

Abstract
Here, we show that tamoxifen resistance is induced by cancer-associated fibroblasts (CAFs). Coculture of estrogen receptor positive (ER+) MCF7 cells with fibroblasts induces tamoxifen and fulvestrant resistance with 4.4 and 2.5-fold reductions, respectively, in apoptosis compared with homotypic MCF7 cell cultures. Treatment of MCF7 cells cultured alone with high-energy mitochondrial “fuels” (L-lactate or ketone bodies) is sufficient to confer tamoxifen resistance, mimicking the effects of coculture with fibroblasts. To further demonstrate that epithelial cancer cell mitochondrial activity is the origin of tamoxifen resistance, we employed complementary pharmacological and genetic approaches. First, we studied the effects of two mitochondrial “poisons,” namely metformin and arsenic trioxide (ATO), on fibroblast-induced tamoxifen resistance. We show here that treatment with metformin or ATO overcomes fibroblast-induced tamoxifen resistance in MCF7 cells. Treatment with the combination of tamoxifen plus metformin or ATO leads to increases in glucose uptake in MCF7 cells, reflecting metabolic uncoupling between epithelial cancer cells and fibroblasts. In coculture, tamoxifen induces the upregulation of TIGAR (TP53-induced glycolysis and apoptosis regulator), a p53 regulated gene that simultaneously inhibits glycolysis, autophagy and apoptosis and reduces ROS generation, thereby promoting oxidative mitochondrial metabolism. To genetically mimic the effects of coculture, we next recombinantly overexpressed TIGAR in MCF7 cells. Remarkably, TIGAR overexpression protects epithelial cancer cells from tamoxifen-induced apoptosis, providing genetic evidence that increased mitochondrial function confers tamoxifen resistance. Finally, CAFs also protect MCF7 cells against apoptosis induced by other anticancer agents, such as the topoisomerase inhibitor doxorubicin (adriamycin) and the PARP-1 inhibitor ABT-888. These results suggest that the tumor microenvironment may be a general mechanism for conferring drug resistance. In summary, we have discovered that mitochondrial activity in epithelial cancer cells drives tamoxifen resistance in breast cancer and that mitochondrial “poisons” are able to re-sensitize these cancer cells to tamoxifen. In this context, TIGAR may be a key “druggable” target for preventing drug resistance in cancer cells, as it protects cancer cells against the onset of stress-induced mitochondrial dys-function and aerobic glycolysis.
Keywords: drug resistance, tamoxifen, metformin, tumor stroma, microenvironment, Warburg Effect, aerobic glycolysis, mitochondrial oxidative phosphorylation, TIGAR, glucose uptake, oxidative stress, reactive oxygen species (ROS), cancer associated fibroblasts
Introduction
Breast cancer is one of the most common causes of cancer death in women1 accounting for one third of cancer diagnoses and 15% of cancer deaths in the United States.2 More than 70% of breast cancers are estrogen receptor positive (ER+).3-5 Anti-hormonal therapy has improved the prognosis of ER+ breast cancer, but late recurrences are very frequent. At least a fourth of patients with operable ER+ breast cancer will have recurrence of their disease at 10 years6 and up to a third of patients will recur by 15 years.7 The vast majority of recurrences are metastases and metastatic ER+ breast cancer is an incurable disease that ultimately leads to the patient’s death. Therefore, improved treatment strategies for ER+ breast cancer are urgently needed. The aim of this study was to identify mechanism(s) of anti-estrogen resistance and discover new therapeutic targets to overcome drug resistance in ER+ breast cancers.
Many mechanisms have been described for acquired anti-estrogen resistance in breast cancers; for recent reviews, please refer to.5,8 Most of the studies on acquired anti-estrogen resistance have been performed with tamoxifen and have focused on the role of epithelial cancer cells. The main mechanism(s) for tamoxifen resistance include: i) activation of ER-independent pro-survival pathways, such as ERBB2,9,10 EGFR,11 IGFR12 and c-Src13-15; ii) altered expression of ER co-regulators, such as increased AIB1/SRC316,17; iii) altered regulation of downstream effectors of the ER involved in cell cycle and apoptosis regulation, such as NFkB,18 Erk,19 PI3K,20 c-Myc.21 Bcl-2,22,23 cyclinD1,24,25 cyclin E,26 p27,27 p2128 and PUMA29; iv) changes in ER expression19; v) mutations in the ER gene30 and vi) single nucleotide polymorphisms in cytochrome P450 2D6 (CYP2D6), which is associated with altered tamoxifen metabolism.31
However, little is known about the role of epithelial cancer cell metabolism in tamoxifen resistance. By generating high levels of reactive oxygen species (ROS), tamoxifen impairs mitochondrial function.32 Tamoxifen-induced oxidative stress can increase the expression of the redox sensitive transcription factor AP-1 and lead to tamoxifen resistance.33 A role in tamoxifen resistance has been found also for the prolyl hydroxylase domain proteins (PHD). PHDs induce the degradation of HIF1-α, one of the major transcription factors involved in cell metabolism and inhibition of mitochondrial activity. Overexpression of PHD1 promotes breast cancer growth and tamoxifen resistance34 and loss of PHD1 activity inhibits estrogen-dependent breast cancer tumorigenesis.35 These results suggest that tamoxifen resistance is associated with high mitochondrial activity.
The role of estrogen in the regulation of the breast stroma is not fully characterized. It is known that mammary extracellular matrix (ECM) composition, including fibronectin and its binding to integrins, varies with the reproductive cycle and estrogen levels.36,37
It was also shown that the tumor stroma regulates the activation of ER and progesterone receptor (PR), promoting tumor growth. The cancer-associated fibroblasts (CAFs) upregulate aromatase expression and increase estrogen levels in the tumor microenvironment.38 CAFs secrete fibroblast growth factor, which activates the progesterone receptor.39 Finally, culturing cancer cells with fibroblasts derived from ER(-)/PR(-) tumors leads to tamoxifen resistance, with activation of AKT and MAPK, and hyper-phosphorylation of ER.40
Among the stromal cells, CAFs have been shown to play a key role in supporting tumor growth, and determining tumor prognosis. For example, a loss of caveolin-1 (Cav-1) in stromal fibroblasts is associated with poor prognosis in breast cancer patients.41-44 Breast cancer patients with a loss of stromal Cav-1 have resistance to tamoxifen, with shorter progression free survival (PFS). In one study, loss of stromal Cav-1 was associated with PFS of 28.6% at 5 years, compared with 90.2% for patients where stromal Cav-1 was present.45
Coculture of the ER+ MCF7 cells with immortalized human fibroblasts generates CAFs, accompanied by Cav-1 downregulation.46 This in vitro coculture system mimics the behavior of breast cancers in vivo, with a tumor promoting microenvironment and aggressive epithelial cancer cells.46-48 Cav-1 downregulation in fibroblasts generates high levels of ROS, with increased mitophagy and autophagy, and higher levels of ketone bodies.48,49 It has been shown that autophagy leads to ketogenesis50 and that high levels of ketone bodies induce autophagy.51
Cav-1 low fibroblasts display an aberrant form of metabolism, with increased aerobic glycolysis, mitochondrial impairment, and increased expression of monocarboxylate transporter 4 (MCT4) which is involved in the secretion of lactate and ketone bodies.47,48,52 Conversely, epithelial cancer cells in proximity to Cav-1 low fibroblasts show decreased glycolysis, increased mitochondrial activity, and increased expression of the MCT1 transporter involved in the uptake of lactate and ketone bodies.47,48,52 Thus, loss of stromal Cav-1 leads to metabolic-coupling between epithelial cancer cells and CAFs, with the unidirectional transfer of catabolites from highly glycolytic fibroblasts to the oxidative epithelial cancer cells. Functionally, this metabolic-coupling protects cancer cells against apoptosis.
Since loss of stromal Cav-1 is a predictive biomarker for tamoxifen resistance,41 our current study aimed to investigate if CAFs can confer resistance to tamoxifen and other chemotherapeutic agents. To study tamoxifen resistance, we employed an in vitro epithelial-fibroblast coculture model, with stromal Cav-1 downregulation. In this context, our results directly show that fibroblasts are sufficient to confer tamoxifen and drug resistance.
