FAAP20 binding but does enhance the binding interaction through the UBZ4 domain of FAAP20.
Furthermore, monoubiquitinated Flag-Rev1 coimmunoprecipitated strongly with wild-type FAAP20 but only weakly with the FAAP20 UBZ4 mutant (Fig. 4d). Collectively, these data indicate that monoubiquitination of Rev1 enhances the interaction with FAAP20 via its interaction with the UBZ4 domain of FAAP20, and thereby contributing to the stable association of the two proteins at sites of replication stress. Accordingly, the hypersensitivity of the Rev1 UBM mutant to DNA damage may also result from a defective FAAP20 interaction as well as from a defect in PCNA-Ub mediated recruitment25.
FAAP20 and Rev1 colocalize at sites of replication stress
To further characterize the interaction between FAAP20 and Rev1 in vivo, we examined the subcellular localization of FAAP20. GFP-tagged FAAP20, expressed in U2OS or HeLa cells, was primarily localized in the nucleus (Fig. 5a and data not shown). Interestingly, we observed nuclear foci of GFP-FAAP20 in some cells, which increased in a damage-inducible manner, similar to the increase in Rev1 (Fig. 5a,b). Mutation of the UBZ4 domain abrogated the ability of FAAP20 to form nuclear foci, suggesting that the interaction with ubiquitin is necessary for foci formation (Fig. 5b). The UBZ mutant FAAP20 protein was instead localized more to the cytoplasm. The foci were also visualized in FA patient-derived FANCA-deficient fibroblasts suggesting that foci formation is not dependent on FANCA or on FANCD2 monoubiquitination (Supplementary Fig. 5a).
Figure 5. FAAP20 and Rev1 colocalize at sites of replication stress.

(a) Fluorescence microscopy of U2OS cells, transfected with GFP-tagged FAAP20 and treated with 15 J m−2 UVC for 14 hr. Cells with more than five foci were quantified. Data shown are mean ± s.d. from three independent experiments. * p < 0.01 (b) GFP-fluorescence of undamaged U2OS cells transfected with GFP-FAAP20 wild-type or UBZ4 mutant. Immunoblot shows comparable expression levels. (c) Immunostaining of PCNA in U2OS cells, transfected with GFP-tagged Rev1 or FAAP20 and treated with 15 J m−2 UVC. A representative image is shown, in which 84.1 % of the FAAP20 foci colocalize with PCNA foci. (d) Anti-myc immunostaining in U2OS cells, co-transfected with GFP-Rev1 and myc-tagged FAAP20 wild-type or mutant and treated with 15 J m−2 UVC. Immunostaining of cells transfected with GFP-Rev1 alone served as negative control for antibody specificity. A representative image is shown, in which 83.8 % of the FAAP20 foci colocalize with Rev1 foci. (e) The mutation frequency in damaged (1,000 J m−2 UVC) supF plasmid was determined recovered from siRNA treated 293T cells. Data shown are mean ± s.d. from three independent experiments. * p < 0.05 compared with UVC-treated control.
The similarity of Rev1 and FAAP20 foci assembly prompted us to test whether FAAP20 foci also represent sites of replication stress. As previously reported in cells with cisplatin-induced intrastrand crosslinks20, Rev1 showed extensive colocalization with PCNA upon UVC irradiation, indicating that Rev1 is recruited to the region of stalled DNA replication by recognizing monoubiquitinated PCNA (Fig. 5c). Importantly, UVC-induced GFP-FAAP20 foci also exhibited colocalization with PCNA suggesting that the DNA lesion marked by PCNA contains both Rev1 and FAAP20 together (Fig. 5c). Rev1 and FAAP20 foci also colocalized following UVC treatment (Fig. 5d). We also observed the induction of PCNA monoubiquitination and colocalization of FAAP20 with PCNA foci as well as Rev1-PCNA colocalization following a high dose of MMC treatment, implying that the FAAP20-Rev1 interaction is also relevant to replication-dependent ICL repair process (Supplementary Fig. 5b,c). Taken together, these results suggest that FAAP20 is relocalized to DNA lesions in response to replication stress where it interacts with Rev1 and promotes its foci assembly.