The bioenergetic state of MCF7 cells is regulated by crosstalk with adjacent fibroblasts and loss of stromal Cav-1 leads to increased MCF7 cell mitochondrial activity. To search for potential strategies to overcome fibroblast-induced tamoxifen resistance, we sought to target the mitochondrial activity of cancer cells. Thus, we investigated the use of metformin and arsenic trioxide (ATO), which are drugs that impair mitochondrial function, as a strategy to overcome anti-estrogen resistance in epithelial cancer cells. Metformin is an FDA approved drug for the treatment of type 2 diabetes mellitus and ATO is approved for the treatment of acute promyelocytic leukemia. There is extensive clinical experience and a good safety profile for both agents. Our results directly show that treatment with ATO and with the combination of metformin and tamoxifen overcomes fibroblast-induced tamoxifen resistance.
Metformin is an inhibitor of mitochondrial electron transport chain complex I, which leads to decreased ATP production and increased AMP,53,54 thus inducing activation of AMPK.55 In mouse embryonal fibroblasts, and several cancer cell lines, AMPK activation by metformin leads to inhibition of mTOR and inhibition of proliferation.56-59
ATO is reactive with thiol containing proteins, which play an important role as antioxidants. ATO therefore generates high levels of ROS and uncouples mitochondrial oxidative phosphorylation.60,61 The anticancer mechanisms of ATO have been ascribed to the generation of ROS and the impairment of the TCA cycle and mitochondrial respiration.60,62
Since a loss of Cav-1 in stromal cells leads to high mitochondrial activity in adjacent epithelial cancer cells, we reasoned that the mechanisms of tamoxifen resistance may be due to the metabolic reprogramming of tumor cells, toward oxidative phosphorylation. Consistent with our hypothesis, we show that treatment with “mitochondrial poisons,” i.e., metformin and ATO, overcomes tamoxifen resistance and disrupts the metabolic-coupling between fibroblasts and cancer cells. In further support of our hypothesis, we show here that tamoxifen induces the upregulation of TIGAR (TP53-induced glycolysis and apoptosis regulator), a p53 regulated gene that simultaneously inhibits glycolysis, autophagy and apoptosis and reduces ROS generation, thereby promoting oxidative mitochondrial metabolism.63,64 To investigate if TIGAR-mediated increases in mitochondrial activity protect cancer cells from apoptosis, we recombinantly overexpressed TIGAR in MCF7 cells. Indeed, we show here that TIGAR overexpression confers drug resistance upon MCF7 cells, and protects them against the onset of aerobic glycolysis.
Results
Fibroblasts induce tamoxifen- and fulvestrant-resistance in MCF7 cells
Clinical prognostic studies have shown that breast cancer patients with a loss of stromal Cav-1 are resistant to tamoxifen.45 We set out to evaluate if anti-estrogen resistance could be recapitulated in an in vitro breast cancer model where MCF7 cells are cocultured with fibroblasts. We have previously shown that in this co-culture model, MCF7 cells induce Cav-1 downregulation in adjacent fibroblasts, and that conversely, fibroblasts protect MCF7 cells against apoptosis. Figure 1A shows that, upon tamoxifen treatment, MCF7 cells in coculture display a 4.4-fold reduction in apoptosis compared with MCF7 cells in single culture (compare third and forth bar) (p = 0.0002). Tamoxifen treatment does not significantly increase apoptosis of cocultured MCF7 cells, compared with control cocultures treated with vehicle alone (compare second and forth bar). Thus, MCF7 cells in coculture with fibroblasts are protected from tamoxifen-induced apoptosis.
Figure 1.

Fibroblasts induce resistance to hormonal therapy in MCF7 cells. (A) MCF7 cells were plated in homotypic culture or in coculture with fibroblasts at a ratio of 1:5 MCF7 cells to fibroblasts. The next day, cells were treated with 10 µM tamoxifen or vehicle alone (ethanol, control media) for 24 h. Apoptosis was measured with annexin V staining. Note that MCF7 cells in coculture are resistant to tamoxifen-induced apoptosis (4.4-fold reduction), compared with MCF7 single cells treated with tamoxifen (compare third and fourth bar) (p = 0.0002). (B) MCF7 cells were plated in homotypic culture or in coculture with fibroblasts at a ratio of 1:5 MCF7 cells to fibroblasts. The next day, cells were treated with 10 µM fulvestrant or vehicle alone (DMSO, control media) for 48 h. Apoptosis was measured with annexin V staining. Note that MCF7 cells in coculture show a ~2.5-fold reduction of fulvestrant-induced apoptosis compared with single cell culture treated with fulvestrant (p = 0.01). These results suggest that fibroblasts induce tamoxifen-resistance.
To confirm that fibroblasts induce anti-estrogen resistance, fulvestrant (ICI, 182,700), a pure anti-estrogen drug, was used. Fulvestrant is an analog of 17-β-estradiol with an alkinyl-sulphinyl side chain, which binds to the estrogen receptor and leads to blocking and degradation of the receptor.65 Fulvestrant has been shown to be more effective than tamoxifen in the treatment of advanced breast cancer.66 Homotypic cultures or cocultures of MCF7 cells were treated with 10 µM fulvestrant for 48 h, and subjected to apoptosis analysis. Figure 1B shows that fibroblasts protect MCF7 cells against fulvestrant-induced apoptosis, with a 2.5-fold reduction in the rate of apoptosis, compared with homotypic MCF7 cell cultures treated with fulvestrant (p = 0.01). Consistent with our previously published data,47 MCF7 cells are protected against apoptosis by fibroblasts. Figure 1A and B show that untreated MCF7 cells in coculture display an approximately 3-fold reduction in apoptosis compared with untreated single cell type MCF7 cells. Therefore, coculture with fibroblasts is sufficient to overcome the antitumor effects of tamoxifen and fulvestrant.
It is important to note that although fibroblasts induce resistance to tamoxifen and fulvestrant in MCF7 cells, this does not occur to the same degree. Tamoxifen-treated cocultured MCF7 cells have similar apoptosis rates of untreated cocultured MCF7 cells (Fig. 1A). On the other hand, fulvestrant-treated MCF7 cells in coculture show a 2.1-fold higher apoptosis rate, compared with untreated MCF7 cells in coculture (Fig. 1B) (p = 0.0006).
Lactate and ketone bodies induce tamoxifen-resistance in MCF7 cells
We have previously shown that Cav-1 (−/−) stromal cells secrete elevated levels of high-energy metabolites, such as lactate and ketone bodies,49 and that these metabolites increase cancer cell mitochondrial activity. We next asked if lactate and ketone bodies may serve to protect cancer cells against tamoxifen-induced apoptosis. To this end, single cell cultures of MCF7 cells were incubated with L-lactate, β-hydroxy-butyrate, butanediol or control media, prior to treatment with tamoxifen or vehicle alone.
Figure 2 shows that, as expected, tamoxifen treatment induces the apoptosis of MCF7 cells. However, treatment with L-lactate, β-hydroxy-butyrate, or butanediol, decreases tamoxifen-induced apoptosis by 3-, 3.7- and 2.6-fold, respectively (p < 0.003 for all the comparisons). Thus, the administration of lactate or ketone bodies mimics coculture with fibroblasts, and decreases tamoxifen-induced MCF7 cell apoptosis.
Figure 2.

Lactate and ketone bodies confer tamoxifen-resistance in MCF7 cells. MCF7 cells were plated in homotypic culture. The next day, cells were incubated with 10 mM β-hydroxybutyrate (BHB), 10 mM butanediol (BND) or 10 mM L-lactate. After 24 h, 10 µM tamoxifen or vehicle alone (ethanol) was added for an additional 24 h. Apoptosis was measured with annexin V and PI staining. Note that as expected, tamoxifen induces a ~2-fold increase in early apoptosis (annexin V + and PI -), compared with vehicle alone treated cells. However, treatment with lactate and ketone bodies abolishes tamoxifen-induced cell death, suggesting that lactate and ketone bodies confer tamoxifen-resistance (p < 0.03, vs. all conditions).
Metformin overcomes tamoxifen resistance in MCF7 cells in coculture, by disrupting the metabolic coupling of fibroblasts with MCF7 cells
We have previously shown that stromal and breast cancer cells are metabolically-coupled and that in a coculture model, MCF7 cells show increased mitochondrial capacity. In order to overcome the tamoxifen-resistance induced by fibroblasts, we sought to treat MCF7-fibroblast cocultures with the mitochondrial complex I inhibitor metformin. Figure 3A shows that the combination of metformin and tamoxifen is able to overcome the apoptosis resistance of MCF7 cells in coculture, with a 2.4-fold increase in total cell death (left panel) and a 2.1-fold increase in apoptosis rate (right panel), compared with control coculture conditions (p < 0.002). MCF7 cells in coculture treated with either tamoxifen or metformin alone are protected against apoptosis, but the combination of metformin and tamoxifen shows synergy in inducing MCF7 cell apoptosis.
Figure 3.

Metformin overcomes the tamoxifen-resistance of cocultured MCF7 cells. (A) Apoptosis: MCF7 cells were plated in coculture with fibroblasts at a ratio of 1:5 MCF7 cells to fibroblasts. The next day, 1 µM tamoxifen, 100 µM metformin or tamoxifen-metformin combination were administered for 24 h. Apoptosis was measured with annexin V and PI staining. In the left panel, the percentage of MCF7 cells undergoing cell death (annexin V+ and/or PI+) is displayed. In the right panel, the percentage of annexin V+ MCF7 cells is displayed. Note that the treatment of cocultured MCF7 cells with tamoxifen or metformin does not induce cell death. However, the treatment with the combination of tamoxifen and metformin induces a more than 2-fold increase in apoptotic cell death of cocultured MCF7 cells, compared with control cells, indicating that the combination of tamoxifen and metformin is sufficient to overcome the fibroblast-induced tamoxifen resistance (p = 0.006 and p = 0.002, for the left and right panels, respectively). (B) Glucose uptake: The glucose uptake assay was performed on cocultures of fibroblasts and MCF7 cells, using 2-NBDG fluorescence uptake. The ratio of glucose uptake (fibroblasts to MCF7 cells) was calculated as a measure of metabolic coupling between fibroblasts and MCF7 cells. Note that in untreated cocultures, the ratio of glucose uptake between fibroblasts and MCF7 cells is about 2, indicating that fibroblasts take up about two times more glucose than MCF7 cells. This is consistent with the idea of a metabolic coupling between fibroblasts and MCF7 cells. However, the combination of metformin with tamoxifen normalizes the glucose uptake ratio in cocultured cells. Note that upon treatment with the combination of metformin and tamoxifen, the glucose uptake ratio is about 1.2, compared with 1.8 under control conditions (p = 1.6 x 10−5). These results indicate that the combination of metformin with tamoxifen disrupts the metabolic coupling between fibroblasts and MCF7 cells.
To dissect the mechanisms by which metformin overcomes the tamoxifen-resistance of MCF7 cell cocultures, we evaluated the possibility that treatment with the combination of metformin and tamoxifen disrupts metabolic-coupling between fibroblasts and MCF cells. To this end, a glucose uptake assay was performed on cocultures of RFP (+) MCF7 cells and fibroblasts. The ratio of glucose uptake between fibroblasts and MCF7 cells was assessed, as a measurement of metabolic coupling. Figure 3B shows that in untreated cocultures, the glucose uptake ratio is of 1.8, indicating that in cocultures, fibroblasts take up roughly twice the amount of glucose, relative to MCF7 cells. However, treatment with the combination of tamoxifen and metformin decreases the glucose uptake ratio to 1.2, suggesting a disruption in metabolic-coupling. Thus, the resistance of cocultured MCF7 cells to the single agent tamoxifen can be overcome by the combination of tamoxifen and metformin, and this leads to similar glucose uptake in both fibroblasts and MCF7 cells.
ATO overcomes fibroblast-induced apoptotic-resistance in MCF7 cells, by increasing glucose uptake in resistant cancer cells
We have previously demonstrated that MCF7 cells in coculture with fibroblasts have increased mitocondrial activity.47 ATO leads to the generation of hydrogen peroxide, thus inhibiting the mitochondrial electron transport chain and increasing glycolysis.67 To further test if inhibition of mitochondrial activity could overcome tamoxifen resistance, MCF7-fibroblast cocultures were treated with 12 µM tamoxifen, 20 µM ATO or tamoxifen-ATO combination. Figure 4A shows that ATO treatment increases the total cell death (left panel) rate and the apoptosis (right panel) rate of apoptosis-resistant MCF7 cells in coculture by 2.1 and 2.6-fold. Interestingly, ATO did not have an additive effect with tamoxifen in inducing apoptosis of MCF7 cells in coculture. ATO alone or in combination with tamoxifen leads to similar apoptosis rates in MCF7 cells in coculture.
Figure 4.

ATO overcomes resistance to apoptosis of cocultured MCF7 cells. (A) Apoptosis: MCF7 cells were plated in coculture with fibroblasts at a ratio of 1:5 MCF7 cells to fibroblasts. After 24 h, 12 µM tamoxifen, 20 µM ATO or the combination of tamoxifen and ATO were administered for 24 h. Apoptosis was measured with annexin V and PI staining. In the left panel, the percentage of cell death (annexin V+ and/or PI+) of MCF7 cells is displayed. In the right panel, the percentage of annexin V+ MCF7 cells is displayed. Note that treatment with ATO overcomes the resistance to apoptosis of cocultured MCF7 cells. Cocultured MCF7 cells treated with ATO or with the combination of ATO and tamoxifen display more than 2-fold increase in apoptotic cell death compared with control cocultured cells. (B and C) Glucose uptake: MCF7-RFP (+) cells were plated in coculture with fibroblasts at a ratio of 1:5 MCF7 cells to fibroblasts. After 24 h, 12 µM tamoxifen, 10 µM ATO or both were administered for 24 h. The glucose uptake of MCF7-RFP (+) cells was measured using 2-NBDG fluorescence uptake by flow cytometry. (B) Note that ATO induces a 2-fold increase of glucose uptake by cocultured MCF7 compared with control untreated and tamoxifen-treated cells (p = 6 ×10−6). (C) The ratio of glucose uptake (fibroblasts to MCF7 cells) was calculated as a measure of the metabolic coupling between fibroblasts and MCF7 cells. Note that treatment with ATO and with the combination of tamoxifen and ATO reverses the metabolic coupling between MCF7 cells and fibroblasts in coculture. The glucose uptake ratio between fibroblasts and MCF7 cells nearly equalizes upon treatment with ATO and with the tamoxifen and ATO combination, changing from 1.8 under control conditions to 1.2. p = 0.002 CTR vs ATO, p = 0.0009 CTR vs ATO+T.
To dissect the mechanisms by which ATO overcomes the apoptosis/tamoxifen-resistance of cocultured MCF7 cells, we measured glucose uptake of cocultured MCF7 cells upon treatment with tamoxifen, ATO and the tamoxifen-ATO combination, using NBDG fluorescence. Figure 4B shows that ATO increases glucose uptake by 2-fold in MCF7 cells (p = 0.0001). Interestingly, the combination of ATO and tamoxifen does not significantly change glucose uptake, compared with the single agent ATO (Fig. 4B). To assess metabolic-coupling between fibroblasts and MCF7 cells, the glucose uptake ratio of fibroblasts-to-MCF7 cells in coculture was calculated. Figure 4C shows that ATO normalizes the glucose uptake ratio between fibroblasts and MCF7 cells. The glucose uptake ratio is about 1.8 under control conditions, and decreases to 1.2 after treatment with ATO or with the combination of ATO and tamoxifen.
Therefore, under untreated coculture conditions, glucose uptake is nearly twice as high in fibroblasts, compared with MCF7 cells. However, treatment with the drugs that greatly induce apoptosis in cocultured MCF7 cells (such as the tamoxifen-metformin combination or ATO) increases glucose uptake in MCF7 cells. This likely reflects metabolic-uncoupling between fibroblasts and MCF7 cells.
TIGAR expression is upregulated in MCF7 cells in cocultures treated with tamoxifen, and in epithelial cancer cells of human breast cancers lacking stromal Cav-1
Previous studies have shown that TIGAR is an anti-glycolytic and anti-apoptotic protein. To dissect the molecular mechanisms driving fibroblast-induced tamoxifen-resistance, we hypothesized that tamoxifen may induce TIGAR upregulation in MCF7 cells in coculture. To this end, fibroblast-MCF7 cell cocultures were treated with tamoxifen or vehicle alone and immunostained with TIGAR (red) and Keratin 8/18 (green, used as a marker of MCF7 cells) antibodies. Figure 5A shows that TIGAR is preferentially upregulated in MCF7 cells in coculture, especially after tamoxifen treatment, indicating that TIGAR upregulation may be one possible mechanisms underlying tamoxifen resistance.
Figure 5.