FAAP20 promotes translesion synthesis through Rev1
Efficient Rev1 foci formation is required for Rev1-mediated translesion synthesis25. To interrogate the effect of FAAP20 deficiency on the function of Rev1, we employed the supF shuttle vector-based mutagenesis assay that measures TLS activity in mammalian cells28,29. Cells transfected with Rev1 siRNA that was previously shown to induce MMC-induced chromosomal aberrations19 exhibited reduced mutation frequency in the UVC-damaged supF structural gene, confirming the importance of Rev1 in error-prone DNA damage bypass (Fig. 5e). Either FAAP20 or FANCA knockdown also rendered cells hypomutable for UVC-induced mutagenesis, but not as significantly as Rev1 knockdown, suggesting that PCNA-Ub-dependent Rev1 regulation also contributes to Rev1-mediated mutagenesis. Importantly, knockdown of both FAAP20 and Rev1 did not further decrease the mutation frequency compared with Rev1 depletion, indicating that FAAP20 promotes translesion synthesis through downstream Rev1 (Fig. 5e). Sequencing analysis revealed mutation spectra of point mutations consistent with error-prone TLS from both control and FAAP20 knockdown cells (Supplementary Fig. 6a). These data further demonstrate the importance of the FA pathway in regulating the error-prone damage bypass mediated by the FAAP20-Rev1 axis.
DISCUSSION
The FA pathway is branched for TLS regulation
The FA pathway coordinates multiple DNA repair pathways including HR, TLS, and NER (nucleotide excision repair) to preserve genomic stability in response to DNA replication stress30. Although the FA core complex is known to regulate Rev1-dependent TLS pathways, the mechanism whereby the FA core complex interacts with Rev1 has remained unknown. Here, we show that FAAP20, a novel subunit of the FA core complex, connects the FA core complex and Rev1 TLS polymerases. FAAP20 binds to FANCA and maintains the integrity of the FA core, an E3 ligase complex required for FANCD2 monoubiquitination. The C-terminal UBZ4 domain of FAAP20 also interacts with the monoubiquitinated form of Rev1 and stabilizes Rev1 nuclear foci where PCNA/Rev1 DNA damage bypass complexes replicate over a lesion that blocks DNA replication.
Our study indicates that, when a replication complex encounters a DNA lesion, the FA core complex performs a dual role in DNA repair (Fig. 6). On the one hand, the complex functions as E3 ligase to monoubiquitinate the FANCD2/I heterodimer, thereby initiating homologous recombination repair. HR repairs double strand breaks and protects cells from inappropriate DNA repair by NHEJ (Nonhomologous end joining) that can lead to chromosomal translocations31. On the other hand, the complex binds to Rev1 and promotes efficient Rev1 foci assembly at the stalled replication fork. Rev1 assembly is further stabilized by PCNA-Ub binding. In this way, the core complex channels the DNA lesion into the TLS polymerase bypass pathway to promote the completion of DNA replication. The FAAP20 subunit of the FA core complex therefore appears to be the factor that connects the FA pathway with error-prone lesion bypass through its unique ubiquitin-binding domain.
Figure 6. A branched model of the FA pathway.