Tamoxifen induces TIGAR upregulation in cocultured MCF7 cells. (A) Fibroblast-MCF7 cell cocultures were cultured for 1 d with 12 µM tamoxifen or with vehicle alone. Then, cells were fixed and immuno-stained with anti-TIGAR (red) and anti K8–18 (green) antibodies. Nuclei were counterstained with DAPI (blue). TIGAR staining (red only) is shown in the top panels to better appreciate the tamoxifen-induced TIGAR upregulation in MCF7 cells. Original magnification 40x. (B) TIGAR is highly expressed in human epithelial breast cancer samples with loss of stromal Cav-1. Paraffin-embedded tissue sections from human breast cancer samples lacking Cav-1 were immunostained with antibodies directed against TIGAR. Slides were counterstained with hematoxylin. Note that TIGAR is highly expressed preferentially in epithelial breast cancer cells while stromal cells lack TIGAR expression. Original magnification, 40x.
To validate these results, immunohistochemistry with TIGAR antibodies was performed on human breast cancer samples lacking stromal Cav-1. Previous studies have shown that a loss of stromal Cav-1 correlates with tamoxifen-resistance in human breast cancers. Figure 5B shows elevated TIGAR expression in human epithelial breast cancer cells, whereas TIGAR expression is much lower in adjacent stromal cells. This provides further evidence for metabolic heterogeneity within the different cellular compartments of breast cancers, with a loss of stromal Cav-1.
TIGAR overexpression protects MCF7 cells from tamoxifen-induced apoptosis
To establish a direct cause-effect relationship between TIGAR expression and tamoxifen resistance, we generated MCF7 cells recombinantly overexpressing TIGAR (TIGAR-MCF7). Control MCF7 cells transfected with the empty vector (LV-105-MCF7) alone were generated in parallel. TIGAR overexpression was confirmed by immunoblot analysis, using TIGAR specific antibodies (Fig. 6).
Figure 6.
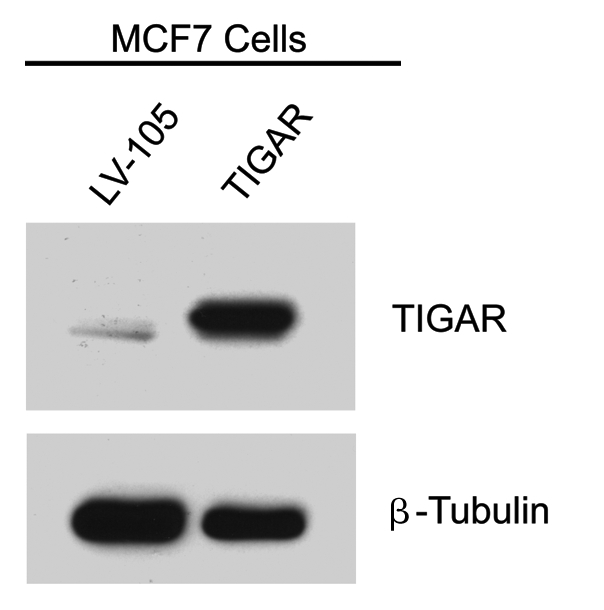
Generation of TIGAR overexpressing MCF7 cells. MCF7 cells recombinantly overexpressing TIGAR and empty vector control (LV-105) were generated and subjected to protein gel analysis with antibodies against TIGAR. As expected, the TIGAR overexpressing MCF7 cell line shows higher levels of TIGAR than the control. β-tubulin was used as an equal loading control.
We next examined if TIGAR overexpression mediates tamoxifen resistance in MCF7 cells, since TIGAR’s metabolic functions are well-characterized and include protection against apoptosis, inhibition of glycolysis, with increased mitochondrial activity. To this end, TIGAR-MCF7 cells and empty vector control cells were treated with tamoxifen or with vehicle alone, and apoptosis was analyzed by annexin V and PI staining. Figure 7 shows that, after tamoxifen treatment, MCF7 cells overexpressing TIGAR are protected against apoptosis compared with empty-vector control cells. Notably, TIGAR-MCF7 cells display a 1.9-fold decrease in total cell death (right panel) and a 1.8-fold decrease in apoptosis (left panel), compared with vector alone control cells.
Figure 7.

TIGAR overexpression mediates tamoxifen-resistance in MCF7 cells. TIGAR-MCF7 cells and control MCF7 cells were treated with 12 µM tamoxifen or vehicle alone for 24 h. Apoptosis was measured by annexin V and PI. Left panel: Percentage of annexin V+ MCF7 cells. Right panel: Percentage of MCF7 cell death (annexin V+ and/or PI+). Note that upon tamoxifen treatment, TIGAR overexpressing MCF7 cells display a nearly 2-fold decrease in apoptosis as compared with Lv-105 control MCF7 cells, indicating that TIGAR expression is one of the mechanisms mediating tamoxifen resistance in breast cancer cells. (p = 0.002 and p = 0.0001 respectively).
We next asked if TIGAR expression mediates resistance against other drugs, such as ATO. Figure 8A shows that TIGAR-MCF7 cells are resistant to ATO-induced cell death, and display a 1.8-fold decrease in apoptosis. Previous studies have shown that TIGAR prevents glycolysis. To evaluate if TIGAR-mediated apoptosis-resistance correlates with decreased glycolytic power, we next measured lactate production in cell culture media of TIGAR-MCF7 cells after ATO treatment. Interestingly, Figure 8B shows that ATO treatment increases lactate production in vector alone control MCF7 cells by 2.5-fold, indicating that ATO-induced cells death correlates with increased aerobic glycolysis in cancer cells. However, upon ATO treatment, TIGAR-MCF7 cells display a 1.5-fold reduction in lactate production, as compared with vector alone controls cells under identical conditions. However, no significant differences in lactate production were found between TIGAR-MCF7 cells and control MCF7 cells under basal untreated conditions. These results indicate that TIGAR overexpression may protect against drug-induced apoptosis, by decreasing the glycolytic power of cancer cells.
Figure 8.

TIGAR overexpressing MCF7 cells are resistant to ATO-induced apoptosis. (A) Apoptosis. TIGAR-MCF7 cells and control MCF7 cells were treated for 24 h with 10 µM ATO and apoptosis was measured by annexin V and PI. Note that MCF7 cells overexpressing TIGAR show a 1.8-fold decrease in apoptosis as compared with control LV-MCF7 cells, suggesting that TIGAR induces drug resistance (p = 0.008). (B) Lactate Assay. Lactate production was measured on the cell culture media of TIGAR-MCF7 cells and control LV-MCF7 cells treated with 10 µM ATO or vehicle alone for 24 h. Note that ATO treatment induces a significant increase in lactate production in control Lv-MCF7 cells. However, upon ATO treatment, MCF7 cells overexpressing TIGAR generate less lactate than LV-MCF7 cells. After ATO treatment, control LV-MCF7 cells show a much larger increase in lactate production, compared with MCF7 cells overexpressing TIGAR (p = 0.0045).
Fibroblasts induce resistance of MCF7 cells to doxorubicin and the PARP inhibitor ABT-888
To evaluate if fibroblasts induce resistance to other anticancer agents doxorubicin, a topoisomerase inhibitor which generates high levels of ROS, and ABT-888, a PARP-1 inhibitor, were used. MCF7 cells were plated as single cell cultures or as cocultures with fibroblasts and treated with either doxorubicin, ABT-888, or vehicle alone.
Figure 9A shows that MCF7 cells in homotypic culture display an 11.6-fold reduction and a 6.8-fold reduction in cell viability (as assessed by cell numbers) after treatment with doxorubicin and ABT-888 respectively, compared with control (vehicle alone) MCF7 cells. However, in coculture with fibroblasts, treatment with doxorubicin, and ABT-888 does not affect the viability of MCF7 cells (Fig. 9B). These results indicate that fibroblasts confer resistance to a variety of drugs, not only to anti-estrogen therapy.
Figure 9.

Fibroblasts induce drug resistance in MCF7 cells. To evaluate if fibroblasts protect cancer cells against cell death induced by known chemotherapy agents, MCF7 cells were cultured alone or cocultured with GFP (+) fibroblasts with 50 nM doxorubicin and 30 nM ABT-888 (a PARP inhibitor) for 24 h. To quantify cell viability, GFP (-) cell (MCF7 cells) numbers were measured using flow cytometry. (A) Homotypic cultures: Note that treatment with doxorubicin and ABT-888 greatly decreases the MCF7 cell viability, by 11.6 and 6.8-fold as compared with vehicle alone control (p = 0.001 and p = 0.007 respectively). (B) Cocultures: Note that treatment with doxorubicin and ABT-888 does not affect the viability of MCF7 cells in coculture with fibroblasts, suggesting that fibroblasts induce drug-resistance (p = 0.96 and p = 0.71 respectively).
Discussion
We have found that fibroblasts with Cav-1 downregulation induce tamoxifen and fulvestrant resistance in MCF7 breast cancer cells and that this resistance is mediated by the low glycolytic activity of MCF7 cells. We have also shown here that fibroblasts induce resistance to other anticancer drugs such as doxorubicin (adriamycin) and the PARP-1 inhibitor, ABT-888. Previous studies have shown that the downregulation of stromal Cav-1 induces metabolic- coupling, between epithelial cancer cells and fibroblasts. Herein, we show that MCF7 cells with high mitochondrial activity and low glucose uptake display tamoxifen resistance. Experimentally, we induced mitochondrial activity and decreased glucose uptake in MCF7 cells using three independent methods: coculture with fibroblasts, administration of lactate and ketone bodies, and genetic overexpression of the anti-glycolytic protein TIGAR (summarized schematically in Fig. 10).
Figure 10.

Energy transfer confers chemo-resistance in cancer cells. Schematic diagram summarizing our current findings. See text for details. Note that glycolytic fibroblasts provide nutrients (such as L-lacate and ketones) to fuel oxidative mitochondrial metabolism in epithelial cancer cells. This, in turn, drives protection against stress-induced apoptosis that is normally triggered by anticancer drugs, conferring drug-resistance. Specific MCT transporters allow the shuttling of L-lactate and ketones from fibroblasts (MCT4) to cancer cells (MCT1) (see reference 52 for details). Thus, fibroblasts and cancer cells are metabolically-coupled, in a form of “parasitic cancer metabolism.” This mechanism provides important new “druggable” targets for overcoming chemo-resistance, such as MCTs and TIGAR, among others. MCT, monocarboxylate transporters.
Our study adds to the body of literature showing that fibroblasts induce cancer chemotherapy resistance. It has previously been shown that fibroblasts derived from ER-/PR- tumors induce tamoxifen resistance in a pre-malignant model of human MCF10AT cells (MCF10A cells transformed with T24-Ha-Ras) and MCF7 cells.40 The stroma has also been shown to induce tamoxifen resistance in a mouse model of spontaneous breast cancer.68 Mesenchymal stem cells protect leukemia cells from chemotherapy-induced apoptosis (araC, vincristine and mitoxantrone).69 Murine fibroblasts in a Transwell coculture system induced etoposide resistance in a human pancreatic cancer cell line which is chemosensitive in homotypic culture.70 Also, a study has shown that the elimination of CAFs using a DNA vaccine against Fibroblast Activation Protein (FAP) synergizes with doxorubicin in decreasing tumor weight and lung metastasis surface area in a murine breast cancer model.71
Here, using a pharmacologic approach, we demonstrate that mitochondrial activity in epithelial cancer cells drives tamoxifen resistance. Importantly, the use of two mitochondrial inhibitors (metformin and ATO) is sufficient to overcome resistance and induce cell death of cocultured MCF7 cells. Metformin and ATO decrease mitochondrial activity and increase glucose uptake in MCF7 cells. The combination of tamoxifen with metformin or ATO equalizes the glucose uptake ratio between fibroblasts and cancer cells in coculture, suggesting that high-energy catabolites are no longer transferred from fibroblasts to epithelial cancer cells and there is metabolic uncoupling.
Retrospective studies have shown that diabetics treated with metformin show a cancer risk reduction of approximately 40% compared with diabetics not treated with metformin.72-76 It also appears that metformin sensitizes to chemotherapy. In a retrospective study examining the rates of pathologic complete remission (pCR) after neoadjuvant chemotherapy, it was found that 24% of diabetic patients taking metformin achieved pCR, compared with 8% of those who were not receiving metformin.77 In summary, the use of metformin in diabetic patients is associated with an improved outcome and our study provides a rationale for its use in breast cancer.
It is encouraging to note that in addition to the studies performed with diabetic patients, a pilot clinical study in non-diabetic patients showed that metformin at a low dose of 250 mg daily can prevent aberrant colon crypt formation.78 The doses used in the studies described here are comparable. Although many studies have shown the antitumor properties of metformin, the mechanisms of action have not been clearly defined yet.79 We are now able to define a mitochondrial-based mechanism of action involved in metformin’s ability to overcome tamoxifen resistance. It is also noteworthy that metformin has a good safety profile with few side effects,80 both crucial requirements for a drug used in the adjuvant setting.
Despite its good prospects for the treatment of breast cancer, it was unclear which subgroup of patients would benefit from metformin treatment. The use of metformin in breast cancer has been studied in more detail in patients with diabetes. Metformin’s safety record, the fact that it has been extensively studied, its use for the management of other diseases such as polycystic ovary syndrome,81 and that it has well-described actions independent of insulin, makes it an attractive breast cancer drug even in non-diabetic patients. Our data indicate that the subgroup of patients with a loss stromal Cav-1 may benefit from the treatment of the combination of tamoxifen with metformin.
ATO is also a promising drug for breast cancer treatment. ATO is an FDA-approved drug for the treatment of acute promyelocytic leukemia and is well tolerated over prolonged periods of time,82,83 although its safety profile is not as good as that of metformin. The doses of ATO used in our study are similar to physiologically achievable doses in humans.61 ATO is known to induce oxidative stress and generates high levels of hydrogen peroxide because of thiol protein dysfunction which alters mitochondria. Metformin and ATO in combination with antiestrogens shows promise in decreasing the rates of recurrence of breast cancer, but additional pharmacokinetic and pharmodynamic studies are needed prior to large scale clinical trials evaluating effectiveness.
Most drug specific predictors to guide chemotherapy selection have not been useful, as seen for example with the role of TOP2A amplification and anthracycline sensitivity.84 However, a stromal genetic signature has been shown to predict resistance to neoadjuvant breast cancer chemotherapy.85 Stromal Cav-1 status may guide the decision to add chemotherapy to adjuvant hormonal therapy in ER+ breast cancer, since a loss of stromal Cav-1 is associated with tamoxifen resistance. Even though the exact role of stromal Cav-1 status in conjuction with genetic profiling remains to be defined, it may provide complimentary information to that obtained for example with the 21-gene assay recurrence score Oncotype DX, which is well established and part of routine care.86
TIGAR may be another predictive biomarker of tamoxifen resistance. We have discovered that TIGAR expression in epithelial cancer cells induces tamoxifen resistance. Mechanistically, the loss of Cav-1 in fibroblasts leads to the upregulation of TIGAR in epithelial cancer cells, with consequent decreases in ROS levels, inhibition of glycolysis, autophagy and apoptosis. This is the first report showing that tamoxifen resistance is mediated by a glycolysis inhibitor, such as TIGAR.
Our data suggests that the efficacy of anticancer therapies, such as tamoxifen, not only depends on epithelial tumor cell characteristics, but also on the host tumor microenvironment. To improve outcomes in breast cancer, it is crucial to decrease rates of acquired tamoxifen resistance. Our study suggests that patients with loss of stromal Cav-1 may benefit from combinations of tamoxifen and metformin or ATO, to decrease the rate of recurrence. We have discovered that drugs that promote glycolysis and inhibit mitochondrial function in epithelial cancer cells are possible treatment options for this tamoxifen resistant subtype of breast cancer.
Conclusions
A loss of stromal caveolin-1 (Cav-1) is a biomarker for predicting poor clinical outcome and tamoxifen resistance in human breast cancers. Coculture with breast cancer MCF7 cells transforms normal human fibroblasts into cancer-associated fibroblasts (CAFs) with Cav-1 downregulation. In addition, coculture leads to metabolic-coupling between CAFs and cancer cells. CAFs show increased glucose uptake and glycolytic activity, impaired mitochondrial function, and increased generation of lactate and ketone bodies. Also, Cav-1 knockdown in fibroblasts is sufficient to generate glycolytic CAFs. Conversely, epithelial cancer cells in coculture display increased mitochondrial activity and protection from apoptosis. The administration of lactate and ketone bodies to epithelial cancer cells induces mitochondrial activity, in the same way as coculture with Cav-1 deficient fibroblasts. Since a loss of Cav-1 in fibroblasts protects epithelial cancer cells from apoptosis, we hypothesized that the increased mitochondrial activity of epithelial cancer cells is the underlying cause of tamoxifen resistance. This is clinically relevant because anti-estrogen therapy in breast cancer induces the apoptosis of epithelial cancer cells, but acquired resistance develops frequently.