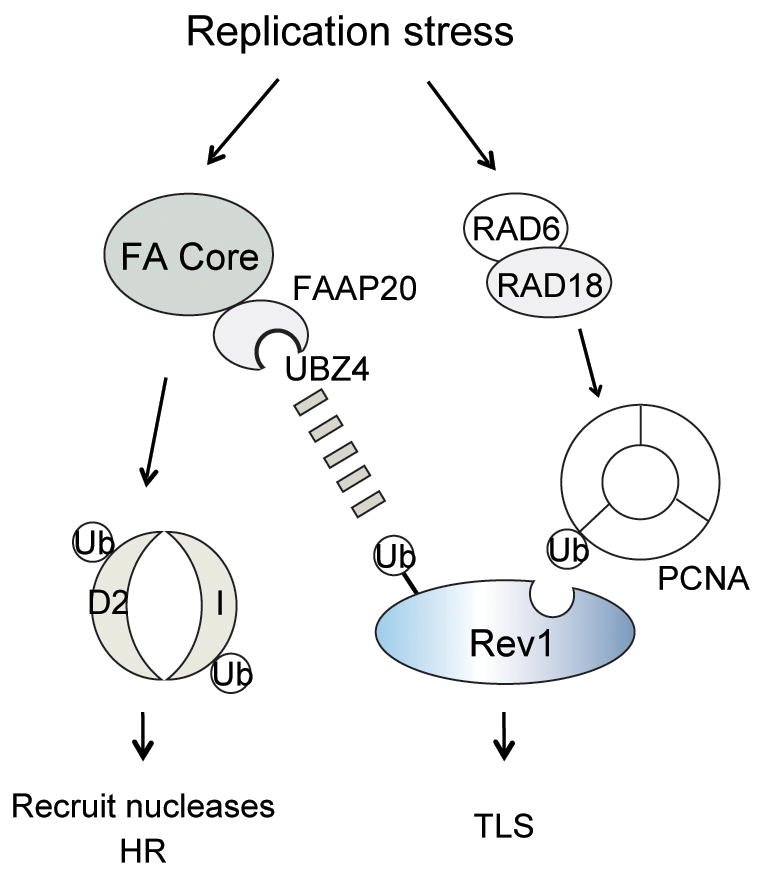
Upon replication stress, the FA core complex monoubiquitinates FANCD2/I to recruit nucleases and promote homologous recombination. FAAP20 stabilizes the FA core complex. The FAAP20-containing FA core complex is also required for efficient Rev1 foci assembly where the ubiquitin-binding property of FAAP20 contributes to stabilizing the PCNA-associated Rev1 complex at the stalled replication fork to direct lesion bypass. FAAP20-Rev1 interaction may be further enhanced by uncharacterized interaction.
Rev1 is recruited to TLS sites by PCNA monoubiquitination
Previous studies have indicated that Rev1 is recruited to sites of stalled DNA replication via its direct interaction with monoubiquitinated PCNA, the processivity clamp for DNA TLS polymerases25. Following DNA damage, PCNA is monoubiquitinated by the RAD6/RAD18 enzyme complex, and PCNA-Ub recruits Rev1 via its C-terminal ubiquitin binding motif (UBM)32. Abrogation of PCNA monoubiquitination by genetic disruption of RAD18 results in a partial but not complete loss of Rev1-dependent DNA damage tolerance, suggesting that mechanisms independent of PCNA monoubiquitination could regulate Rev1 function33,34. Our results indicate that the FA core complex stabilization of Rev1, mediated through FAAP20, may perform this PCNA-independent mechanism.
Several other recent studies indicate that the FA pathway is highly integrated with RAD18-mediated PCNA monoubiquitination. First, RAD18 stimulates the FA core complex-dependent monoubiquitination of FANCD235–38. RAD18 can stimulate FA core complex-dependent FANCD2 monoubiquitination, even in the absence of PCNA monoubiquitination39,40. Second, monoubiquitinated FANCD2 colocalizes with PCNA-Ub in replication foci41, and FANCD2 contains a PIP (PCNA-interacting protein) box within its primary sequence42. Finally, FANCD2-Ub and PCNA-Ub are coordinately deubiquitinated by the same deubiquitinating (DUB) enzyme complex, USP1 and UAF143 .