Here, we have discovered that mitochondrial activity in epithelial cancer cells drives tamoxifen resistance in breast cancer and that mitochondrial inhibition is able to re-sensitize cancer cells to tamoxifen. The protective effect of fibroblasts on cancer cells is not exclusive of tamoxifen since similar effects are seen with doxorubicin and ABT-888. The discovery that CAFs are able to induce anti-estrogen resistance in breast cancer by increasing mitochondrial activity in epithelial cancer cells has important implications for establishing new drug screening strategies. The efficacy of anticancer drugs needs to be evaluated in the context of their ability to target CAFs and metabolically uncouple fibroblasts from epithelial cancer cells.
Materials and Methods
Materials
Antibodies were obtained as follows: anti-cytokeratin 8/18 (Fitzgerald Industries International, 20R-CP004), anti TIGAR (Abcam, ab37910), β-tubulin (Sigma). Other reagents were as follows: 4,6-diamidino-2-phenylindole (DAPI), Prolong Gold Antifade mounting reagent, propidium iodide (PI), and 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG, N13195), were from Invitrogen. Annexin V-APC was from BD (550474). L-lactate, β-hydroxybutyric acid, butanediol, tamoxifen, metformin, ATO and doxorubicin were purchased from Sigma. Fulvestrant was purchased from Tocris. ABT-888 was purchased from ChemieTek. The L-lactate assay kit (ECLC-100) was purchased from Bioassay Systems.
Cell cultures
Cell culture experiments were performed, as previously described with minor modifications.46 Human skin fibroblasts immortalized with human telomerase reverse transcriptase (hTERT-BJ1 cells) were purchased originally from Clontech, Inc. The breast cancer cell line, MCF7, was from ATCC. GFP (+) and RFP (+) MCF7 cells tagged with GFP and RFP were used in coculture experiments and flow cytometry was used to distinguish fibroblasts and MCF7 cells. All cells were maintained in DMEM, with 10% Fetal Bovine Serum (FBS) and Penicillin 100 units/mL-Streptomycin 100 µg/mL (Pen-Strep, Invitrogen). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Fibroblasts and MCF7 cells were co-plated or plated as a homotypic cell culture in 12-well plates in 1 ml of complete media. In coculture experiments, fibroblasts were plated first and MCF7 cells were plated within 2 h of fibroblast plating. The total number of cells per well in coculture was 1 x 105 cells. Coculture experiments were performed at a 5:1 fibroblast-to-epithelial cell ratio. As controls, homotypic cultures of fibroblasts and MCF7 cells were plated in parallel, using the same number of a given cell population as the corresponding co-cultures. The day after plating, the media was changed to DMEM with 10% NuSerum (a low protein alternative to FBS; BD Biosciences) and Pen-Strep and the different drugs were added at that point. Cells were maintained in this media for one to five days, until further analysis. To study the effect of tamoxifen, ATO and metformin, MCF7 cells alone and cocultures were cultured in 10% Nuserum and treated with the drugs for 24 h. L-Lactate, β-hydroxy-butyrate or butanediol were administered to MCF7 cells cultured in 10% NuSerum for 48 h.
Propidium Iodide and Annexin-V cell death detection
Cell death was quantified by flow cytometry using propidium iodide (PI) and Annexin-V-APC, as previously described with minor modifications.47 Briefly, MCF7-GFP cells were plated in 12 well plates with fibroblasts. The day after, media was changed to DMEM with 10% NuSerum, and drugs were added. After 24–48 h (as indicated in the figure legends), cells were collected by centrifugation and re-suspended in 500 μL of Annexin-V Binding Buffer. Then, the annexin V-APC conjugate (BD Biosciences, 550474) (4 μL) and PI (1 μL) was added and incubated in the dark at room temperature for 5 min. Cells were then analyzed by flow cytometry using a GFP signal detector (to detect MCF7-GFP), a PE Texas Red signal detector and an APC signal detector. GFP negative cells were considered fibroblasts.
Measurement of glucose uptake
2-NBDG was used to measure glucose uptake in single type cell cultures and cocultures of RFP (+) MCF7 cells and fibroblasts. Briefly, cultures were incubated with 200 µM 2-NBDG at 37°C for 30 min. Subsequently, cells were harvested and suspended in a 0.01 M Hepes (pH 7.4), 0.14 M NaCl and 2.5 mM CaCl2 solution and analyzed by flow cytometry using RFP signal detector (to detect MCF7-RFP) and a fluorescein signal detector (to measure glucose uptake). RFP negative cells were considered fibroblasts.
Immunocytochemistry (ICC)
Cocultures were fixed after 24 h of treatment with tamoxifen or vehicle alone (ethanol). Then, the ICC protocol was performed as previously described with minor modifications.87 Briefly, cells were fixed for 30 min at room temperature in 2% paraformaldehyde diluted in PBS, after which they were permeabilized with cold methanol at -20°C for 5 min. The cells were rinsed with PBS with 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM), and incubated with ammonium chloride (NH4Cl) in PBS to quench free aldehyde groups. After rinsing with PBS/CM, cells were blocked with immuno-fluorescence (IF) buffer (PBS/CM, 1% BSA, 0.1% Tween 20) for 1 h at room temperature. Primary antibodies were incubated in IF buffer for 1 h at room temperature. After washing with IF buffer (3x, 10 min each), cells were incubated for 30 min at room temperature with fluorochrome-conjugated secondary antibodies diluted in IF buffer. Finally, slides were washed at room temperature with IF buffer (3x, 10 min each), rinsed with PBS/CM and counterstained with DAPI (10 µ/mL), rinsed in PBS/CM and mounted with Prolong Gold Anti-fade Reagent.
Confocal microscopy
Images were collected with a Zeiss LSM510 meta confocal system, using a 405-nm Diode excitation laser with a band pass filter of 420–480-nm, a 488-nm Argon excitation laser with a band pass filter of 505–550-nm, and a 543-nm HeNe excitation laser with a 561–604-nm filter. Images were acquired with a 40x objective.
Immunohistochemistry
Paraffin-embedded sections from human breast cancer samples were immuno-stained as previously described.88 Briefly, sections were deparaffinized, rehydrated and washed in PBS. Antigen retrieval was performed in 10 mM sodium citrate, pH 6.0 for 10 min using a pressure cooker. Then, sections were blocked with 3% hydrogen peroxide for 10 min followed by an incubation using an avidin-biotin blocking kit (Dako) to block endogenous biotin. After incubation with 10% goat serum for 1 h, sections were incubated with primary antibodies overnight at 4°C. Antibody binding was detected using a biotinylated secondary antibody (Vector Labs, Burlingame, CA) followed by strepavidin-HRP (Dako, Carpinteria, CA). Immunoreactivity was revealed using 3, 3′ diaminobenzidine. Negative controls were performed in parallel.
TIGAR recombinant overexpression
TIGAR (EX-W1314-Lv105) and control (EX-Lv105) vectors were purchased from GeneCopoeia and lenti-viruses were prepared according to the manufacturer’s protocol. Virus-containing media were centrifuged, filtered (0.45 μM PES low protein filter) and stored in 1 mL aliquots at -80°C. MCF7 cells (120,000 cells/well) were plated in 12 well dishes in growth media. After 24 h, the media was removed and replaced with 250 μl DMEM, 5% FBS, 150 μl of virus-containing media and 5 μg/ml polybrene. Twenty-four hours post infection, the media containing virus was removed and replaced with growth media. The cells were selected with puromycin (2 μg/ml) for three days after infection. For drug treatments, cells were plated in DMEM 10% FBS. After 24 h, the media was changed to 10% NuSerum and supplemented with tamoxifen, ATO or vehicle alone. Cells were treated for 24 h prior to Annexin V and PI staining.
L-Lactate assay
The L-lactate assay was performed as per manufacturer’s recommendations with minor modifications and as previously described.89 Briefly, Lv-105-(empty vector) or TIGAR-MCF7 cells were seeded (1 × 105 per well) in 12-well plates in 1000 µL of DMEM 10% FBS. The next day, the media was changed to 600 µL of DMEM 10% NuSerum and treated with 10 µM ATO or vehicle alone (sodium hydroxide). After 24 h, the media of each well was collected and spun down to remove debris before measuring lactate concentration using the EnzyChromTM L-Lactate Assay Kit (cat#ECLC-100, BioAssay Systems). After removing the media, the remaining attached cells were harvested to determine their protein content. Finally, the amount of L-lactate in the media was normalized for total cell protein content.
Protein gel blot analysis
Cells were lysed in lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, and 60 mM n-octyl-glucoside), supplemented with protease (Roche Applied Science) and phosphatase inhibitors (Sigma). After rotation at 4°C for 40 min, samples were centrifuged at 10,000 x g for 10 min at 4°C to remove insoluble debris. Protein concentrations were determined using the BCA reagent (Pierce, Rockford, IL). Cell lysates were then separated by SDS-PAGE and transferred to nitrocellulose. Membranes were washed in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20), and blocked with TBST supplemented with 5% nonfat dry milk (Carnation). Then, membranes were incubated with primary antibodies in TBST and 1% bovine serum albumin, washed, and incubated with horseradish peroxidase-conjugated secondary antibodies. To visualize bound antibodies, membranes were incubated with the Supersignal chemiluminescence substrate (Pierce).
Treatment with Doxorubicin and ABT-888
MCF7 cells were plated alone or in coculture with GFP (+) fibroblasts in complete media. Next day, the media was changed to DMEM 10% NuSerum and cells were treated with 50 nM doxorubicin, 30 nM ABT-888 (a PARP inhibitor) or vehicle alone for 24 h. Cell viability was assessed by evaluating the number of GFP (-) cell (MCF7 cells) using flow cytometry.
Acknowledgments
M.P.L. and his laboratory were supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. A.K.W was supported by a Young Investigator Award from Breast Cancer Alliance, Inc. and a Susan G. Komen Career Catalyst Grant. F.S. was supported by grants from the W.W. Smith Charitable Trust, the Breast Cancer Alliance (BCA), and a Research Scholar Grant from the American Cancer Society (ACS). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072, and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.).
This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Dr. Ubaldo E. Martinez-Outschoorn would like to thank Dr. Jose Martinez for his continued help and enthusiastic support.
Glossary
Abbreviations:
- 2-NBDG
NBD-2-deoxy-glucose
- ATO
arsenic trioxide
- CAFs
cancer-associated fibroblasts
- Cav-1
caveolin-1
- ECM
extracellular matrix
- ER
estrogen receptor
- MCT
monocarboxylate transporter
- PFS
progression free survival
- PHD
prolyl hydroxylase domain proteins
- PI
propidium iodide
- PR
progesterone receptor
- ROS
reactive oxygen species
- TIGAR
TP53-induced glycolysis and apoptosis regulator
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/17780
References
- 1.Key TJ, Verkasalo PK, Banks E. Epidemiology of breast cancer. Lancet Oncol. 2001;2:133–40. doi: 10.1016/S1470-2045(00)00254-0. [DOI] [PubMed] [Google Scholar]
- 2.Lacey JV, Jr., Devesa SS, Brinton LA. Recent trends in breast cancer incidence and mortality. Environ Mol Mutagen. 2002;39:82–8. doi: 10.1002/em.10062. [DOI] [PubMed] [Google Scholar]
- 3.Li CI, Daling JR, Malone KE. Incidence of invasive breast cancer by hormone receptor status from 1992 to 1998. J Clin Oncol. 2003;21:28–34. doi: 10.1200/JCO.2003.03.088. [DOI] [PubMed] [Google Scholar]
- 4.Ravdin PM, Cronin KA, Howlader N, Berg CD, Chlebowski RT, Feuer EJ, et al. The decrease in breast-cancer incidence in 2003 in the United States. N Engl J Med. 2007;356:1670–4. doi: 10.1056/NEJMsr070105. [DOI] [PubMed] [Google Scholar]
- 5.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 6.Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M, et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11:1135–41. doi: 10.1016/S1470-2045(10)70257-6. [DOI] [PubMed] [Google Scholar]
- 7.Early Breast Cancer Trialists' Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 8.Higgins MJ, Rae JM, Flockhart DA, Hayes DF, Stearns V. Pharmacogenetics of tamoxifen: who should undergo CYP2D6 genetic testing? J Natl Compr Canc Netw. 2009;7:203–13. doi: 10.6004/jnccn.2009.0014. [DOI] [PubMed] [Google Scholar]
- 9.Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, et al. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003;144:1032–44. doi: 10.1210/en.2002-220620. [DOI] [PubMed] [Google Scholar]
- 10.Arpino G, Wiechmann L, Osborne CK, Schiff R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev. 2008;29:217–33. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–33. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 12.Nicholson RI, Hutcheson IR, Knowlden JM, Jones HE, Harper ME, Jordan N, et al. Nonendocrine pathways and endocrine resistance: observations with antiestrogens and signal transduction inhibitors in combination. Clin Cancer Res. 2004;10:346S–54S. doi: 10.1158/1078-0432.CCR-031206. [DOI] [PubMed] [Google Scholar]
- 13.Dorssers LC, van Agthoven T, Dekker A, van Agthoven TL, Kok EM. Induction of antiestrogen resistance in human breast cancer cells by random insertional mutagenesis using defective retroviruses: identification of bcar-1, a common integration site. Mol Endocrinol. 1993;7:870–8. doi: 10.1210/me.7.7.870. [DOI] [PubMed] [Google Scholar]
- 14.Brinkman A, de Jong D, Tuinman S, Azaouagh N, van Agthoven T, Dorssers LC. The substrate domain of BCAR1 is essential for anti-estrogen-resistant proliferation of human breast cancer cells. Breast Cancer Res Treat. 2010;120:401–8. doi: 10.1007/s10549-009-0403-4. [DOI] [PubMed] [Google Scholar]
- 15.Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat. 2006;97:263–74. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- 16.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–61. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 17.Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, et al. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663–6. doi: 10.1038/nature07483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer. 2007;7:59. doi: 10.1186/1471-2407-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23:2469–76. doi: 10.1200/JCO.2005.01.172. [DOI] [PubMed] [Google Scholar]
- 20.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406–13. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller TW, Balko JM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, et al. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin Cancer Res. 2011;17:2024–34. doi: 10.1158/1078-0432.CCR-10-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nehra R, Riggins RB, Shajahan AN, Zwart A, Crawford AC, Clarke R. BCL2 and CASP8 regulation by NF-kappaB differentially affect mitochondrial function and cell fate in antiestrogen-sensitive and -resistant breast cancer cells. FASEB J. 2010;24:2040–55. doi: 10.1096/fj.09-138305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Planas-Silva MD, Bruggeman RD, Grenko RT, Smith JS. Overexpression of c-Myc and Bcl-2 during progression and distant metastasis of hormone-treated breast cancer. Exp Mol Pathol. 2007;82:85–90. doi: 10.1016/j.yexmp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Prall OW, Rogan EM, Musgrove EA, Watts CK, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol Cell Biol. 1998;18:4499–508. doi: 10.1128/mcb.18.8.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stendahl M, Kronblad A, Ryden L, Emdin S, Bengtsson NO, Landberg G. Cyclin D1 overexpression is a negative predictive factor for tamoxifen response in postmenopausal breast cancer patients. Br J Cancer. 2004;90:1942–8. doi: 10.1038/sj.bjc.6601831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhillon NK, Mudryj M. Ectopic expression of cyclin E in estrogen responsive cells abrogates antiestrogen mediated growth arrest. Oncogene. 2002;21:4626–34. doi: 10.1038/sj.onc.1205576. [DOI] [PubMed] [Google Scholar]
- 27.Carroll JS, Lynch DK, Swarbrick A, Renoir JM, Sarcevic B, Daly RJ, et al. p27(Kip1) induces quiescence and growth factor insensitivity in tamoxifen-treated breast cancer cells. Cancer Res. 2003;63:4322–6. [PubMed] [Google Scholar]
- 28.Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci USA. 2000;97:9042–6. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts CG, Millar EK, O'Toole SA, McNeil CM, Lehrbach GM, Pinese M, et al. Identification of PUMA as an estrogen target gene that mediates the apoptotic response to tamoxifen in human breast cancer cells and predicts patient outcome and tamoxifen responsiveness in breast cancer. Oncogene. 2011;30:3186–97. doi: 10.1038/onc.2011.36. [DOI] [PubMed] [Google Scholar]
- 30.Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27:3423–9. doi: 10.1200/JCO.2008.17.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer. 2009;9:576–86. doi: 10.1038/nrc2683. [DOI] [PubMed] [Google Scholar]
- 32.Nazarewicz RR, Zenebe WJ, Parihar A, Larson SK, Alidema E, Choi J, et al. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007;67:1282–90. doi: 10.1158/0008-5472.CAN-06-3099. [DOI] [PubMed] [Google Scholar]