We also observed monoubiquitination of PCNA and colocalization of FAAP20 and PCNA in MMC-treated cells, suggesting that the interaction between Rev1 and FAAP20 may also regulate the TLS step of ICL repair. Intriguingly, a recent study proposed a role of hSNM1A in initiating ICL repair with XPF-ERCC1 nucleases44. Given that RAD18-dependent PCNA monoubiquitination recruits hSNM1A to the damaged replication forks23, these findings further support a role of PCNA-Ub in coordinating ICL repair.
Stimulation of TLS by the FA core complex
The FA core complex is required not only for the assembly of Rev1 foci but also for efficient lesion bypass and error-prone point mutagenesis19. How the FA core complex, and the FAAP20 subunit, stimulates TLS repair remains unclear, and several mechanisms are possible. First, the mere stabilization of Rev1 at the site of DNA damage may be sufficient for the promotion of TLS. Second, FAAP20 binding may stimulate the deoxycytidyl (dCMP) nucleotidyl transferase activity of Rev1 or may promote the binding of other TLS polymerases. Third, the FA core complex has other enzymatic activities which may further contribute to TLS. For example, the translocase activity of the FANCM subunit or the ubiquitin ligase activity of the FANCL subunit may be required for TLS activation. Future studies will examine the possible requirement of these enzymatic functions in the activation of TLS.
Supplementary Material
Acknowledgments
We would like to thank Lisa A. Moreau for chromosome aberration assay, Drs. Kay Hofmann (Miltenyi Biotec GmbH, Germany), Ivan Dikic (Institute of Biochemistry II, Germany), Alan R. Lehmann (University of Sussex, UK), and Emily H.-Y. Cheng (Memorial Sloan-Kettering Cancer Center, USA) for reagents. We also thank members of D’Andrea laboratory for valuable input and helpful discussions. H.K. is a recipient of the Leukemia and Lymphoma Society Career Development Fellowship. K.Y is a Harvard University Presidential Scholar. This work was supported by R01DK43389 and R01HL52725 to A.D.D.
Footnotes
AUTHOR CONTRIBUTIONS
H.K. and A.D.D. designed and interpreted experiments. H.K. and D.D. conducted experiments. K.Y. performed bioinformatics analysis. H.K. and A.D.D. wrote the manuscript. A.D.D. directed the project.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
METHODS
Methods are available in the online version of the paper at http://www.nature.com/nsmb/.
References
- 1.Joenje H, Patel KJ. The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet. 2001;2:446–57. doi: 10.1038/35076590. [DOI] [PubMed] [Google Scholar]
- 2.Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–94. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciccia A, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Smogorzewska A, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joo W, et al. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–6. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto KN, et al. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci U S A. 2011;108:6492–6. doi: 10.1073/pnas.1018487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cybulski KE, Howlett NG. FANCP/SLX4: A Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011;10:1757–63. doi: 10.4161/cc.10.11.15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang M, D'Andrea AD. A new nuclease member of the FAN club. Nat Struct Mol Biol. 2010;17:926–928. doi: 10.1038/nsmb0810-926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bienko M, et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–4. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann K. Ubiquitin-binding domains and their role in the DNA damage response. DNA Repair (Amst) 2009;8:544–56. doi: 10.1016/j.dnarep.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Knipscheer P, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papadopoulo D, Guillouf C, Mohrenweiser H, Moustacchi E. Hypomutability in Fanconi anemia cells is associated with increased deletion frequency at the HPRT locus. Proc Natl Acad Sci U S A. 1990;87:8383–7. doi: 10.1073/pnas.87.21.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirchandani KD, D'Andrea AD. The Fanconi anemia/BRCA pathway: a coordinator of cross-link repair. Exp Cell Res. 2006;312:2647–53. doi: 10.1016/j.yexcr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 14.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 15.Lehmann AR, et al. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amst) 2007;6:891–9. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Waters LS, et al. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73:134–154. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niedzwiedz W, et al. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell. 2004;15:607–20. doi: 10.1016/j.molcel.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- 19.Mirchandani KD, McCaffrey RM, D'Andrea AD. The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA Repair (Amst) 2008;7:902–11. doi: 10.1016/j.dnarep.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hicks JK, et al. Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol. 2010;30:1217–30. doi: 10.1128/MCB.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang TT, D'Andrea AD. Regulation of DNA repair by ubiquitylation. Nat Rev Mol Cell Biol. 2006;7:323–34. doi: 10.1038/nrm1908. [DOI] [PubMed] [Google Scholar]
- 22.Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461–7. doi: 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- 23.Yang K, Moldovan GL, D'Andrea AD. RAD18-dependent recruitment of SNM1A to DNA repair complexes by a ubiquitin-binding zinc finger. J Biol Chem. 2010;285:19085–91. doi: 10.1074/jbc.M109.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo C, et al. REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell. 2006;23:265–71. doi: 10.1016/j.molcel.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 25.Guo C, et al. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol Cell Biol. 2006;26:8892–900. doi: 10.1128/MCB.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood A, Garg P, Burgers PMJ. A ubiquitin-binding motif in the translesion DNA polymerase Rev1 mediates its essential functional interaction with ubiquitinated proliferating cell nuclear antigen in response to DNA damage. J Biol Chem. 2007;282:20256–20263. doi: 10.1074/jbc.M702366200. [DOI] [PubMed] [Google Scholar]
- 27.Hershko A, Rose IA. Ubiquitin-aldehyde: a general inhibitor of ubiquitin-recycling processes. Proc Natl Acad Sci U S A. 1987;84:1829–1833. doi: 10.1073/pnas.84.7.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parris CN, Seidman MM. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene. 1992;117:1–5. doi: 10.1016/0378-1119(92)90482-5. [DOI] [PubMed] [Google Scholar]
- 29.Choi JH, Pfeifer GP. The role of DNA polymerase eta in UV mutational spectra. DNA Repair. 2005;4:211–220. doi: 10.1016/j.dnarep.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bunting SF, Nussenzweig A. Dangerous liaisons: Fanconi anemia and toxic nonhomologous end joining in DNA crosslink repair. Mol Cell. 2010;39:164–166. doi: 10.1016/j.molcel.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 33.Ross AL, Simpson LJ, Sale JE. Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res. 2005;33:1280–1289. doi: 10.1093/nar/gki279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arakawa H, et al. A Role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4:e366. doi: 10.1371/journal.pbio.0040366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song IY, et al. Rad18-mediated translesion synthesis of bulky DNA adducts is coupled to activation of the Fanconi anemia DNA repair pathway. J Biol Chem. 2010;285:31525–36. doi: 10.1074/jbc.M110.138206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams SA, Longerich S, Sung P, Vaziri C, Kupfer GM. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood. 2011;117:5078–87. doi: 10.1182/blood-2010-10-311761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park HK, Wang H, Zhang J, Datta S, Fei P. Convergence of Rad6/Rad18 and Fanconi anemia tumor suppressor pathways upon DNA damage. PLoS One. 2010;5:e13313. doi: 10.1371/journal.pone.0013313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng L, Huntoon CJ, Karnitz LM. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J Cell Biol. 2010;191:249–57. doi: 10.1083/jcb.201005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palle K, Vaziri C. Rad18 E3 ubiquitin ligase activity mediates Fanconi anemia pathway activation and cell survival following DNA Topoisomerase 1 inhibition. Cell Cycle. 2011;10:1625–38. doi: 10.4161/cc.10.10.15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keaton MA, Dutta A. Rad18 emerges as a critical regulator of the Fanconi Anemia pathway. Cell Cycle. 2011;10:1625–38. doi: 10.4161/cc.10.15.15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 42.Howlett NG, Harney JA, Rego MA, Kolling FW, Glover TW. Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J Biol Chem. 2009;284:28935–28942. doi: 10.1074/jbc.M109.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang K, et al. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011;25:1847–1858. doi: 10.1101/gad.17020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang AT, et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.