- 33.Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst. 2000;92:1926–34. doi: 10.1093/jnci/92.23.1926. [DOI] [PubMed] [Google Scholar]
- 34.Seth P, Krop I, Porter D, Polyak K. Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression) Oncogene. 2002;21:836–43. doi: 10.1038/sj.onc.1205113. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, et al. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell. 2009;16:413–24. doi: 10.1016/j.ccr.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woodward TL, Mienaltowski AS, Modi RR, Bennett JM, Haslam SZ. Fibronectin and the alpha(5)beta(1) integrin are under developmental and ovarian steroid regulation in the normal mouse mammary gland. Endocrinology. 2001;142:3214–22. doi: 10.1210/en.142.7.3214. [DOI] [PubMed] [Google Scholar]
- 37.Schedin P, Mitrenga T, McDaniel S, Kaeck M. Mammary ECM composition and function are altered by reproductive state. Mol Carcinog. 2004;41:207–20. doi: 10.1002/mc.20058. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi Y, Hayashi S. Estrogen-related cancer microenvironment of breast carcinoma. Endocr J. 2009;56:1–7. doi: 10.1507/endocrj.K08E-099. [DOI] [PubMed] [Google Scholar]
- 39.Giulianelli S, Cerliani JP, Lamb CA, Fabris VT, Bottino MC, Gorostiaga MA, et al. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int J Cancer. 2008;123:2518–31. doi: 10.1002/ijc.23802. [DOI] [PubMed] [Google Scholar]
- 40.Shekhar MP, Santner S, Carolin KA, Tait L. Direct involvement of breast tumor fibroblasts in the modulation of tamoxifen sensitivity. Am J Pathol. 2007;170:1546–60. doi: 10.2353/ajpath.2007.061004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witkiewicz AK, Casimiro MC, Dasgupta A, Mercier I, Wang C, Bonuccelli G, et al. Towards a new “stromal-based” classification system for human breast cancer prognosis and therapy. Cell Cycle. 2009;8:1654–8. doi: 10.4161/cc.8.11.8544. [DOI] [PubMed] [Google Scholar]
- 42.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–43. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koo JS, Park S, Kim SI, Lee S, Park BW. The impact of caveolin protein expression in tumor stroma on prognosis of breast cancer. Tumour Biol. 2011;32:787–99. doi: 10.1007/s13277-011-0181-6. [DOI] [PubMed] [Google Scholar]
- 44.Qian N, Ueno T, Kawaguchi-Sakita N, Kawashima M, Yoshida N, Mikami Y, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci. 2011;102:1590–6. doi: 10.1111/j.1349-7006.2011.01985.x. [DOI] [PubMed] [Google Scholar]
- 45.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 47.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005;280:25864–70. doi: 10.1074/jbc.M502456200. [DOI] [PubMed] [Google Scholar]
- 52.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11:554–65. doi: 10.1016/j.cmet.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 55.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 57.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–73. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 59.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–12. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 60.Mitchell RA, Chang BF, Huang CH, DeMaster EG. Inhibition of mitochondrial energy-linked functions by arsenate. Evidence for a nonhydrolytic mode of inhibitor action. Biochemistry. 1971;10:2049–54. doi: 10.1021/bi00787a013. [DOI] [PubMed] [Google Scholar]
- 61.Miller WH, Jr., Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–903. [PubMed] [Google Scholar]
- 62.Flora SJ. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2011;51:257–81. doi: 10.1016/j.freeradbiomed.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 63.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 64.Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–26. doi: 10.1038/emboj.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howell A. Pure oestrogen antagonists for the treatment of advanced breast cancer. Endocr Relat Cancer. 2006;13:689–706. doi: 10.1677/erc.1.00846. [DOI] [PubMed] [Google Scholar]
- 66.Howell A, Robertson JF, Abram P, Lichinitser MR, Elledge R, Bajetta E, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol. 2004;22:1605–13. doi: 10.1200/JCO.2004.02.112. [DOI] [PubMed] [Google Scholar]
- 67.Chen SJ, Zhou GB, Zhang XW, Mao JH, de The H, Chen Z. From an old remedy to a magic bullet: molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood. 2011;117:6425–37. doi: 10.1182/blood-2010-11-283598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pontiggia O, Rodriguez V, Fabris V, Raffo D, Bumaschny V, Fiszman G, et al. Establishment of an in vitro estrogen-dependent mouse mammary tumor model: a new tool to understand estrogen responsiveness and development of tamoxifen resistance in the context of stromal-epithelial interactions. Breast Cancer Res Treat. 2009;116:247–55. doi: 10.1007/s10549-008-0113-3. [DOI] [PubMed] [Google Scholar]
- 69.Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008;68:5198–205. doi: 10.1158/0008-5472.CAN-08-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Müerköster S, Wegehenkel K, Arlt A, Witt M, Sipos B, Kruse ML, et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004;64:1331–7. doi: 10.1158/0008-5472.CAN-03-1860. [DOI] [PubMed] [Google Scholar]
- 71.Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE. 2009;4:e7965. doi: 10.1371/journal.pone.0007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson JA, Pollak M. Insulin, glucose and the increased risk of cancer in patients with type 2 diabetes. Diabetologia. 2010;53:2086–8. doi: 10.1007/s00125-010-1855-0. [DOI] [PubMed] [Google Scholar]
- 73.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes Care. 2010;33:1674–85. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bodmer M, Meier C, Krahenbuhl S, Jick SS, Meier CR. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care. 2010;33:1304–8. doi: 10.2337/dc09-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–5. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bosco JL, Antonsen S, Sorensen HT, Pedersen L, Lash TL. Metformin and incident breast cancer among diabetic women: a population-based case-control study in Denmark. Cancer Epidemiol Biomarkers Prev. 2011;20:101–11. doi: 10.1158/1055-9965.EPI-10-0817. [DOI] [PubMed] [Google Scholar]
- 77.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hosono K, Endo H, Takahashi H, Sugiyama M, Sakai E, Uchiyama T, et al. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res (Phila) 3:1077–83. doi: 10.1158/1940-6207.CAPR-10-0186. [DOI] [PubMed] [Google Scholar]
- 79.Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev Res (Phila) 3:1060–5. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bolen S, Feldman L, Vassy J, Wilson L, Yeh HC, Marinopoulos S, et al. Systematic review: comparative effectiveness and safety of oral medications for type 2 diabetes mellitus. Ann Intern Med. 2007;147:386–99. doi: 10.7326/0003-4819-147-6-200709180-00178. [DOI] [PubMed] [Google Scholar]
- 81.Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ. 2003;327:951–3. doi: 10.1136/bmj.327.7421.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shen ZX, Shi ZZ, Fang J, Gu BW, Li JM, Zhu YM, et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2004;101:5328–35. doi: 10.1073/pnas.0400053101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ravandi F, Estey E, Jones D, Faderl S, O'Brien S, Fiorentino J, et al. Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol. 2009;27:504–10. doi: 10.1200/JCO.2008.18.6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harris LN, Broadwater G, Abu-Khalaf M, Cowan D, Thor AD, Budman D, et al. Topoisomerase II{alpha} amplification does not predict benefit from dose-intense cyclophosphamide, doxorubicin, and fluorouracil therapy in HER2-amplified early breast cancer: results of CALGB 8541/150013. J Clin Oncol. 2009;27:3430–6. doi: 10.1200/JCO.2008.18.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 86.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 87.Sotgia F, Del Galdo F, Casimiro MC, Bonuccelli G, Mercier I, Whitaker-Menezes D, et al. Caveolin-1−/− null mammary stromal fibroblasts share characteristics with human breast cancer-associated fibroblasts. Am J Pathol. 2009;174:746–61. doi: 10.2353/ajpath.2009.080658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sotgia F, Casimiro MC, Bonuccelli G, Liu M, Whitaker-Menezes D, Er O, et al. Loss of caveolin-3 induces a lactogenic microenvironment that is protective against mammary tumor formation. Am J Pathol. 2009;174:613–29. doi: 10.2353/ajpath.2009.080653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]